- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Computational Analysis of Maize Enhancer Regulatory Elements Using ATAC-STARR-seq
Published: Mar 5, 2024 DOI: 10.21769/BioProtoc.4953 Views: 340
Reviewed by: G. Alex MasonPrashanth N Suravajhala

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The blueprints for development, response to the environment, and cellularfunction are largely the manifestation of distinct gene expression programscontrolled by the spatiotemporal activity of cis-regulatory elements. Althoughbiochemical methods for identifying accessible chromatin—a hallmark ofactive cis-regulatory elements—have been developed, approaches capable ofmeasuring and quantifying cis-regulatory activity are only beginning to berealized. Massively parallel reporter assays coupled to chromatin accessibilityprofiling present a high-throughput solution for testing thetranscription-activating capacity of millions of putatively regulatory DNAsequences in parallel. However, clear computational pipelines for analyzingthese high-throughput sequencing-based reporter assays are lacking. In thisprotocol, I layout and rationalize a computational framework for the processingand analysis of the transposase accessible chromatin profiling followed byself-transcribed active regulatory region sequencing (ATAC-STARR-seq) data froma recent study in Zea mays. The approach described herein canbe adapted to other sequencing-based reporter assays and is largely modelorganism–agnostic with appropriate input substitutions.
Keywords: STARR-seqBackground
Eukaryotic cells exhibit remarkable functional and morphological diversity despite containing a generally invariant copy of the same genomic sequence. Cellular heterogeneity arises in part due to the activities of cis-regulatory elements (CREs), short DNA-binding motifs recognized by sequence-specific transcription factors (TFs). CREs are often found in clusters termed cis-regulatory modules (CRMs) that dictate highly dynamic spatiotemporal patterns of gene expression via the cooperative activities of DNA-bound TFs (Schmitz et al., 2022). For proper activation of transcription, the cell strictly regulates CRM activity by controlling TF access of CRM sequences through nucleosome dynamics. Genome-wide approaches, such as the assay for transposase accessible chromatin sequencing (ATAC-seq), have been developed to profile accessible chromatin regions (ACRs) (Buenrostro et al., 2013; Minnoye et al., 2021). In general, CRMs that localize to accessible chromatin reflect active regulatory elements (Marand et al., 2017; Schmitz et al., 2022). Thus, activation and silencing of gene expression is effectively controlled by the relative chromatin accessibility of cognate CRMs.
CREs can be classified into distinct functional groups based on their regulatory effect on transcription, including enhancers, silencers, promoters, and insulators (Schmitz et al., 2022). Of these, enhancers are of particular interest due to their transcription-activating properties that function independently of location and orientation of their target genes, in contrast to the stereotypical locations of promoters surrounding gene transcription start sites (TSSs) (Marand et al., 2017; Schmitz et al., 2022). While analysis of chromatin accessibility in distinct tissues and cell types has been central to the identification of CRMs (Marand et al., 2021), chromatin profiling techniques are largely qualitative and lack the ability to quantitatively estimate regulatory activity. To overcome these challenges, massively parallel reporter assays (MPRAs) have been developed to quantify the transcription-activating properties of diverse sequences (Melnikov et al., 2012; Arnold et al., 2013). In particular, self-transcribing active regulatory region sequencing (STARR-seq) demonstrates the greatest potential for broad application by eliminating the need for homogenous cell lines available only in mammalian models typical of other MPRA methods (Arnold et al., 2013; Ricci et al., 2019; Sun et al., 2019; Jores et al., 2020). Although STARR-seq was originally designed to profile the entire genome for regulatory activity, recent implementations have successfully utilized ATAC-seq libraries as input (ATAC-STARR-seq), reducing the search space to potential regulatory regions and offsetting sequencing costs and library complexity requirements (Figure 1). Despite its promise as a powerful approach towards understanding cis-regulatory activity, computational analysis of ATAC-STARR-seq data remains challenging, particularly due to a lack of dedicated software and computational pipelines.
Here, I present a computational pipeline for analysis of ATAC-STARR-seq data generated in Zea mays L., cultivar B73 (Ricci et al., 2019). After processing and evaluation of data quality, I demonstrate how ATAC-STARR-seq data analysis allows for the interrogation of new biological questions. The pipeline can be run entirely from the code below or through freely available bash, perl, and R scripts hosted at https://github.com/Bio-protocol/Maize_ATAC_STARR_seq.
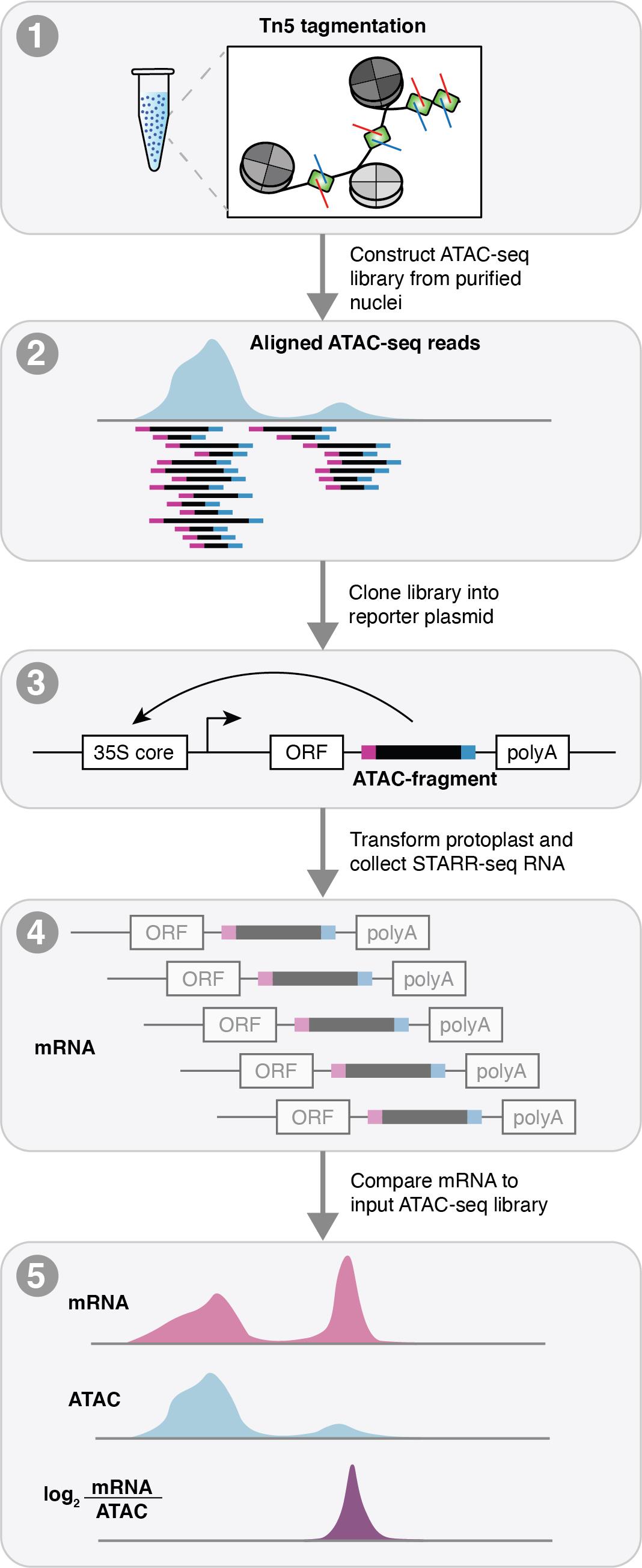
Figure 1. Schematic of assay for transposase accessible chromatin profiling followed by self-transcribed active regulatory region sequencing (ATAC-STARR-seq). ATAC-STARR-seq begins by first generating an ATAC-seq library. The ATAC fragments are then cloned into a reporter assay and transformed into maize protoplasts. Transformed protoplasts are then split into two pools: the first for sequencing the input fragments (ATAC-seq DNA) and the second for purifying transcribed (mRNA) ATAC-seq fragments that facilitate their own transcription from the reporter construct. Raw sequenced reads for the ATAC-seq input and mRNA output are processed and aligned to the maize reference genome and compared to provide estimates of cis-regulatory activity.
Equipment
This pipeline assumes that a user has knowledge of shell commands and is comfortable working on a Linux-based operating system.
Computational requirements
The following procedure can be run on any Linux-like system. However, this protocol and publicly available code is written for executing commands via a high-performance computing (HPC) cluster managed by a SLURM scheduler. Still, the code presented here can be readily converted to TORQUE or other HPC systems. The pipeline assumes a working Perl interpreter version 5.30.0 or greater and R version 3.6.2 or greater.
Software
The following analytical procedure makes use of several standard computational tools that are assumed to be available in the user’s shell environment:
BWA MEM (Li and Durbin, 2009) v0.7.17; http://bio-bwa.sourceforge.net/bwa.shtml
SAMtools (Li et al., 2009) v1.14; http://www.htslib.org
BEDtools (Quinlan and Hall, 2010) v2.27.1; https://bedtools.readthedocs.io/en/latest/
SRA-toolkit (Leinonen et al., 2011) v2.11.1; https://github.com/ncbi/sra-tools
fastp (Chen et al., 2018) v0.20.0; https://github.com/OpenGene/fastp
pigz v2.4; https://zlib.net/pigz/
MACS2 (Liu, 2014) v2.2.7.1; https://pypi.org/project/MACS2/
UCSC binaries (Kent et al., 2010) v1.04.0; http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/
tabix (Li, 2011) v0.2.6; http://www.htslib.org/doc/tabix.html
IGV (Thorvaldsdottir et al., 2013) v2.11.1; https://software.broadinstitute.org/software/igv
MEME (Grant et al., 2011) v5.4.1; https://meme-suite.org/meme/index.html
CrossMap (Zhao et al., 2014) v0.5.1; http://crossmap.sourceforge.net/
DeepTools (Ramirez et al., 2014) v3.5.1; https://deeptools.readthedocs.io/en/develop/index.html
Input data
The starting input for this computational pipeline uses paired-end sequencing data from an ATAC-STARR-seq experiment performed on maize protoplasts (Ricci et al., 2019). The ATAC-STARR-seq experiment consisted of a DNA input (ATAC-seq library) and a mRNA readout (self-transcribed regulatory regions) to identify genomic regions exhibiting transcription-activating regulatory activity.
Transfected ATAC-seq DNA-input FASTQ
Transcribed ATAC-seq mRNA FASTQ
Procedure
An overview of the ATAC-STARR-seq pipeline is presented in Figure 2.
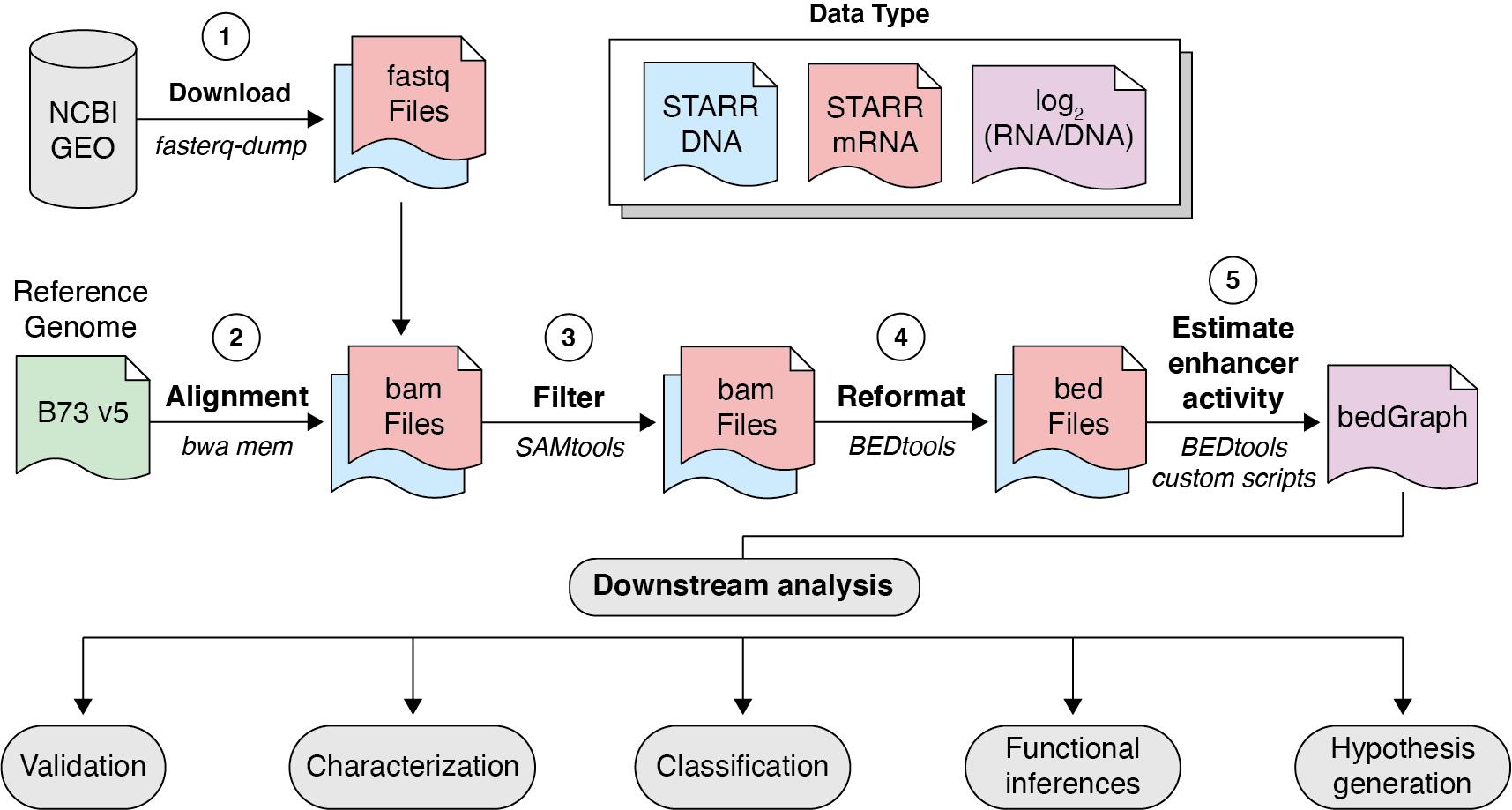
Figure 2. Computational workflow for the assay for transposase accessible chromatin profiling followed by self-transcribed active regulatory region sequencing (ATAC-STARR-seq) data. Raw sequence ATAC-STARR-seq data are first acquired from NCBI GEO, processed, and aligned to the reference genome with BWA MEM, filtered via SAMtools, reformatted as fragments with BEDtools, and compared via BEDtools and custom R scripts to provide estimates of enhancer activity for downstream analysis.
Download and prepare the requisite data and reference genome sequence
Raw mRNA ATAC-STARR-seq data generated from transfection of Zea mays leaf ATAC-seq fragments in Z. mays protoplasts, and the accompanying ATAC-seq input fragments, are publicly available on NCBI GEO. ATAC-STARR-seq mRNA and DNA input can be downloaded with fasterq-dump available from the SRA-toolkit package:
# set variables and download FASTQ files
mkdir FASTQ_files
cd FASTQ_files
fasterq-dump -o B73_maize_DNA_input.fastq SRR10964904
fasterq-dump -o B73_maize_mRNA_output.fastq SRR10964905
To save disk space, we will compress the FASTQ files with pigz. By default, pigz uses all available processors or eight if the number of available processors is unknown. Alternatively, users can use the unix tool, gzip, to compress the STARR mRNA and ATAC input DNA FASTQ files.
# compress fastq files
pigz *.fastq
# NOT RUN
# Tip: gzip can be used as an alternative to pigz (parallel gzip)
# gzip *.fastq
Download the B73 reference genome sequence and gene annotation. The original article mapped raw reads to version 4 of the B73 maize reference genome. In this case study, I map and analyze maize ATAC-STARR-seq data to version 5 of the B73 maize reference genome to showcase how updated reference genomes and read mapping strategies enable informative reanalysis of publicly available data sets (Hufford et al., 2021).
# download reference data
cd ../
mkdir Genome_Reference
cd Genome_Reference
wget https://download.maizegdb.org/Zm-B73-REFERENCE-NAM-5.0/Zm-B73-REFERENCE-NAM-5.0.fa.gz
wget https://download.maizegdb.org/Zm-B73-REFERENCE-NAM-5.0/Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.gff3.gz
To map data to the reference genome, we first need to decompress the FASTA file. Constructing reference genome indices is a prerequisite for BWA alignment and allows for faster post-alignment processing of BAM/SAM/BED formatted files with command line tools such as SAMtools and BEDtools.
# create indices for reference genome FASTA
gunzip Zm-B73-REFERENCE-NAM-5.0.fa.gz
samtools faidx Zm-B73-REFERENCE-NAM-5.0.fa
bwa index Zm-B73-REFERENCE-NAM-5.0.fa
Trim adapters and remove low quality reads
Illumina platforms may produce reads with adapter sequences on the 3’ ends if the DNA insert is shorter than the number of cycles. Additionally, the fidelity of sequencing by synthesis deteriorates with each additional cycle due to phasing, the desynchronization of cycles that results from unremoved terminator caps ultimately leading to greater uncertainty of base calls in later cycles. Removing adapter contamination and low-quality bases increases the total number of alignable reads, particularly important when analyzing a relatively lower sequence complexity experiment, such as ATAC-STARR-seq. We will use fastp to remove sequencing adapters and low-quality reads for the mRNA output and DNA input. A script to perform read trimming can be found here: https://github.com/Bio-protocol/Maize_ATAC_STARR_seq/blob/master/workflow/step01_trim_raw_reads.sh
# set variables
cd ../
fastqdir=$PWD/FASTQ_files
dna=B73_maize_DNA_input
rna=B73_maize_mRNA_output
threads=16
# trim and filter DNA input reads
fastp -j $dna.json -h $dna.html -w $threads \
-i $fastqdir/${dna}_1.fastq.gz -I $fastqdir/${dna}_2.fastq.gz \
-o $fastqdir/${dna}_1.trim.fastq.gz -O $fastqdir/${dna}_2.trim.fastq.gz
# trim and filter mRNA output reads
fastp -j $rna.json -h $rna.html -w $threads \
-i $fastqdir/${rna}_1.fastq.gz -I $fastqdir/${rna}_2.fastq.gz \
-o $fastqdir/${rna}_1.trim.fastq.gz -O $fastqdir/${rna}_2.trim.fastq.gzbS - | samtools sort - > $outdir/B73_maize_mRNA_output.raw.bam
# tidy up log files
mkdir fastp_log_files
mv *.json *.html fastp_log_files
Align and process sequenced reads
After trimming and quality filtering reads, we align the input DNA and output mRNA reads to the maize B73 version 5 reference genome. To speed up the alignment and downstream processing, we are using 24 CPUs (-t 24) to align the input and output reads. However, users should modify this value to reflect the number of available cores on their system. Additionally, we mark split hits as secondary alignments (-M) to be filtered out downstream, as the maize genome is highly repetitive. The output of BWA MEM is piped to SAMtools view for compression (SAM to BAM) and sorted by alignment coordinate prior to further processing to minimize the footprint on disk. A script to perform these steps can be found here: https://github.com/Bio-protocol/Maize_ATAC_STARR_seq/blob/master/workflow/step02_align_STARR_data.sh
cd ../
mkdir BAM_files
outdir=$PWD/BAM_files
refdir=$PWD/Genome_Reference
fastqdir=$PWD/FASTQ_files
# align DNA input and pipe to samtools for SAM to BAM conversion
bwa mem -M -t 24 $refdir/Zm-B73-REFERENCE-NAM-5.0.fa $fastqdir/B73_maize_DNA_input_1.trim.fastq.gz $fastqdir/B73_maize_DNA_input_2.trim.fastq.gz | samtools view -bS - | samtools sort - > $outdir/B73_maize_DNA_input.raw.bam
# align RNA output and pipe to samtools for SAM to BAM conversion
bwa mem -M -t 24 $refdir/Zm-B73-REFERENCE-NAM-5.0.fa $fastqdir/B73_maize_mRNA_output_1.trim.fastq.gz $fastqdir/B73_maize_mRNA_output_2.trim.fastq.gz | samtools view -bS - | samtools sort - > $outdir/B73_maize_mRNA_output.raw.bam
To ensure that only high-quality alignments remain, here we remove non-properly paired reads (-f 3), secondary hits (-F 256), alignments with low mapping quality (-q 10), and multiple mapped reads (grep -v -E -e '\bXA:Z:’) using a combination of SAMtools and unix commands. The header, which contains information on the reference genome and the read mapping parameters, is retained in the output by setting the -h flag in the SAMtools view command.
# filter DNA input alignments
samtools view -h -q 10 -f 3 -F 256 $outdir/B73_maize_DNA_input.raw.bam
| grep -v -E -e '\bXA:Z:' | samtools view -bSh - > $outdir/B73_maize_DNA_input.mq10.pp.unique.bam
# filter mRNA output alignmentssamtools view -h -q 10 -f 3 -F 256 $outdir/B73_maize_ mRNA_output.raw.bam
| grep -v -E -e '\bXA:Z:' | samtools view -bSh - > $outdir/B73_maize_mRNA_output.mq10.pp.unique.bamA major difference between analysis of ATAC-seq and STARR-seq data is how assay information is captured by sequencing. For paired-end ATAC-seq, since Tn5 inserts sequencing adapters adjacent to its bound genomic location, chromatin accessibility is encoded as the 5’ ends of sequenced reads. In contrast, STARR-seq produces mRNA transcripts from fragments that are capable of activating their own transcription; thus, the entire STARR-seq mRNA and DNA fragment is informative for analysis. The following commands extract the coordinates of sequenced fragments by leveraging the CIGAR strings in BAM paired-end alignments for DNA input and mRNA output. A script to extract fragments can be found here: https://github.com/Bio-protocol/Maize_ATAC_STARR_seq/blob/master/workflow/step03_extract_fragments.sh
# create output directory
mkdir BED_files
# variables
outdir=$PWD/BAM_files
beddir=$PWD/BED_files
dna=B73_maize_DNA_input
rna=B73_maize_mRNA_output
# extract DNA input fragments (ignore the warnings from bedtools with respect to “missing” mate pairs, these reflect one of the pairs that had its mate filtered in prior steps)
echo " extracting fragments from STARR DNA input ..."
samtools sort -n $outdir/$dna.mq10.pp.unique.bam \
| bedtools bamtobed -bedpe -i - \
| sort -k1,1 -k2,2n - \
| cut -f1,2,6 - > $beddir/$dna.fragments.bed
# extract mRNA output fragmentsecho " extracting fragments from STARR mRNA output ..."
samtools sort -n $outdir/$rna.mq10.pp.unique.bam \
| bedtools bamtobed -bedpe -i - \
| sort -k1,1 -k2,2n - \
| cut -f1,2,6 - > $beddir/$rna.fragments.bed
Identify regions with enriched activity over background
To identify regions of the genome with the capacity to activate transcription, we assess enrichment of mRNA reads relative to the input ATAC-seq fragments using MACS2. As MACS2 is a general peak caller, we need to adjust the default settings to tailor the analysis specifically for ATAC-STARR-seq data. Since this experiment did not use unique molecular identifiers, and the number of transcripts from fragment is a direct reflection of its regulatory activity, duplicate mRNA fragments are retained (--keep-dup all). To directly use coverages determined by the input fragment coordinates, we turn off the default fragment shifting model (--nomodel) and set the input type to BEDPE (-f BEDPE). Additionally, we reduce the maximum gap size between candidate peaks to allow for the identification of fine-mapped regulatory elements within a broader regulatory region (--max-gap 50) by setting the minimum peak size to 300 (--min-length 300). Finally, we use the background coverage rates in place of the local bias, which aids in peak detection (--nolambda). A script to perform peak calling can be found here: https://github.com/Bio-protocol/Maize_ATAC_STARR_seq/blob/master/workflow/step04_call_peaks.sh
# prepare output directory and input files
mkdir Peak_data
# generate input filesuniq $beddir/$rna.fragments.bed > $beddir/$rna.fragments.uniq.bed
uniq $beddir/$dna.fragments.bed > $beddir/$dna.fragments.uniq.bed
cat $beddir/$rna.fragments.uniq.bed $beddir/$dna.fragments.uniq.bed | sort -k1,1 -k2,2n - > $beddir/ALL.fragments.uniq.bed
# find regulatory regionsecho " calling regulatory regions without duplicate removal ..."
macs2 callpeak -t $beddir/$rna.fragments.bed \
-c $beddir/ALL.fragments.uniq.bed \
--keep-dup all \
--max-gap 50 \
--min-length 300 \
--nolambda \
--nomodel \
-f BEDPE \
-g 1.6e9 \
--bdg \
-n STARR_wdups
# find regulatory regionsecho " calling regulatory regions with duplicate removal ..."
macs2 callpeak -t $beddir/$rna.fragments.uniq.bed \
-c $beddir/$dna.fragments.uniq.bed \
--keep-dup all \
--max-gap 50 \
--min-length 300 \
--nolambda \
--nomodel \
-f BEDPE \
-g 1.6e9 \
--bdg \
-n STARR_nodups
# find regulatory regions using all unique fragmentsecho " calling regulatory regions by aggregating all unique fragments ..."
macs2 callpeak -t $beddir/ALL.fragments.uniq.bed \
--keep-dup all \
--max-gap 50 \
--min-length 300 \
--nolambda \
--nomodel \
-f BEDPE \
-g 1.69e9 \
--bdg \
-n STARR_ALL
# clean-up outputmv STARR_* Peak_data
# merge peakscd Peak_data
cat STARR_wdups_peaks.narrowPeak STARR_nodups_peaks.narrowPeak STARR_ALL_peaks.narrowPeak | sort -k1,1 -k2,2n - | bedtools merge -i - > STARR_merged_peaks.bed
To estimate regulatory activity at fine scale, first we need to create a list of unique intervals based on mRNA and DNA fragments. Next, we count the intersection of mRNA and DNA fragments for each unique interval. Finally, we remove all the temporary files to reduce the footprint on disk. A script to estimate enhancer activity can be found here: https://github.com/Bio-protocol/Maize_ATAC_STARR_seq/blob/master/workflow/step05_estimate_enhancer_activity.sh
# variables
beddir=$PWD/BED_files
dna=B73_maize_DNA_input
rna=B73_maize_mRNA_output
ref=./Genome_Reference/Zm-B73-REFERENCE-NAM-5.0.fa.fai
# sort the referencesort -k1,1 -k2,2n $ref > $ref.sorted
# merge RNA/DNAcat $beddir/$rna.fragments.uniq.bed $beddir/$dna.fragments.uniq.bed \
| sort -k1,1 -k2,2n - \
| bedtools genomecov -i - -bga -g $ref.sorted \
| sort -k1,1 -k2,2n - \
| cut -f1-3 - > $beddir/Unique_genomic_intervals.bed
# count fragmentsbedtools intersect -a $beddir/Unique_genomic_intervals.bed \
-b $beddir/$rna.fragments.bed \
-c -sorted -g $ref.sorted > $beddir/$rna.activity.raw.bed
bedtools intersect -a $beddir/$rna.activity.raw.bed \-b $beddir/$dna.fragments.bed \
-c -sorted -g $ref.sorted > $beddir/B73_maize_mRNA_DNA.activity.raw.bed
# clean up temporary filesrm $beddir/Unique_genomic_intervals.bed
rm $beddir/$rna.activity.raw.bed
We and others define enhancer activity as the enrichment of mRNA transcripts that are produced by DNA fragments in the assay in terms of log2(mRNA/DNA) at unique fragment intervals. Prior to taking the log2 ratio of mRNA to DNA, we normalize both the input and output to per million to account for differences in sequencing depth and complexity. A pseudocount of one is added to any interval with at least one RNA or DNA fragment. The following code can also be run from the command line using >Rscript Estimate_Enhancer_Activity.R with the following script: https://github.com/Bio-protocol/Maize_ATAC_STARR_seq/blob/master/workflow/bin/Estimate_Enhancer_Activity.R
# open an interactive R session to estimate enhancer activity
cd $beddir
R
# load dataa <- read.table("B73_maize_mRNA_DNA.activity.raw.bed")
# reformatrownames(a) <- paste(a$V1,a$V2,a$V3,sep="_")
a[,1:3] <- NULL
a <- as.matrix(a)
colnames(a) <- c("mRNA", "DNA")
a <- a[rowSums(a)!=0,]
a <- a + 1
# normalizea <- a %*% diag(x=1e6/colSums(a))
colnames(a) <- c("mRNA", "DNA")
a <- as.data.frame(a)
# estimate enhancer activitya$enhancer_activity <- log2(a$mRNA/a$DNA)
# reformat outputrownames(a) <- gsub("scaf_","scaf", rownames(a))
df <- data.frame(do.call(rbind, strsplit(rownames(a), "_")))
df$X1 <- gsub("scaf", "scaf_", as.character(df$X1))
mrna <- df
dna <- df
df$X4 <- a$enhancer_activity
mrna$X4 <- a$mRNA
dna$X4 <- a$DNA
# cap negative activity at 0df$X4 <- ifelse(df$X4 < 0, 0, df$X4)
# save enhancer activity BEDGRAPH file to diskwrite.table(df, file="B73_maize.enhancer_activity.bdg",quote=F, row.names=F, col.names=F, sep="\t")
write.table(mrna, file="B73_maize.mRNA.bdg",quote=F, row.names=F, col.names=F, sep="\t")
write.table(dna, file="B73_maize.DNA.bdg",quote=F, row.names=F, col.names=F, sep="\t")
# exit interactive modeq()
To visualize enhancer activity and the normalized mRNA and DNA fragments at any given locus, per million coverage values in bedGraph format from the previous step can be converted into bigwig files (using bedGraphToBigWig from UCSC Utils) for facile visualization using the Integrated Genomics Viewer (IGV) or JBrowse instances.
bedGraphToBigWig
B73_maize.enhancer_activity.bdg ../Genome_Reference/Zm-B73-REFERENCE-NAM-5.0.fa.fai.sorted B73_maize.enhancer_activity.bw
bedGraphToBigWig
B73_maize.mRNA.bdg ../Genome_Reference/Zm-B73-REFERENCE-NAM-5.0.fa.fai.sorted B73_maize.mRNA.bw
bedGraphToBigWig
B73_maize.DNA.bdg ../Genome_Reference/Zm-B73-REFERENCE-NAM-5.0.fa.fai.sorted B73_maize.DNA.bwTo view the enhancer activity, mRNA, and DNA fragment bigwig files, download and install IGV ( https://software.broadinstitute.org/software/igv/download) on your local machine.
Unpack, bgzip, and index the gene product annotation. Then, load all bigwig, narrowPeak, and genome annotation files using File > Load from File… in IGV. An example of an IGV screenshot is shown in Figure 3.
# change directory
cd ../Genome_Reference
# unzipgunzip Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.gff3.gz
# sort by coordinate and remove whole chromosome intervalssort -k1,1 -k4,4n Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.gff3 | grep -v assembly - > Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.sorted.gff3
# compress with bgzipbgzip Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.sorted.gff3
# index with tabixtabix -p gff Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.sorted.gff3.gz
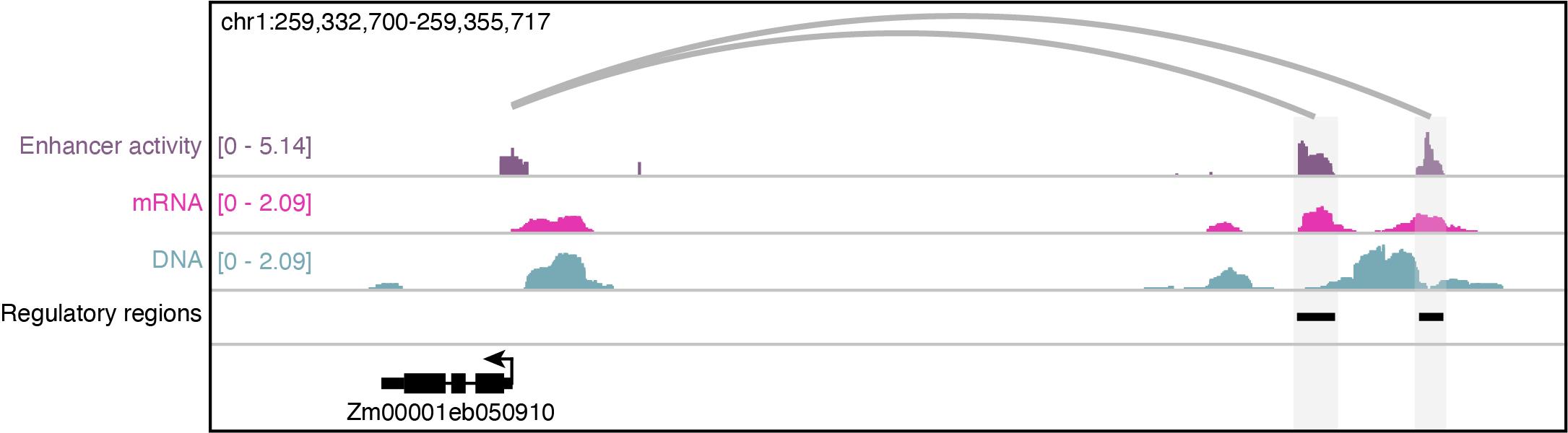
Figure 3. Visualization of the assay for transposase accessible chromatin profiling followed by self-transcribed active regulatory region sequencing (ATAC-STARR-seq) data in Zea mays. Normalized (reads per million) coverages of the DNA ATAC-seq input (blue), self-transcribed mRNA fragments (pink), and enhancer activity [purple; log2(mRNA/DNA)] of a 23 kb window. Regulatory regions are shown as black bars, while the grey loops reflect predicted enhancer-gene links.
Create a list of control regions
To assess enrichment of STARR regulatory regions determined by MACS2 relative to random control regions, we first need to identify regions of the genome that can be uniquely mapped given the sequencing output and read lengths. Although there are numerous methods for estimating mappability, I illustrate a simple approach using synthetic reads tailored to the sequencing parameters of the present experiment. First, we generate the same number of random read pairs with the same sequencing length (36 nucleotides) for mRNA and DNA input using the wgsim tool that is supplied to SAMtools. The synthetic reads are then remapped and uniformly processed as the original STARR-seq sequencing experiments to identify regions that are uniquely mappable. By constraining randomized control region selection to uniquely mappable genomic intervals, we ensure that downstream comparisons will not be biased by mappability and repeat composition. A script to construct control regions can be found here: https://github.com/Bio-protocol/Maize_ATAC_STARR_seq/blob/master/workflow/step06_create_control_regions.sh
# move into the “Genome_Reference” directory
cd ./Genome_Reference
# estimate read countsmRNA_counts=$(samtools view -c ./BAM_files/B73_maize_mRNA_output.raw.bam)
DNA_counts=$(samtools view -c ./BAM_files/B73_maize_DNA_input.raw.bam)
# generate simulated reads matching the mRNA output using wgsim from the SAMtools packagewgsim -1 36 -2 36 -d 300 -N $mRNA_counts Zm-B73-REFERENCE-NAM-5.0.fa simulated_STARR_mRNA_r1.fq simualted_STARR_mRNA_r2.fq
# generate simulated reads matching the DNA inputwgsim -1 36 -2 36 -d 300 -N $DNA_counts Zm-B73-REFERENCE-NAM-5.0.fa simulated_STARR_DNA_r1.fq simualted_STARR_DNA_r2.fq
# compress synthetic fastq filespigz *.fq
Remap the synthetic reads using the same pipeline as for the original STARR-seq data.
# variables
outdir=$PWD/BAM_files
refdir=$PWD/Genome_Reference
ref=$refdir/Zm-B73-REFERENCE-NAM-5.0.fa.fai.sorted
fastqdir=$refdir
# align synthetic mRNA output and pipe to samtools for SAM to BAM conversionbwa mem -M -t 24 $refdir/Zm-B73-REFERENCE-NAM-5.0.fa \
$fastqdir/simulated_STARR_mRNA_r1.fq.gz \
$fastqdir/simualted_STARR_mRNA_r2.fq.gz \
| samtools view -bS - \
| samtools sort - > $outdir/simulated_STARR_mRNA.raw.bam
# align synthetic DNA input and pipe to samtools for SAM to BAM conversionbwa mem -M -t 24 $refdir/Zm-B73-REFERENCE-NAM-5.0.fa \
$fastqdir/simulated_STARR_DNA_r1.fq.gz \
$fastqdir/simulated_STARR_DNA_r2.fq.gz \
| samtools view -bS - \
| samtools sort - > $outdir/simulated_STARR_DNA.raw.bam
Process and filter reads using the original STARR-seq pipeline.
# filter synthetic mRNA alignments
echo " filtering synthetic STARR mRNA alignments ..."
samtools view -h -q 10 -f 3 $outdir/simulated_STARR_mRNA.raw.bam \
| grep -v -E -e '\bXA:Z:' \
| samtools view -bSh - > $outdir/simulated_STARR_mRNA.mq10.pp.unique.bam
# filter synthetic DNA alignmentsecho " filtering STARR DNA alignments ..."
samtools view -h -q 10 -f 3 $outdir/simulated_STARR_DNA.raw.bam \
| grep -v -E -e '\bXA:Z:' \
| samtools view -bSh - > $outdir/simulated_STARR_DNA.mq10.pp.unique.bam
Identify uniquely mappable regions.
# extract mRNA output fragments
echo " extracting fragments from simulated STARR mRNA output ..."
samtools sort -n $outdir/simulated_STARR_mRNA.mq10.pp.unique.bam \
| bedtools bamtobed -bedpe -i - \
| sort -k1,1 -k2,2n - \
| cut -f1,2,6 - > $refdir/simulated_STARR_mRNA.fragments.bed
# extract DNA input fragmentsecho " extracting fragments from simulated STARR DNA input ..."
samtools sort -n $outdir/simulated_STARR_DNA.mq10.pp.unique.bam \
| bedtools bamtobed -bedpe -i - \
| sort -k1,1 -k2,2n - \
| cut -f1,2,6 - > $refdir/simulated_STARR_DNA.fragments.bed
# merge all fragments (sorting by coordinate at this step may take a while)cat $refdir/simulated_STARR_mRNA.fragments.bed $refdir/simulated_STARR_DNA.fragments.bed \
| sort -k1,1 -k2,2n \
| bedtools merge -i - > $refdir/mappable_genomic_regions.bed
Construct control regions using only mappable regions and excluding putative regulatory regions output by MACS2.
# create controls
peaks=$PWD/Peak_data/STARR_merged_peaks.bed
bedtools shuffle -i $peaks \
-g $ref \
-incl $refdir/mappable_genomic_regions.bed \
-excl $peaks \
| sort -k1,1 -k2,2n - > $PWD/Peak_data/STARR_CONTROL.bed
Compare enhancer activity
Determine enhancer activity for predicted enhancers output by MACS2 as well as the negative control regions.
# create directory to contain analysis
cd ../
mkdir 01_Peak_Analysis
cd 01_Peak_Analysis
# map maximum enhancer activity to putative regulatory regions (wdups)bedtools map -a ../Peak_data/STARR_merged_peaks.bed -b ../BED_files/B73_maize.enhancer_activity.bdg -o max -c 4 > STARR_merged_peaks.enhancer_activity.bed
# map maximum enhancer activity to controlbedtools map -a ../Peak_data/STARR_CONTROL.bed -b ../BED_files/B73_maize.enhancer_activity.bdg -o max -c 4 > STARR_CONTROL.enhancer_activity.bed
To remove regulatory regions with enhancer activity similar to background, we filter STARR regulatory regions using an empirical false discovery rate (eFDR) based on the matched control regions. A user-specified eFDR threshold identifies the enhancer activity value in the control set that removes 1-eFDR percent of control regions. In this example, we set the FDR to a widely used rate of 0.05. The following code performs and plots eFDR filtering and enhancer activity distributions and can be run from the command line using >Rscript eFDR_Filter_STARR_Peaks.R. Filtering STARR peaks based on eFDR thresholds derived from the control regions is visualized in Figure 4A. An R script of the following code can be found here: https://github.com/Bio-protocol/Maize_ATAC_STARR_seq/blob/master/workflow/bin/eFDR_Filter_STARR_Peaks.R
# start an interactive R session
> R
# load librarieslibrary(scales)
# load datastarr <- read.table("STARR_merged_peaks.enhancer_activity.bed")
con <- read.table(”STARR_CONTROL.enhancer_activity.bed")
# set missing to 0starr$V4[starr$V4=='.'] <- 0
con$V4[con$V4=='.'] <- 0
# convert to numericstarr$V4 <- as.numeric(as.character(starr$V4))
con$V4 <- as.numeric(as.character(con$V4))
# get empirical thresholdsfdr <- 0.05
threshold <- quantile(con$V4, (1-fdr))
# filter STARR regulatory regionsfiltered <- subset(starr, starr$V4 >= threshold)
# estimate fraction of retained regionsfrac <- signif(nrow(filtered)/nrow(starr), digits=4)
# set up multipanel plot areapdf("Density_eFDR_STARR_Peak_Filtering.pdf", width=5, height=5)
# plot control/observed enhancer activities for STARR peaks with duplicatesden.starr <- density(starr$V4)
den.con <- density(con$V4)
plot(NA,
xlab="Enhancer Activity",
ylab="Density",
xlim=c(range(range(den.starr$x), range(den.con $x))),
ylim=c(range(range(den.starr$y), range(den.con$y))))
polygon(x=c(min(den.starr$x), den.starr$x, max(den.starr$x)),
y=c(0, den.starr$y, 0), col=alpha("darkorchid4", 0.5), border=NA)
polygon(x=c(min(den.con$x), den.con$x, max(den.con$x)),
y=c(0, den.con$y, 0), col=alpha("grey80", 0.5), border=NA)
abline(v=threshold, col="red", lty=2)
mtext(paste0("STARR peaks pass = ",frac," (", nrow(filtered), "/", nrow(starr),")"))
legend("right", legend=c("STARR Peaks", "Control Peaks", paste0("eFDR = ", fdr)), col=c("darkorchid4", "grey75", "red"), border=c(NA, NA, "red"), pch=c(16, 16, NA), lty=c(NA, NA, 2))
# close devicedev.off()
# save filtered STARR regulatory regionswrite.table(filtered, file="STARR_merged_peaks.enhancer_activity.eFDR05.bed", quote=F, row.names=F, col.names=F, sep="\t")
# exit Rq()
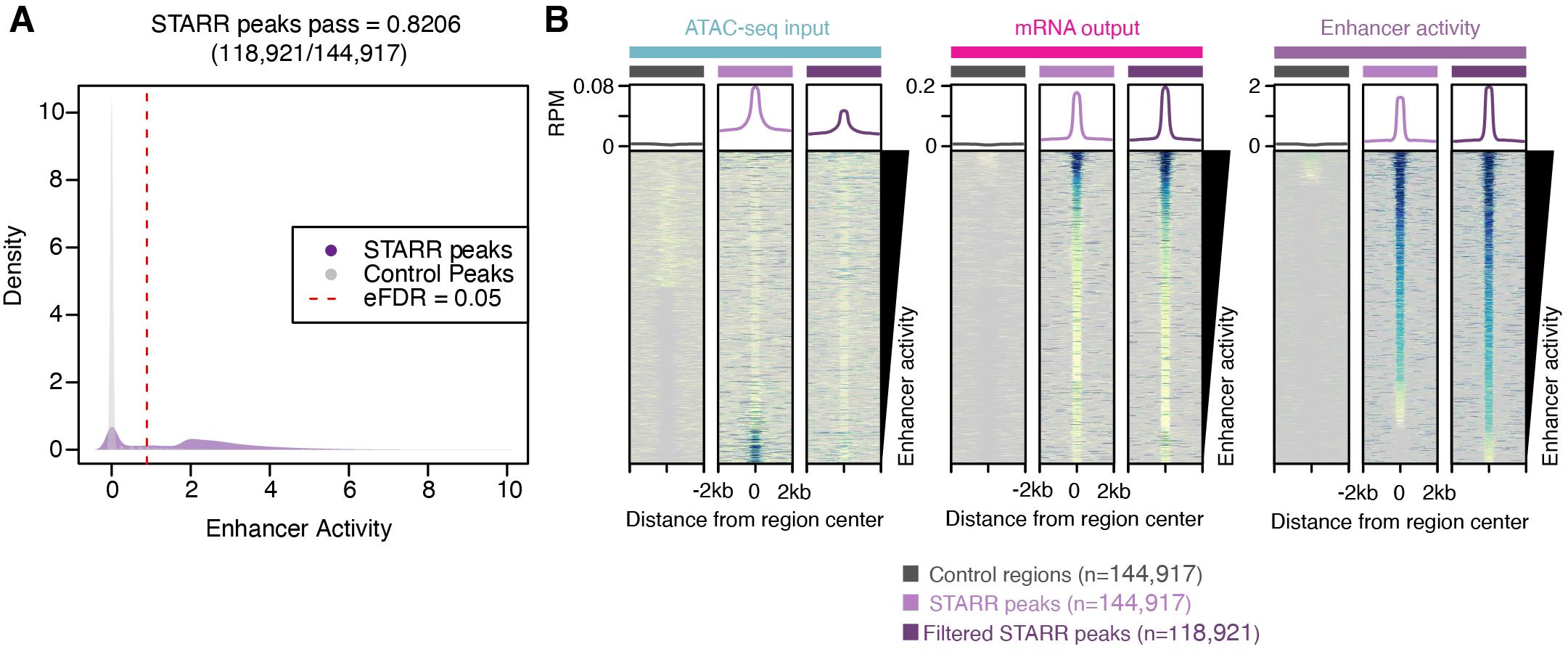
Figure 4. Analysis of self-transcribed active regulatory region (STARR) regulatory region enhancer activity. A. Distribution of enhancer activity for STARR peaks (purple) and random control regions (grey). Dashed red line indicates the 95% quantile of enhancer activity of random control regions. B. Average (top) and individual site heatmaps of reads per million (RPM) for ATAC-seq input (left), mRNA output (middle), and enhancer activity (right) for control regions, all STARR peaks, and the filtered STARR peak set.We can now assess the relative enhancer activities across all regions for the filtered and unfiltered STARR peaks and controls using DeepTools (Figure 4B). A script to plot heatmaps via DeepTools can be found here: https://github.com/Bio-protocol/Maize_ATAC_STARR_seq/blob/master/workflow/step07_plot_enhancer_activity.sh
# load function
getmaps(){
# inputina=$1
id=$2
dat=../BED_files/*.bw
# outputouta=$id.mat.gz
outm=$id.mat.txt
fig=$id.pdf
# parametersthreads=48
window=2000
bin=20
cols=YlGnBu
# create matrix
computeMatrix reference-point --referencePoint center \
-S $dat \
-b $window -a $window \
-R $ina \
--missingDataAsZero \
-o $outa \
--outFileNameMatrix $outm \
-p $threads --binSize $bin
# plot heatmapplotHeatmap --matrixFile $outa \
--colorMap YlGnBu \
-out $id.heatmap.pdf
}
export -f getmaps
# run for each filegetmaps STARR_merged_peaks.enhancer_activity.bed STARR_peaks
getmaps STARR_merged_peaks.enhancer_activity.eFDR05.bed STARR_peaks_filtered
getmaps STARR_CONTROL.enhancer_activity.bed control_regions
Identification of large regulatory domains in the maize genome
One question these data allow us to ask is whether a relationship exists between the size of a regulatory region and its enhancer activity. The so called super enhancers in mammalian systems describe hyperactive transcription-activating regulatory domains associated with cell identity that exhibit increased density of TF binding sites compared to typical enhancers (Hnisz et al., 2013). Integration of the STARR peaks and enhancer activities with other datasets allows us to determine whether similar hyperactive regulatory domains exist in maize. To query TF binding site density, we first download the position weight matrices of known TFs from the MEME database and identify putative TF binding sites using fimo (also from the MEME suite) conditioning on a P-value threshold less than 1-5. A script to identify large regulatory domains can be found here: https://github.com/Bio-protocol/Maize_ATAC_STARR_seq/blob/master/workflow/step08_identify_large_regulatory_domains.sh
# make a new directory for the TFBS analysis
cd ../
mkdir 02_Hyperactive_Regulatory_Region_Analysis
cd ./02_Hyperactive_Regulatory_Region_Analysis
# download and decompress motif databaseswget https://meme-suite.org/meme/meme-software/Databases/motifs/motif_databases.12.23.tgz
tar -xvzf motif_databases.12.23.tgz
rm motif_databases.12.23.tgz
# variablesthreads=16
ref=../Genome_Reference/Zm-B73-REFERENCE-NAM-5.0.fa
peaks=../01_Peak_Analysis/STARR_merged_peaks.enhancer_activity.eFDR05.bed
controls=../01_Peak_Analysis/STARR_CONTROL.enhancer_activity.bed
motifs=./motif_databases/ARABD/ArabidopsisDAPv1.meme
# extract fasta sequencesbedtools getfasta -bed $peaks -fi $ref -fo $peaks.fasta
bedtools getfasta -bed $controls -fi $ref -fo $controls.fasta
# identify putative TFBSfimo --oc TFBS_peaks $motifs $peaks.fasta
fimo --oc TFBS_controls $motifs $controls.fasta
# reformat fimo output (filtering p-value > 1e-5) using the perl script provided in the github repository (https://github.com/Bio-protocol/Maize_ATAC_STARR_seq/blob/master/workflow/bin/convertMotifCoord.pl )perl convertMotifCoord.pl TFBS_peaks/fimo.gff | sed -e 's/_tnt//g' - | sort -k1,1 -k2,2n - > TFBS_peaks.motifs.bed
perl convertMotifCoord.pl TFBS_controls/fimo.gff | sed -e 's/_tnt//g' - | sort -k1,1 -k2,2n - > TFBS_controls.motifs.bed
# annotate motif coverage/counts for STARR and control peaksbedtools annotate -i ../01_Peak_Analysis/STARR_merged_peaks.enhancer_activity.eFDR05.bed -files TFBS_peaks.motifs.bed -both | sort -k1,1 -k2,2n - > STARR_merged_peaks.enhancer_activity.eFDR05.ann.bed
bedtools annotate -i ../01_Peak_Analysis/STARR_CONTROL.enhancer_activity.bed -files TFBS_controls.motifs.bed -both | sort -k1,1 -k2,2n - > STARR_CONTROL.enhancer_activity.ann.bed
# extract genesperl -ne 'if($_ =~ /^#/){next;}chomp;my@col=split("\t",$_);if($col[2] eq 'gene'){print"$_\n";}' ../Genome_Reference/Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.gff3 | sort -k1,1 -k4,4n - > ../Genome_Reference/Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.genes.gff3
# classify genomic context of STARR and control peaks (you can ignore the warnings from bedtools about inconsistent naming conventions, you can thank the genome assembly team for these annoying, but harmless warnings)bedtools closest -a STARR_merged_peaks.enhancer_activity.eFDR05.ann.bed -b ../Genome_Reference/Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.genes.gff3 -D b > STARR_merged_peaks.enhancer_activity.eFDR05.ann2.bed
bedtools closest -a STARR_CONTROL.enhancer_activity.ann.bed -b ../Genome_Reference/Zm-B73-REFERENCE-NAM-5.0_Zm00001eb.1.genes.gff3 -D b > STARR_CONTROL.enhancer_activity.ann2.bed
# clean upmv STARR_merged_peaks.enhancer_activity.eFDR05.ann2.bed STARR_merged_peaks.enhancer_activity.eFDR05.ann.bed
mv STARR_CONTROL.enhancer_activity.ann2.bed STARR_CONTROL.enhancer_activity.ann.bed
We can now investigate the relationship among regulatory region size, motif density, and enhancer activity to identify putative regulatory domains (Figure 5A–5F). To do so, we will start an interactive R session and load the annotated peak and control files from above. A script to automate the following code can be found here: https://github.com/Bio-protocol/Maize_ATAC_STARR_seq/blob/master/workflow/bin/Characterize_Regulatory_Regions.R
# open R
> R
# Analyze regulatory regions
# load libraries
library(vioplot)
library(dplyr)
library(MASS)
library(RColorBrewer)
library(scales)
# load data
starr <- read.table("STARR_merged_peaks.enhancer_activity.eFDR05.ann.bed")
control <- read.table("STARR_CONTROL.enhancer_activity.ann.bed")
# select random control regions to match the filtered STARR peaks
control <- control[sample(nrow(starr)),]
# rename columns for clarity (frac_RR_motif = fraction of regulatory region covered by motifs)
starr[,7:15] <- NULL
control[,7:15] <- NULL
colnames(starr)[4:7] <- c("activity", "motif_counts", "frac_RR_motif", "gene_distance")
colnames(control)[4:7] <- c("activity", "motif_counts", "frac_RR_motif", "gene_distance")
# classify
starr$class <- ifelse((starr$gene_distance < 0 & starr$gene_distance > -200), "TSS", ifelse(starr$gene_distance < -200 & starr$gene_distance > -2000, "promoter", ifelse(starr$gene_distance > 0 & starr$gene_distance < 1000, "TTS", ifelse(starr$gene_distance == 0, "genic", "intergenic"))))
control$class <- ifelse((control$gene_distance < 0 & control$gene_distance > -200), "TSS", ifelse(control$gene_distance < -200 & control$gene_distance > -2000, "promoter", ifelse(control$gene_distance > 0 & control$gene_distance < 1000, "TTS", ifelse(control$gene_distance == 0, "genic", "intergenic"))))
# plot distributionpdf("STARR_peak_control_genomic_distribution.pdf", width=10, height=5)
layout(matrix(c(1:2), nrow=1))
pie(table(starr$class))
pie(table(control$class))
dev.off()
# estimate regulatory region size (in log10 scale)starr$size <- log10(starr$V3-starr$V2)
control$size <- log10(control$V3-control$V2)
# compare sizes between peaks and controls (sanity check)pdf("STARR_peak_control_sizes.pdf", width=5, height=6)
vioplot(starr$size, control$size,
ylab="Interval size (log10)",
col=c("dodgerblue", "grey75"),
names=c(paste0("STARR peaks \n (n=",nrow(starr),")"),
paste0("Control regions \n (n=",nrow(control),")")))
dev.off()
# compare motif countspval <- wilcox.test(starr$motif_counts, control$motif_counts)$p.value
pval <- ifelse(pval==0, 2.2e-16, pval)
mean.peak <- mean(starr$motif_counts)
mean.cont <- mean(control$motif_counts)
# find 95% quantile for control motif countupper.threshold <- quantile(control$motif_counts, 0.95)
# plotpdf("STARR_peak_control_motif_counts.pdf", width=5, height=6)
vioplot(log1p(starr$motif_counts), log1p(control$motif_counts),
ylab="log2(Motif counts + 1)",
col=c("dodgerblue", "grey75"),
names=c(paste0("STARR peaks \n (n=",nrow(starr),")"),
paste0("Control regions \n (n=",nrow(control),")")),
ylim=c(0,8),
areaEqual=T,
h=0.25)
mtext(paste0("Wilcoxon Rank Sum P-value = ", signif(pval, digits=3)))
text(1, 7.5, labels=paste0("Mean = ", signif(mean.peak, digits=3)))
text(2, 7.5, labels=paste0("Mean = ", signif(mean.cont, digits=3)))
points(1, log1p(upper.threshold), col="red", pch="-")
points(2, log1p(upper.threshold), col="red", pch="-")
dev.off()
# split STARR regions by motif counts based on 95% quantile control diststarr$group <- ifelse(starr$motif_counts >= upper.threshold, "high", "low")
pval <- kruskal.test(starr$activity, starr$group)$p.value
pdf("STARR_peak_activity_vs_group.pdf", width=5, height=6)
vioplot(starr$activity~starr$group,
ylab="Enhancer activity",
col=c("dodgerblue4", "dodgerblue"),
names=c(paste0("Motif-enriched \n STARR peaks \n (n=", nrow(starr[starr$group=="high",]),")"),
paste0("Motif-depleted \n STARR peaks \n (n=", nrow(starr[starr$group=="low",]),")")),
areaEqual=F,
xlab="",
h=0.25)
mtext(paste0("Kruskal-Wallis rank sum P-value = ", signif(pval, digits=3)))
dev.off()
# compare STARR region sizepval <- kruskal.test(starr$size, starr$group)$p.value
pval <- ifelse(pval==0, 2.2e-16, pval)
pdf("STARR_peak_size_vs_group.pdf", width=5, height=6)
vioplot(starr$size~starr$group,
ylab="Interval size (log10)",
col=c("dodgerblue4", "dodgerblue"),
names=c(paste0("Motif-enriched \n STARR peaks \n (n=", nrow(starr[starr$group=="high",]),")"),
paste0("Motif-depleted \n STARR peaks \n (n=", nrow(starr[starr$group=="low",]),")")),
areaEqual=F,
xlab="",
h=0.25)
mtext(paste0("Kruskal-Wallis rank sum P-value = ", signif(pval, digits=3)))
dev.off()
# compare motif coveragepval <- kruskal.test(starr$frac_RR_motif, starr$group)$p.value
pval <- ifelse(pval==0, 2.2e-16, pval)
pdf("STARR_peak_motif_coverage_vs_group.pdf", width=5, height=6)
vioplot(starr$frac_RR_motif~starr$group,
ylab="Fraction motif coverage",
col=c("dodgerblue4", "dodgerblue"),
names=c(paste0("Motif-enriched \n STARR peaks \n (n=", nrow(starr[starr$group=="high",]),")"),
paste0("Motif-depleted \n STARR peaks \n (n=", nrow(starr[starr$group=="low",]),")")),
areaEqual=F,
xlab="")
mtext(paste0("Kruskal-Wallis rank sum P-value = ", signif(pval, digits=3)))
dev.off()
# split by groupstarr.me <- subset(starr, starr$group=="high")
starr.md <- subset(starr, starr$group=="low")
write.table(starr.me, file="STARR_starrs_peaks.enhancer_activity.eFDR05.ann.high_motif.bed",
quote=F, row.names=F, col.names=F, sep="\t")
write.table(starr.md, file="STARR_starrs_peaks.enhancer_activity.eFDR05.ann.low_motif.bed",
quote=F, row.names=F, col.names=F, sep="\t")
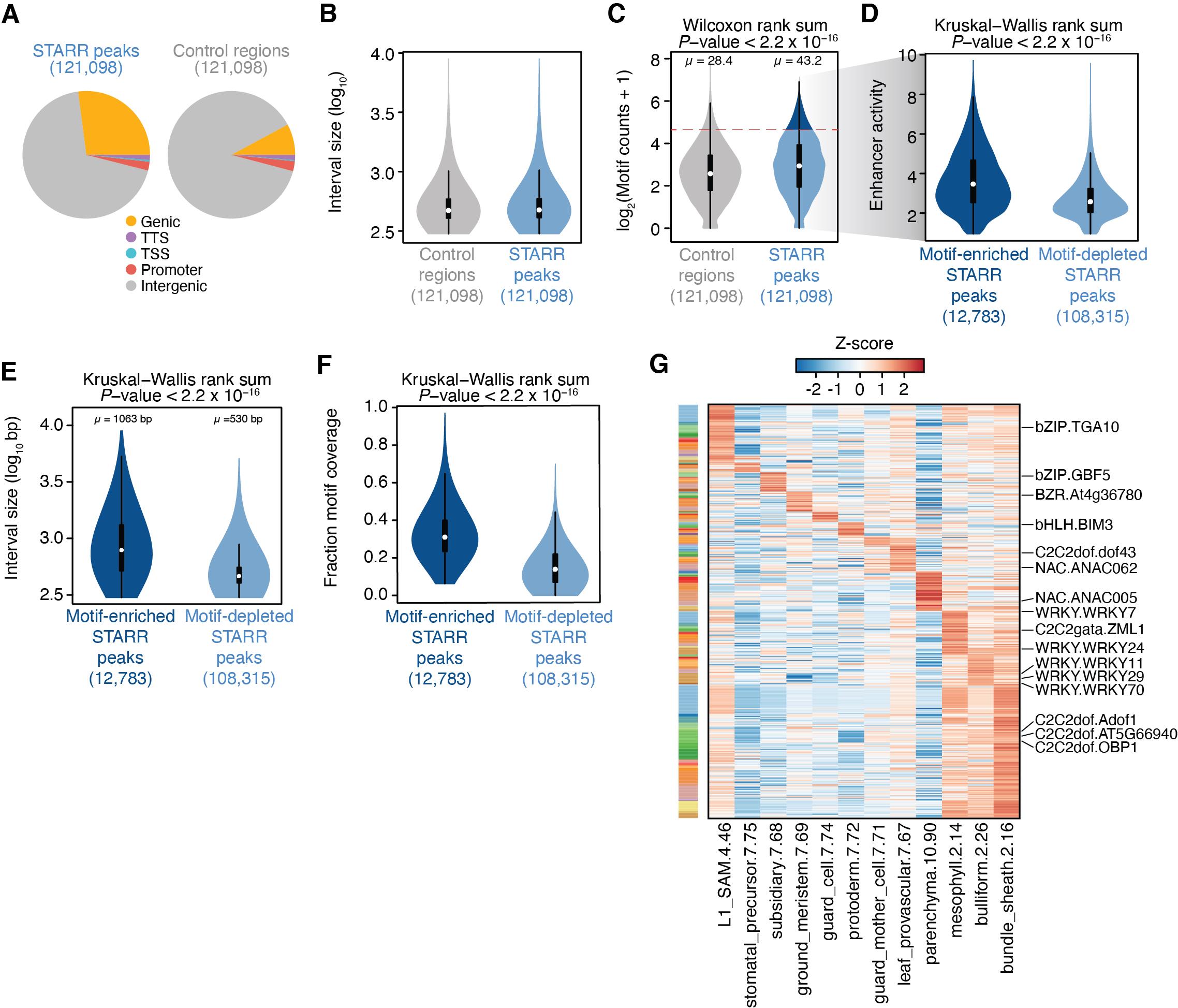
Figure 5. Identification of motif-dense enhancer regulatory domains. A. Genomic distribution of self-transcribed active regulatory region (STARR) peaks (left) and control regions (right). B. Distribution of control region (grey) and STARR peak (blue) interval lengths. C. Distribution of motif counts in control regions (grey) and STARR peaks (blue). The dashed red line indicates the 95% quantile of motif counts from control regions used to classify STARR peaks into high- and low-motif count classes. D. Distribution of enhancer activity for STARR peaks with enriched (dark blue) and depleted (light blue) motif counts. E. Distribution of interval lengths for motif-enriched (dark blue) and motif-depleted (light blue) STARR peaks. F. Distribution of fraction of STARR peaks covered by motif for motif-enriched (dark blue) and motif-depleted (light blue) STARR peaks. G. Heatmap illustrating Z-score-transformed motif-enhancer activities across intergenic motif-enriched STARR peaks scaled by the relative chromatin accessibility in various maize cell types.3. To determine if the large intergenic regulatory domain regions are associated with cell identity, we will compare enhancer activities vs. various cell type–specific ACRs leveraging a recent single-cell ATAC-seq (scATAC-seq) dataset from multiple maize organs (Marand et al., 2021). First, download the matrix containing normalized accessibility counts across accessible chromatin regions for each profiled cell type. We then extract ACR genomic coordinates (which are in version 4 of the B73 reference genome) and convert them to version 5 of the B73 reference genome using the CrossMap tool and chain file.
# download the counts matrix
wget -O maize_scATAC_atlas_ACR_celltype_CPM.txt.gz https://www.ncbi.nlm.nih.gov/geo/download/\?acc\=GSE155178\&format\=file\&file\=GSE155178%5Fmaize%5FscATAC%5Fatlas%5FACR%5Fcelltype%5FCPM%2Etxt%2Egz
# unzipgunzip maize_scATAC_atlas_ACR_celltype_CPM.txt.gz
# download chain filewget https://download.maizegdb.org/Zm-B73-REFERENCE-NAM-5.0/chain_files/B73_RefGen_v4_to_Zm-B73-REFERENCE-NAM-5.0.chain
# extract coordinates and conform chromosome names to V4 referencecut -f1 maize_scATAC_atlas_ACR_celltype_CPM.txt \
| grep '^chr' - \
| perl -ne 'chomp;my@col=split("_",$_);print"$col[0]\t$col[1]\t$col[2]\n";' - \
| sed -e 's/chrB73V4ctg/B73V4_ctg/g' - \
| sed -e 's/chr//g' - \
| sort -k1,1 -k2,2n - > maize_scATAC_atlas_ACRs.bed
# convert ACR coordinates from V4 to V5CrossMap.py bed B73_RefGen_v4_to_Zm-B73-REFERENCE-NAM-5.0.chain maize_scATAC_atlas_ACRs.bed > maize_scATAC_atlas_ACRs.V4_to_V5.txt
# discard unmapped and split projectionsgrep -v 'Unmap\|split' maize_scATAC_atlas_ACRs.V4_to_V5.txt \
| perl -ne 'chomp; my@col=split("\t",$_); print"chr$col[0]_$col[1]_$col[2]\tchr$col[4]_$col[5]_$col[6]\n";' - \
| sort -k1,1 -k2,2n - \
| sed -e 's/chrscaf/scaf/g' - \
| sed -e 's/chrB73V4_ctg/chrB73V4ctg/g' - > maize_scATAC_atlas_ACRs.V4_to_V5.clean.txt
# update ACR coordinates in matrix file using R> R
# read into data framesconv <- read.table("maize_scATAC_atlas_ACRs.V4_to_V5.clean.txt")
mat <- read.table("maize_scATAC_atlas_ACR_celltype_CPM.txt")
# subset mat rows by retained ACRs after projectionshared <- intersect(rownames(mat), as.character(conv$V1))
mat <- mat[shared,]
rownames(conv) <- conv$V1
conv <- conv[shared,]
# update mat rowIDsrownames(mat) <- conv$V2
# save outputwrite.table(mat, file="maize_scATAC_atlas_ACR_celltype_CPM.V5.txt", quote=F, row.names=T, col.names=T, sep="\t")
# exit Rq()
# remove temporary filesrm maize_scATAC_atlas_ACR_celltype_CPM.txt maize_scATAC_atlas_ACRs.bed maize_scATAC_atlas_ACRs.V4_to_V5.txt maize_scATAC_atlas_ACRs.V4_to_V5.clean.txt
4. Intersect the scATAC-seq ACRs with the STARR peaks with enriched motif counts. Load the scATAC-seq matrix and intersected ACRs/STARR peaks files into R to estimate enhancer activity enrichment scaled by relative accessibilities across cell types. As the STARR-seq data was derived from maize seedlings, we further restrict the analysis of scATAC-seq cell types to those derived primarily from maize seedlings. The following code written in R can be executed with the script named “motif_enhancer_activity_maize_celltypes.R” and provides estimates of enhancer activity over various motifs scaled by the relative cell type accessibility, allowing insights into cell type–specific transcription factor regulation of active enhancers (Figure 5G).
# extract ACR coordinates
cut -f1 maize_scATAC_atlas_ACR_celltype_CPM.V5.txt| grep -v 'unknown.5.50' | sed -e 's/scaf_/scaf/g' | perl -ne 'chomp;my@col=split("_",$_);print"$col[0]\t$col[1]\t$col[2]\n";' - | sed -e 's/scaf/scaf_/g' - | sort -k1,1 -k2,2n - > maize_scATAC_atlas_ACRs.V5.bed
# intersect scATAC ACRs with high motif counts STARR peaksbedtools intersect -a STARR_starrs_peaks.enhancer_activity.eFDR05.ann.high_motif.bed -b maize_scATAC_atlas_ACRs.V5.bed -wa -wb > STARR_starrs_peaks.enhancer_activity.eFDR05.ann.high_motif.scATAC_ACRs.bed
# map enhancer activity over motifsbedtools map -a TFBS_peaks.motifs.bed -b ../BED_files/B73_maize.enhancer_activity.bdg -c 4 -o max > TFBS_peaks.motifs.enhancer_activity.bed
# motifs to large regulatory regionsbedtools intersect -a TFBS_peaks.motifs.enhancer_activity.bed -b STARR_starrs_peaks.enhancer_activity.eFDR05.ann.high_motif.scATAC_ACRs.bed -wa -wb > TFBS_peaks.motifs.enhancer_activity.bed
# open R (alternatively, a script to automate the following code can be found here: https://github.com/Bio-protocol/Maize_ATAC_STARR_seq/blob/master/workflow/bin/motif_enhancer_activity_maize_celltypes.R)> R
# estimate enhancer activity cell type specificity
# load libraries
library(RColorBrewer)
library(gplots)
library(edgeR)
# load data
enh <- read.table("STARR_starrs_peaks.enhancer_activity.eFDR05.ann.high_motif.scATAC_ACRs.bed")
acrs <- read.table("maize_scATAC_atlas_ACR_celltype_CPM.V5.txt")
motifs <- read.table("TFBS_peaks.motifs.ENRICHED.enhancer_activity.bed")
# subset for representative leaf-derived clusters
keep <- c("bulliform.2.26",
"bundle_sheath.2.16",
"ground_meristem.7.69",
"guard_cell.7.74",
"guard_mother_cell.7.71",
"L1_SAM.4.46",
"leaf_provascular.7.67",
"mesophyll.2.14",
"parenchyma.10.90",
"protoderm.7.72",
"stomatal_precursor.7.75",
"subsidiary.7.68")
all.acrs <- acrs
# rescale acrs
acrs <- cpm(acrs, log=F)
acrs <- acrs[,keep]
# subset enhancers by genomic featureenh <- subset(enh, enh$V8=="intergenic")
# get overlapping regions from the scATAC matrixenh$ids <- paste(enh$V11,enh$V12,enh$V13,sep="_")
enh <- enh[order(enh$V11, decreasing=T),]
enh <- enh[!duplicated(enh$ids),]
shared <- intersect(enh$ids, rownames(acrs))
rownames(enh) <- enh$ids
enh <- enh[shared,]
enh$starrIDs <- paste(enh$V1, enh$V2, enh$V3,sep="_")
# filter motifsmotifs$starrIDs <- paste(motifs$V5, motifs$V6, motifs$V7, sep="_")
motifs <- motifs[motifs$starrIDs %in% unique(enh$starrIDs),]
# normalize acrsacrs <- t(apply(acrs, 1, function(x){x/max(x)}))
# iterate over each cell typects <- colnames(acrs)
outs <- lapply(cts, function(x){
access <- acrs[rownames(enh),x]
names(access) <- enh$starrIDs
motif.scores <- access[motifs$starrIDs] * as.numeric(as.character(motifs$V15))
mtf <- data.frame(motif=motifs$V4, score=motif.scores)
aves <- aggregate(score~motif, data=mtf, FUN=mean)
score <- aves$score
names(score) <- aves$motif
return(score)
})
outs <- do.call(cbind, outs)
colnames(outs) <- cts
vars <- apply(outs, 1, var)
outs <- outs[vars > 0,]
z <- as.matrix(t(scale(t(outs))))
# # cluster columnsco <- hclust(dist(t(outs)))$order
# reorder rowsz <- z[,co]
row.o <- apply(z, 1, which.max)
z <- z[order(row.o, decreasing=F),]
# capz[z < -3] <- -3
z[z > 3] <- 3
# get familytfs <- data.frame(do.call(rbind, strsplit(rownames(z), "\\.")))
cols2 <- colorRampPalette(brewer.pal(12, "Paired"))(length(unique(tfs$X1)))
tfs$cols2 <- cols2[factor(tfs$X1)]
# visualizepdf("celltype_starr_motif_activity.pdf", width=10, height=10)
heatmap.2(z, scale="none", trace='none',
RowSideColors=tfs$cols,
col=colorRampPalette(rev(brewer.pal(9, "RdBu")))(100),
useRaster=T, Colv=F, Rowv=F, dendrogram="none", margins=c(9,9))
dev.off()
Acknowledgments
This study was funded by support from the National Science Foundation (DBI-1906869) and the National Institute of Health (1K99GM144742) to A.P.M. The ATAC-STARR-seq data analyzed in this study was originally generated by Ricci, Lu, Ji, and colleagues (Ricci et al., 2019).
Competing interests
A.P.M. declares no competing interests.
References
- Arnold, C. D., Gerlach, D., Stelzer, C., Boryn, L. M., Rath, M. and Stark, A. (2013). Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339(6123): 1074-1077.
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. and Greenleaf, W. J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10(12): 1213-1218.
- Chen, S., Zhou, Y., Chen, Y. and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17): i884-i890.
- Grant, C. E., Bailey, T. L. and Noble, W. S. (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27(7): 1017-1018.
- Hnisz, D., Abraham, B. J., Lee, T. I., Lau, A., Saint-Andre, V., Sigova, A. A., Hoke, H. A. and Young, R. A. (2013). Super-enhancers in the control of cell identity and disease. Cell 155(4): 934-947.
- Hufford, M. B., Seetharam, A. S., Woodhouse, M. R., Chougule, K. M., Ou, S., Liu, J., Ricci, W. A., Guo, T., Olson, A., Qiu, Y., et al. (2021). De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. Science 373(6555): 655-662.
- Jores, T., Tonnies, J., Dorrity, M. W., Cuperus, J. T., Fields, S. and Queitsch, C. (2020). Identification of Plant Enhancers and Their Constituent Elements by STARR-seq in Tobacco Leaves[OPEN]. The Plant Cell 32(7): 2120-2131.
- Kent, W. J., Zweig, A. S., Barber, G., Hinrichs, A. S. and Karolchik, D. (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26(17): 2204-2207.
- Leinonen, R., Sugawara, H., Shumway, M. and International Nucleotide Sequence Database, C. (2011). The sequence read archive. Nucleic Acids Res 39(Database issue): D19-21.
- Li, H. (2011). Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics 27(5): 718-719.
- Li, H. and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14): 1754-1760.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16): 2078-2079.
- Liu, T. (2014). Use model-based Analysis of ChIP-Seq (MACS) to analyze short reads generated by sequencing protein-DNA interactions in embryonic stem cells. Methods Mol Biol 1150: 81-95.
- Marand, A. P., Chen, Z., Gallavotti, A. and Schmitz, R. J. (2021). A cis-regulatory atlas in maize at single-cell resolution. Cell 184(11): 3041-3055. e21.
- Marand, A. P., Zhang, T., Zhu, B. and Jiang, J. (2017). Towards genome-wide prediction and characterization of enhancers in plants. Biochim Biophys Acta Gene Regul Mech 1860(1): 131-139.
- Melnikov, A., Murugan, A., Zhang, X., Tesileanu, T., Wang, L., Rogov, P., Feizi, S., Gnirke, A., Callan, C. G., Kinney, J. B., et al. (2012). Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nature Biotechnology 30(3): 271-277.
- Minnoye, L., Marinov, G. K., Krausgruber, T., Pan, L., Marand, A. P., Secchia, S., Greenleaf, W. J., Furlong, E. E. M., Zhao, K., Schmitz, R. J., et al. (2021). Chromatin accessibility profiling methods. Nature Reviews Methods Primers 1(1): 10.
- Quinlan, A. R. and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6): 841-842.
- Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A. and Manke, T. (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42(Web Server issue): W187-191.
- Ricci, W. A., Lu, Z., Ji, L., Marand, A. P., Ethridge, C. L., Murphy, N. G., Noshay, J. M., Galli, M., Mejia-Guerra, M. K., Colome-Tatche, M., et al. (2019). Widespread long-range cis-regulatory elements in the maize genome. Nat Plants 5(12): 1237-1249.
- Schmitz, R. J., Grotewold, E. and Stam, M. (2022). Cis-regulatory sequences in plants: Their importance, discovery, and future challenges. Plant Cell 34(2): 718-741.
- Sun, J., He, N., Niu, L., Huang, Y., Shen, W., Zhang, Y., Li, L. and Hou, C. (2019). Global Quantitative Mapping of Enhancers in Rice by STARR-seq. Genomics Proteomics Bioinformatics 17(2): 140-153.
- Thorvaldsdottir, H., Robinson, J. T. and Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14(2): 178-192.
- Zhao, H., Sun, Z. F., Wang, J., Huang, H. J., Kocher, J. P. and Wang, L. G. (2014). CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30(7): 1006-1007.
Supplementary information
Data and code availability: All data and code have been deposited to GitHub: https://github.com/Bio-protocol/Maize_ATAC_STARR_seq
Article Information
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Marand, A. P. (2024). Computational Analysis of Maize Enhancer Regulatory Elements Using ATAC-STARR-seq. Bio-101: e4953. DOI: 10.21769/BioProtoc.4953.
Category
Bioinformatics and Computational Biology
Plant Science > Plant molecular biology > Genetic analysis
Systems Biology > Genomics > Functional genomics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.
![]() Tips for asking effective questions
Tips for asking effective questions
+ Description
Write a detailed description. Include all information that will help others answer your question including experimental processes, conditions, and relevant images.