- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Classification of a Massive Number of Viral Genomes and Estimation of Time of Most Recent Common Ancestor (tMRCA) of SARS-CoV-2 Using Phylodynamic Analsysis
(*contributed equally to this work) Published: Vol 14, Iss 6, Mar 20, 2024 DOI: 10.21769/BioProtoc.4955 Views: 2654
Reviewed by: Migla MiskinytePooja VermaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2023
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Estimating the time of most recent common ancestor (tMRCA) is important to trace the origin of pathogenic viruses. This analysis is based on the genetic diversity accumulated in a certain time period. There have been thousands of mutant sites occurring in the genomes of SARS-CoV-2 since the COVID-19 pandemic started; six highly linked mutation sites occurred early before the start of the pandemic and can be used to classify the genomes into three main haplotypes. Tracing the origin of those three haplotypes may help to understand the origin of SARS-CoV-2. In this article, we present a complete protocol for the classification of SARS-CoV-2 genomes and calculating tMRCA using Bayesian phylodynamic method. This protocol may also be used in the analysis of other viral genomes.
Key features
• Filtering and alignment of a massive number of viral genomes using custom scripts and ViralMSA.
• Classification of genomes based on highly linked sites using custom scripts.
• Phylodynamic analysis of viral genomes using Bayesian evolutionary analysis sampling trees (BEAST).
• Visualization of posterior distribution of tMRCA using Tracer.v1.7.2.
• Optimized for the SARS-CoV-2.
Graphical overview
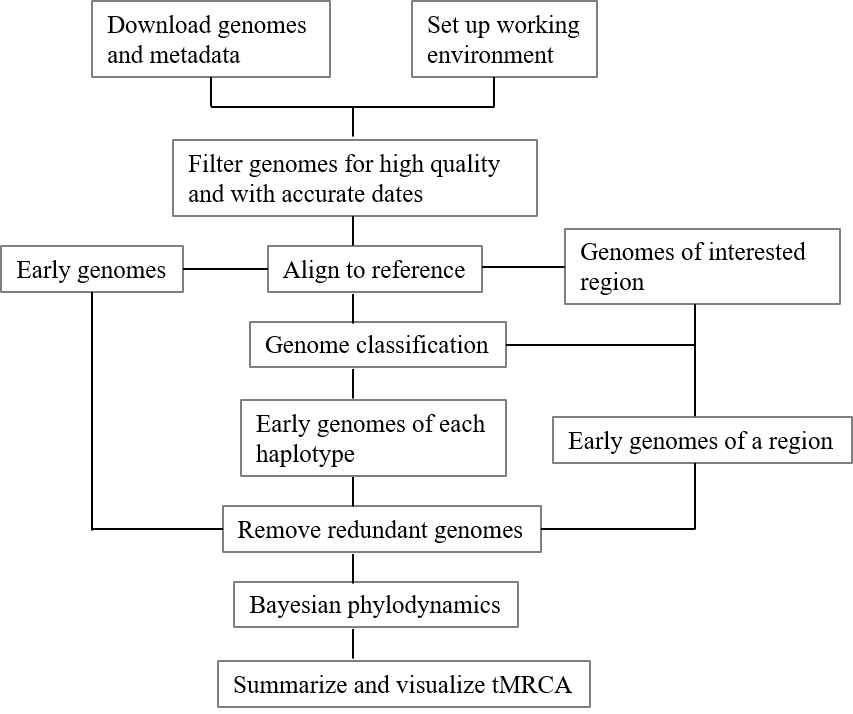
Graphical workflow of time of most recent common ancestor (tMRCA) estimation process
Background
Revealing the origins of pathogenic viruses, crucial for cutting them off from the root and preventing future spillover, requires long-term hard work from scientists all around the world [1]. Although some infectious pathogens can be traced back decades, the debate on their origin continues. For example, AIDS was officially reported on June 5, 1981, by the Centers for Disease Control and Prevention of the USA. Five years later, HIV infection was detected in a human serum sample collected in Léopoldville in early 1959 [2]. Bayesian phylodynamic analyses using recovered viral gene sequences from decades-old paraffin-embedded tissues traced the most recent common ancestor (MRCA) of the M group of HIV back to approximately 1908 (CI 1884–1924), suggesting that HIV has been circulating in the human population for approximately 100 years [3]. MERS-CoV is another example, as it was first reported in a Saudi Arabian man in 2012 [4]. Bats are thought to be the reservoir hosts of MERS-CoV, and dromedary camels are considered to be the major intermediate host [5]; however, the transmission route from animals to humans is not well understood. Researchers tested 189 camel serum samples from 1983 to 1997 and found that 81% had neutralizing antibodies against MERS-CoV, suggesting long-term virus circulation in these animals [6]. Similarly, COVID-19 was first reported on December 27, 2019, in Wuhan, China [7,8], and the Huanan seafood market was suspected to be the place of origin [9]; however, disputes remain. Pekar and colleagues explored the evolutionary dynamics of the first wave of SARS-CoV-2 infections in China using a strict clock Bayesian phylodynamic analysis but failed to capture the index case [10], probably because the redundant sequences were not removed, which usually influences the accuracy of time of MRCA (tMRCA) estimation, as indicated in two recent tMRCA analysis [11,12]. Genome classification plays a critical role in tracing the origin of pathogenic viruses [3,12]. We have previously classified SARS-CoV-2 genomes based on two amino acids, Spike-614 and Orf8-84, and revealed 16 haplotypes. From those, three major haplotypes were found to separately drive the development of the pandemic in China and the world. However, genome classification based on amino acid mutations did not rule out recombination and reverse mutations. In this paper, we provide detailed protocols to filter and classify the massive number of viral genomes according to six highly linked mutations that happened in the early phase of the epidemic by custom scripts and common programs and estimate tMRCA of non-redundant genome subpools.
Materials and reagents
SARS-CoV-2 genome sequences were obtained from the GISAID database [13] on May 1, 2022. Genomes that were collected from hosts other than humans and/or had a length of less than 2,900 nucleotides and more than 0.05% of unknown nucleotides were filtered out. More than 5 million genomes were retained for further analysis.
Equipment
Bioinformatic platform (CentOS, 7.2.1511)
Windows desktop computers (v11, 6/6/2021)
Software and datasets
Perl (v5.34.0, 20/5/2021)
Python (v3.9.0, 5/10/2020)
Gcc (v8.4.1, 28/9/2020)
SeqKit (v2.3.0, 12/8/2022)
minimap2 (v2.24, 26/12/2021)
ViralMSA (v3, 29/6/2007)
cd-hit (v4.8.1, 1/9/2018)
ALTER (v1.3.4, 30/10/2016)
BEAST (v1.10.4, 9/11/2018)
Jdk (v17_windows-x64_bin, 16/6/2021)
Jre (8u341-windows-x64, 5/6/2021)
Tracer (v1.7.2, 5/1/2018)
Global Initiative of Sharing All Influenza Data (GISAID) (https://gisaid.org/) (Access date, 1/5/2022)
All personalized scripts have been deposited in GitHub: https://github.com/XiaowenH/SARS-CoV-2-Classification (Access date, 5/9/2023). Figure 1 is a screenshot of the files in GitHub.
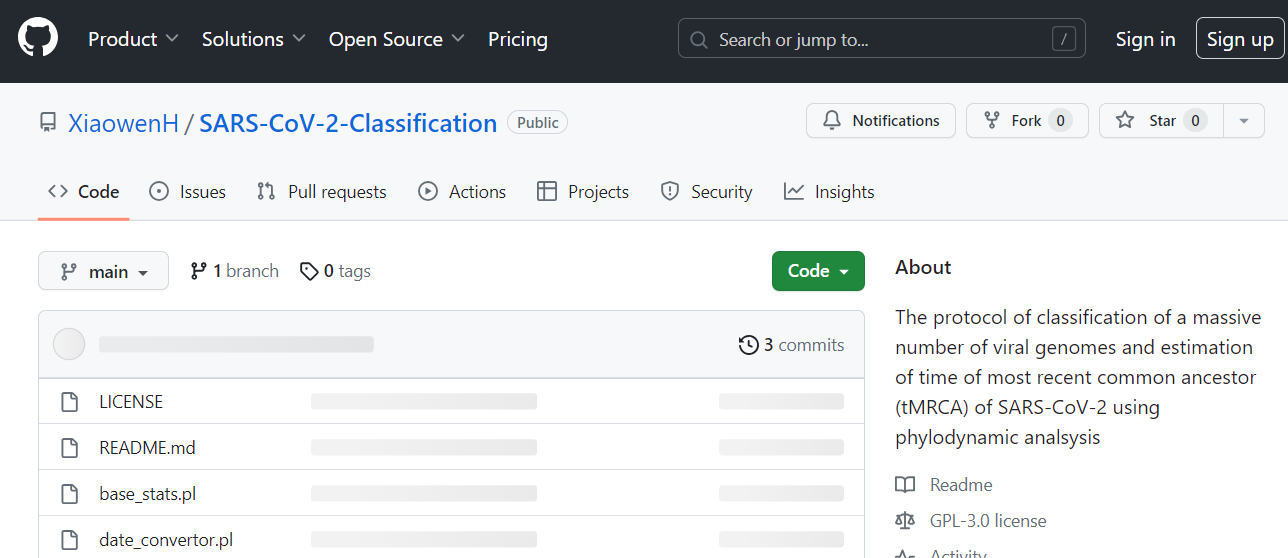
Figure 1. Screenshot of the personalized scripts deposited in GitHub
Procedure
Set up the working environment
Create a Python environment of conda.
$ wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
$ bash Miniconda3-latest-Linux-x86_64.sh -b -p ~/miniconda
$ export PATH="/$HOME/miniconda/bin:$PATH"
$ conda create -n myenv python=3.9
$ conda activate myenv
#Please note that "$HOME" corresponds to the PATH you want to install in the conda environment (the same hereafter).
Install the following software:
Install Seqkit in a Linux platform.
$ conda install -c bioconda seqkit
Install minimap2 in a Linux platform.
$ git clone https://github.com/lh3/minimap2
$ cd minimap2 && make
Install ViralMSA [14] in a Linux platform.
$ wget "https://raw.githubusercontent.com/niemasd/ViralMSA/master/ViralMSA.py"
$ chmod a+x ViralMSA.py
$ sudo mv ViralMSA.py /usr/local/bin/ViralMSA.py
Install CD-HIT in a Linux platform.
$ git clone https://github.com/weizhongli/cdhit.git
$ cd cd-hit
$ make
$ cd cd-hit-auxtools
$ make
Install Alter in a Linux platform.
$ git clone https://github.com/sing-group/ALTER.git
$ cd ALTER
$ /$HOME/apache-maven-3.8.4/bin/mvn
Install BEAST [15] in a Windows platform.
# Download BEAST.v1.10.4 at
http://beast.community/programs
Install BEAGLE in a Windows platform.
# Download BEAGLE at
https://github.com/beagle-dev/beagle-lib
Install Tracer v1.7.2 in a Windows platform.
# Download BEAGLE at
http://beast.community/tracer
Download the viral genomes and metadata
Download SARS-CoV-2 genomes and meta.tsv files from the GISAID database [16] after login (https://gisaid.org/). Note that the content in each column in the meta.tsv may change (e.g., the accession numbers were put in column 3 in the metadata downloaded on May 1, 2022, but in column 5 in the metadata downloaded on May 1, 2023).
Unpack the files.
$ xz -d sequences_fasta_2022_04_29.tar.xz
$ tar -xvf sequences_fasta_2022_04_29.tar
$ xz -d metadata_tsv_2022_04_29.tar.xz
$ tar -xvf metadata_tsv_2022_04_29.tar
Rename the taxa in the Fasta file of the genomes by accession numbers (e.g., EPI_ISL_4405694) using a custom script
$ perl rename_fasta_taxa_to_tsv_acc_column3.pl -t meta.tsv -f sequence.fasta
Fetch GISAID reference sequence
Create a file named “accession.txt,” paste in EPI_ISL_402124, and save.
$ echo ‘EPI_ISL_402124’ >accession.txt
# EPI_ISL_402124 is the reference sequence used by the GISAID database and in many researches.
Fetch the reference sequence from total sequence file.
$ cat sequence.fasta | /$HOME/seqkit grep -f accession.txt -t dna -j 10 -o reference_wiv04.fasta
Retrieve genomes with complete, high coverage sequences and accurate dates and sampled from human hosts
The accession number, host, completeness, and coverage of the genomes are located in columns 3, 8, 18, and 19, respectively, in the metadata of April 29, 2022. The sample collection date is located in column 4. The column number may be different in the metadata downloaded at a different day.
Filter metadata.tsv for accessions with complete genomes, with high coverage, and from human hosts.
$ awk -F '\t' '{if($18 == "True" && $19 == "True" && $8 == "Human") print}' metadata.tsv >global_2022_04_29_human_complete_metadata.tsv
Filter metadata.tsv for accessions with accurate sample collection date using a custom script: dates_filter.pl.
$ perl dates_filter.pl -t global_2022_04_29_human_complete_metadata.tsv -o global_2022_04_29_human_complete_dates_metadata.tsv
Print selected accession numbers to a file from metadata.
$ awk -F '\t' '{print $3}' global_2022_04_29_human_complete_dates_metadata.tsv > global_2022_04_29_human_complete_dates_accessions.txt
Retrieve genome sequences with complete and high coverage and accurate dates.
$ cat sequence.fasta | /$HOME/seqkit grep -f global_2022_04_29_human_complete_dates_accessions.txt -t dna -j 10 -o global_2022_04_29_human_complete_dates_genome.fasta
Align the genome sequences
$ /$HOME/ViralMSA-master/ViralMSA.py -e [your email] -s global_2022_04_29_human_complete_dates_genome.fasta -o output -r reference_wiv04.fasta --omit_ref
All genome classification
Retrieve the six highly linked sites using the custom script fetch_nucleotides_from_alignments.pl (Figure 2).
$ perl fetch_nucleotides_from_alignments.pl -f global_2022_04_29_human_complete_dates_genome.fasta.aln
-r 241-241,3037-3037,8782-8782,14408-14408,23403-23403,28144-28144 -o global_2022_04_29_human_complete_dates_genome.fasta_six_sites.txt

Figure 2. Output format of the six linked sites in the genomes. The six nucleotides of sites 241, 3037, 8782, 14408, 23403, and 28144 were retrieved from SARS-CoV-2 genomes aligned to reference genome wiv04 EPI_ISL_402124.Classify the genomes into haplotypes by the six linked mutation sites (Figure 3).
$ cat global_2022_04_29_human_complete_dates_genome.fasta_six_sites.txt | /$HOME/seqkit grep -s -i –p CCTCAC > global_2022_04_29_human_complete_dates_DS.txt
$ cat global_2022_04_29_human_complete_dates_genome.fasta_six_sites.txt | /$HOME/seqkit grep -s -i -p CCCCAT > global_2022_04_29_human_complete_dates_DL.txt
$ cat global_2022_04_29_human_complete_dates_genome.fasta_six_sites.txt | /$HOME/seqkit grep -s -i -p TTCTGT > global_2022_04_29_human_complete_dates_GL.txt
$ /$HOME/seqkit stats global_2022_04_29_human_complete_dates_DS.txt >haplotype.statistics.txt
$ /$HOME/seqkit stats global_2022_04_29_human_complete_dates_DL.txt >>haplotype.statistics.txt
$ /$HOME/seqkit stats global_2022_04_29_human_complete_dates_GL.txt >>haplotype.statistics.txt
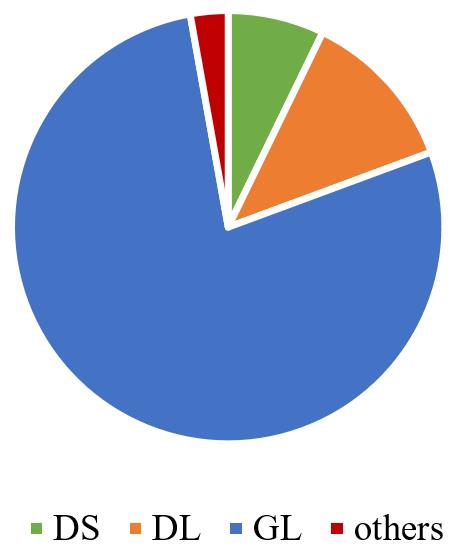
Figure 3. Pie chart of haplotypes in global SARS-CoV-2 genomes. DS (CCTCAC), DL (CCCCAT), and GL (TTCTGT) are the three main haplotypes classified by the six linked sites.Fetch accession numbers of each haplotype.
$ /$HOME/seqkit seq global_2022_04_29_human_complete_dates_DS.txt -n > global_DS.accessions.txt
$ /$HOME/seqkit seq global_2022_04_29_human_complete_dates_DL.txt -n > global_DL.accessions.txt
$ /$HOME/seqkit seq global_2022_04_29_human_complete_dates_GL.txt -n > global_GL.accessions.txt
Retrieve aligned genome sequences of each haplotype.
$ cat global_2022_04_29_human_complete_dates_genome.fasta.aln | /$HOME/seqkit grep -f global_DS.accessions.txt -t dna -j 10 -o global_DS.aln
$ cat global_2022_04_29_human_complete_dates_genome.fasta.aln | /$HOME/seqkit grep -f global_DL.accessions.txt -t dna -j 10 -o global_DL.aln
$ cat global_2022_04_29_human_complete_dates_genome.fasta.aln | /$HOME/seqkit grep -f global_GL.accessions.txt -t dna -j 10 -o global_GL.aln
Classification of early genomes collected in the early phase of the pandemic (from beginning to end of April 2020)
The sample collection date is located in column 4 in the metadata of April 29, 2022.
Retrieve accession numbers of early genomes.
$ awk -F '\t' '{if($4~/2019-12/) print}' global_2022_04_29_human_complete_dates_metadata.tsv > global_early_meta.tsv
$ awk -F '\t' '{if($4~/2020-01/) print}' global_2022_04_29_human_complete_dates_metadata.tsv >> global_early_meta.tsv
$ awk -F '\t' '{if($4~/2020-02/) print}' global_2022_04_29_human_complete_dates_metadata.tsv >> global_early_meta.tsv
$ awk -F '\t' '{if($4~/2020-03/) print}' global_2022_04_29_human_complete_dates_metadata.tsv >> global_early_meta.tsv
$ awk -F '\t' '{if($4~/2020-04/) print}' global_2022_04_29_human_complete_dates_metadata.tsv >> global_early_meta.tsv
$ awk -F '\t' '{print $3}' global_early_meta.tsv >global_early_accessions.txt
Retrieve the aligned sequences of the early genomes.
$ cat global_2022_04_29_human_complete_dates_genome.fasta.aln | /$HOME/seqkit grep -f global_early_accessions.txt -t dna -j 10 -o global_early_all_haplotype.fasta.aln
Retrieve the six highly linked sites using the custom script fetch_nucleotides_from_alignments.pl.
$ perl fetch_nucleotides_from_alignments.pl -f global_early_all_haplotype.fasta.aln
-r 241-241,3037-3037,14408-14408,23403-23403,28144-28144 -o global_early.aln_six_sites.txt
Classify the genomes.
$ cat global_early.aln_six_sites.txt | /$HOME/seqkit grep -s -i -p CCTCAC > global_early_DS.txt
$ cat global_early.aln_six_sites.txt | /$HOME/seqkit grep -s -i -p CCCCAT > global_early_DL.txt
$ cat global_early.aln_six_sites.txt | /$HOME/seqkit grep -s -i -p TTCTGT > global_early_GL.txt
Fetch accession numbers of each haplotype.
$ /$HOME/seqkit seq global_early_DS.txt -n > global_early_DS.accessions.txt
$ /$HOME/seqkit seq global_early_DL.txt -n > global_early_DL.accessions.txt
$ /$HOME/seqkit seq global_early_GL.txt -n > global_early_GL.accessions.txt
Retrieve aligned genome sequences of each haplotype.
$ cat global_early_all_haplotype.fasta.aln | /$HOME/seqkit grep -f global_ early_DS.accessions.txt -t dna -j 10 -o global_ early_DS.aln
$ cat global_early_all_haplotype.fasta.aln | /$HOME/seqkit grep -f global_ early_DL.accessions.txt -t dna -j 10 -o global_ early_DL.aln
$ cat global_early_all_haplotype.fasta.aln | /$HOME/seqkit grep -f global_ early_GL.accessions.txt -t dna -j 10 -o global_ early_GL.aln
Bayesian phylodynamic analysis using the early genomes of three haplotypes as examples
Filter out genomes with unknown higher than 0.05%.
Filter out genomes of DS (CCTCAC) haplotypes with unknown nucleotides higher than 0.05%.
# Calculate nucleotide composition of each sequence in each haplotype
$ perl base_stats.pl global_early_DS.aln
# Pick up accessions with unknowns < 0.05%
$ awk -F '\t' '{if($6 < 0.0005) print$1}' global_early_DS.aln.stats > global_early_DS.aln_0.0005N_ID.txt
# Fetch aligned genome sequences with unknowns <0.05%
$ cat global_early_DS.aln |/$HOME/seqkit grep -f global_early_DS.aln_0.0005N_ID.txt -t dna -j 10 -o global_early_DS_0.0005N.aln
Filter out genomes of DL (CCCCAT) haplotypes with unknown nucleotides higher than 0.05%.
# Calculate nucleotide composition of each sequence in each haplotype
$ perl base_stats.pl global_early_DL.aln
# Pick up accessions with unknowns < 0.05%
$ awk -F '\t' '{if($6 < 0.0005) print$1}' global_early_DL.aln.stats > global_early_DL.aln_0.0005N_ID.txt
# Fetch aligned genome sequences with unknowns <0.05%
$ cat global_early_DL.aln |/$HOME/seqkit grep -f global_early_DL.aln_0.0005N_ID.txt -t dna -j 10 -o global_early_DL_0.0005N.aln
Filter out genomes of GL (TTCTGT) haplotypes with unknown nucleotides higher than 0.05%.
# Calculate nucleotide composition of each sequence in each haplotype.
$ perl base_stats.pl global_early_GL.aln
# Pick up accessions with unknowns < 0.05%
$ awk -F '\t' '{if($6 < 0.0005) print$1}' global_early_GL.aln.stats > global_early_GL.aln_0.0005N_ID.txt
# Fetch aligned genome sequences with unknowns <0.05%
$ cat global_early_GL.aln | /$HOME/seqkit grep -f global_early_GL.aln_0.0005N_ID.txt -t dna -j 10 -o global_early_GL_0.0005N.aln
Remove redundant genomes with a threshold of 0.9997.
$ /$HOME/cd-hit-v4.8.1-2019-0228/cd-hit-est -i global_early_DS_0.0005N.aln -o global_early_DS_0.0005N_CDHit0.9997.fas -M 2500 -c 0.9997 -aL 1 -aS 1 -d 0
$ /$HOME/cd-hit-v4.8.1-2019-0228/cd-hit-est -i global_early_DL_0.0005N.aln -o global_early_DL_0.0005N_CDHit0.9997.fas -M 2500 -c 0.9997 -aL 1 -aS 1 -d 0
$ /$HOME/cd-hit-v4.8.1-2019-0228/cd-hit-est -i global_early_GL_0.0005N.aln -o global_early_GL_0.0005N_CDHit0.9997.fas -M 2500 -c 0.9997 -aL 1 -aS 1 -d 0
Change format to NEX.
$ java -jar /$HOME/ALTER/alter-lib/target/ALTER-1.3.4-jar-with-dependencies.jar -i global_early_DS_0.0005N_CDHit0.9997.fas -ia -o global_early_DS_0.0005N_CDHit0.9997.fas.nex -of NEXUS -oo Windows -op MrBayes
$ java -jar /$HOME/ALTER/alter-lib/target/ALTER-1.3.4-jar-with-dependencies.jar -i global_early_DL_0.0005N_CDHit0.9997.fas -ia -o global_early_DL_0.0005N_CDHit0.9997.fas.nex -of NEXUS -oo Windows -op MrBayes
$ java -jar /$HOME/ALTER/alter-lib/target/ALTER-1.3.4-jar-with-dependencies.jar -i global_early_GL_0.0005N_CDHit0.9997.fas -ia -o global_early_GL_0.0005N_CDHit0.9997.fas.nex -of NEXUS -oo Windows -op MrBayes
Fetch dates for accessions from metadata.
$ awk -F "\t" -v OFS="\t" '{print $3,$4}' global_early_meta.tsv > global_early_dates.txt
Convert dates to decimal dates using a custom script date_convertor.pl.
$ perl date_convertor.pl global_early_dates.txt > global_early_decimal_dates
Create BEAST XML file in Windows platform.
# Open BEAUti-v1.10.4 by double-click its the icon
# Load genome alignment file in NEX format
# Import dates (sample collection dates) in the file global_early_decimal_dates
# Set options for analysis, e.g. set ‘Clocks’ to either strict, or relaxed, set ‘Tree Prior’ to either ‘Bayesian Skyline’ or ‘Bayesian SkyGrid’, set MCMC ‘Length of Chain’ to 1,200 million generations. The run may be stopped when the Explained Sum of Squares (ESS) of all parameters are significant (>200). When the parameters are all set, click ‘Generate BEAST file’.
Run BEAST.
# Open BEAST v1.10.4 by double-click the icon
# Load BEAST XML file, click ‘Run’. The run may take a few days until all ESSs are significant.
Visualize tMRCA by Tracer (Figure 4).
# Open Tracer v1.7.2 by double click the icon
# Import the log file created by BEAST. Posterior distribution of tMRCA can be shown by clicking ‘age(root)’ and ‘Marginal Density’. The mean tMRCA and 95% HPD interval are provided in ‘Estimates’.
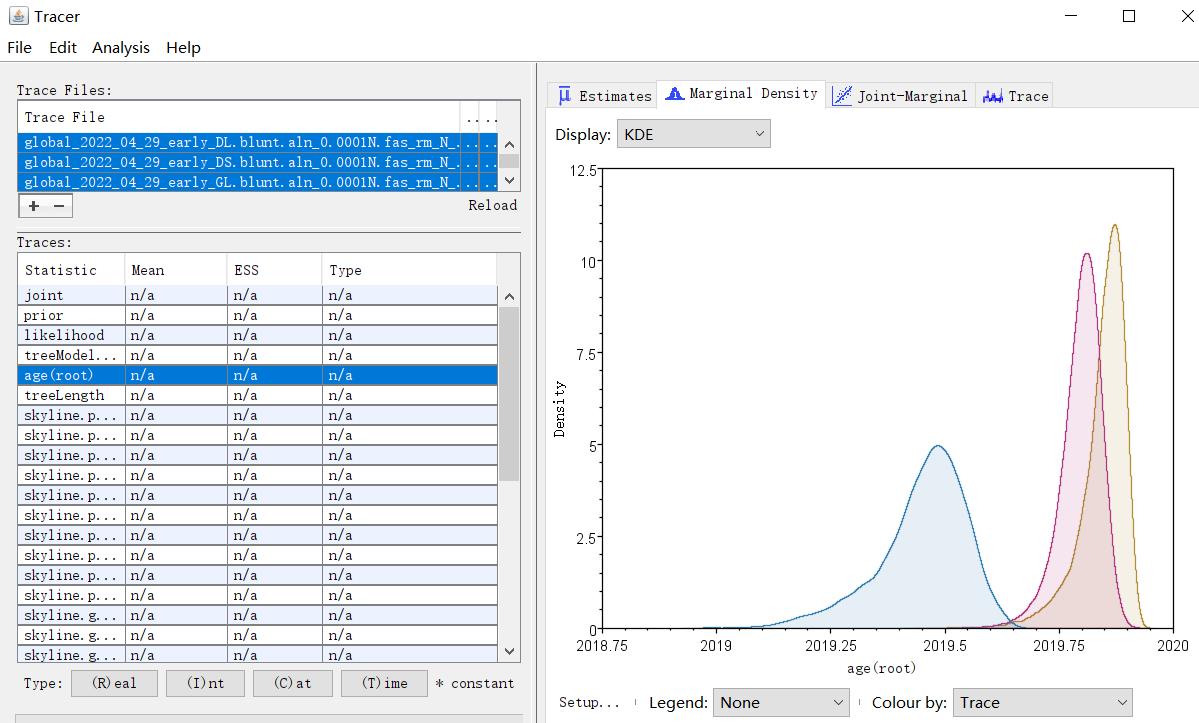
Figure 4. Screenshot of time of most recent common ancestor (tMRCA) estimation of three main haplotypes of the early SARS-CoV-2 genomes as summarized with Tracer. The dates are shown in decimal.
Validation of protocol
This protocol or parts of it has been used and validated in the following research articles:
Hu et al. [17]. Genome characterization based on the Spike-614 and NS8-84 loci of SARS-CoV-2 reveals two major possible onsets of the COVID-19 pandemic. PLoS ONE 18(6): e0279221. https://doi.org/10.1371/journal.pone.0279221 (Figure 4, panel 1; Figure 6).
Guan et al. [12]. Genome analysis reveals much earlier separation and parallel evolution of major haplotypes of SARS-CoV-2 than its occurrence in China. Science in One Health 2(2023), https://doi.org/10.1016/j.soh.2023.100041 (Figure 1; Figure 3; Figure 4).
Acknowledgments
We gratefully acknowledge all data contributors, including the authors and their originating laboratories responsible for obtaining the specimens, and their submitting laboratories for generating the genetic sequence and metadata, and sharing via the GISAID Initiative, on which this research is based. This research was supported by grants from National Key R&D Program of China and the Central Public-interest Scientific Institution Basal Research Fund to J.Z. (1630052020022), and the Project of Science and Technology Department of Sichuan Provincial of China to L.Y. (2019JDJQ0035). This protocol was partially described in PLoS ONE 18(6): e0279221. Doi: 10.1371/journal.pone.0279221 (Hu et al. [17]) and in Science in One Health (2023), Doi: 10.1016/j.soh.2023.100041) (Guan et al. [12]).
Competing interests
The authors declare no competing interests.
Ethical considerations
This protocol is not involved in any experiments.
References
- Tong, Y., Liu, W., Liu, P., Liu, W. J., Wang, Q. and Gao, G. F. (2021). The origins of viruses: discovery takes time, international resources, and cooperation. Lancet 398(10309): 1401–1402.
- Nahmias, A., Weiss, J., Yao, X., Lee, F., Kodsi, R., Schanfield, M., Matthews, T., Bolognesi, D., Durack, D., Motulsky, A., et al. (1986). Evidence for human infection with an HTLV III/LAV-like virus in Central Africa, 1959. Lancet 327(8492): 1279–1280.
- Worobey, M., Gemmel, M., Teuwen, D. E., Haselkorn, T., Kunstman, K., Bunce, M., Muyembe, J. J., Kabongo, J. M., Kalengayi, R. M., Van Marck, E., et al. (2008). Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960. Nature 455(7213): 661–664.
- Zaki, A. M., van Boheemen, S., Bestebroer, T. M., Osterhaus, A. D. and Fouchier, R. A. (2012). Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 367(19): 1814–1820.
- Azhar, E. I., El-Kafrawy, S. A., Farraj, S. A., Hassan, A. M., Al-Saeed, M. S., Hashem, A. M. and Madani, T. A. (2014). Evidence for Camel-to-Human Transmission of MERS Coronavirus. N. Engl. J. Med. 370(26): 2499–2505.
- Müller, M. A., Corman, V. M., Jores, J., Meyer, B., Younan, M., Liljander, A., Bosch, B. J., Lattwein, E., Hilali, M., Musa, B. E., et al. (2014). MERS Coronavirus Neutralizing Antibodies in Camels, Eastern Africa, 1983–1997. Emerging Infectious Diseases 20(12): 2093–2095.
- Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., Zhao, X., Huang, B., Shi, W., Lu, R., et al. (2020). A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 382(8): 727–733.
- Wu, F., Zhao, S., Yu, B., Chen, Y. M., Wang, W., Song, Z. G., Hu, Y., Tao, Z. W., Tian, J. H., Pei, Y. Y., et al. (2020). A new coronavirus associated with human respiratory disease in China. Nature 579(7798): 265–269.
- Li, Q., Guan, X., Wu, P., Wang, X., Zhou, L., Tong, Y., Ren, R., Leung, K. S., Lau, E. H., Wong, J. Y., et al. (2020). Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia. N. Engl. J. Med. 382(13): 1199–1207.
- Pekar, J., Worobey, M., Moshiri, N., Scheffler, K. and Wertheim, J. O. (2021). Timing the SARS-CoV-2 index case in Hubei province. Science 372(6540): 412–417.
- Cheng, C. and Zhang, Z. (2023). SARS-CoV-2 shows a much earlier divergence in the world than in the Chinese mainland. Sci. China Life Sci. 66(6): 1440–1443.
- Guan, S., Hu, X., Yi, G., Yao, L. and Zhang, J. (2023). Genome analysis of SARS-CoV-2 haplotypes: separation and parallel evolution of the major haplotypes occurred considerably earlier than their emergence in China. Science in One Health 2: 100041.
- Khare, S., Gurry, C., Freitas, L., Schultz, M. B., Bach, G., Diallo, A., Akite, N., Ho, J., Lee, R. T., Yeo, W., et al. (2021). GISAID's role in pandemic response. China CDC Wkly 3(49): 1049–1051.
- Moshiri, N. (2020). ViralMSA: massively scalable reference-guided multiple sequence alignment of viral genomes. Bioinformatics(Oxford, England) 37(5): 714–716.
- Suchard, M. A., Lemey, P., Baele, G., Ayres, D. L., Drummond, A. J. and Rambaut, A. (2018). Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4(1): vey016.
- Shu, Y. and McCauley, J. (2017). GISAID: Global initiative on sharing all influenza data – from vision to reality. Eurosurveillance 22(13): e30494.
- Hu, X., Mu, Y., Deng, R., Yi, G., Yao, L. and Zhang, J. (2023). Genome characterization based on the Spike-614 and NS8-84 loci of SARS-CoV-2 reveals two major possible onsets of the COVID-19 pandemic. PLoS One 18(6): e0279221.
Article Information
Copyright
© 2024 The Author(s); This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/).
How to cite
Hu, X., Guan, S., He, Y., Yi, G., Yao, L. and Zhang, J. (2024). Classification of a Massive Number of Viral Genomes and Estimation of Time of Most Recent Common Ancestor (tMRCA) of SARS-CoV-2 Using Phylodynamic Analsysis. Bio-protocol 14(6): e4955. DOI: 10.21769/BioProtoc.4955.
Category
Bioinformatics and Computational Biology
Systems Biology > Genomics > Phylogenetics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.