- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

The Development of an Advanced Model for Multilayer Human Skin Reconstruction In Vivo
Published: Vol 14, Iss 2, Jan 20, 2024 DOI: 10.21769/BioProtoc.4919 Views: 2979
Reviewed by: Pilar Villacampa AlcubierreEVANGELOS THEODOROUAnonymous reviewer(s)
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Human skin reconstruction on immune-deficient mice has become indispensable for in vivo studies performed in basic research and translational laboratories. Further advancements in making sustainable, prolonged skin equivalents to study new therapeutic interventions rely on reproducible models utilizing patient-derived cells and natural three-dimensional culture conditions mimicking the structure of living skin. Here, we present a novel step-by-step protocol for grafting human skin cells onto immunocompromised mice that requires low starting cell numbers, which is essential when primary patient cells are limited for modeling skin conditions. The core elements of our method are the sequential transplantation of fibroblasts followed by keratinocytes seeded into a fibrin-based hydrogel in a silicone chamber. We optimized the fibrin gel formulation, timing for gel polymerization in vivo, cell culture conditions, and seeding density to make a robust and efficient grafting protocol. Using this approach, we can successfully engraft as few as 1.0 × 106 fresh and 2.0 × 106 frozen-then-thawed keratinocytes per 1.4 cm2 of the wound area. Additionally, it was concluded that a successful layer-by-layer engraftment of skin cells in vivo could be obtained without labor-intensive and costly methodologies such as bioprinting or engineering complex skin equivalents.
Key features
• Expands upon the conventional skin chamber assay method (Wang et al., 2000) to generate high-quality skin grafts using a minimal number of cultured skin cells.
• The proposed approach allows the use of frozen-then-thawed keratinocytes and fibroblasts in surgical procedures.
• This system holds promise for evaluating the functionality of skin cells derived from induced pluripotent stem cells and replicating various skin phenotypes.
• The entire process, from thawing skin cells to establishing the graft, requires 54 days.
Graphical overview
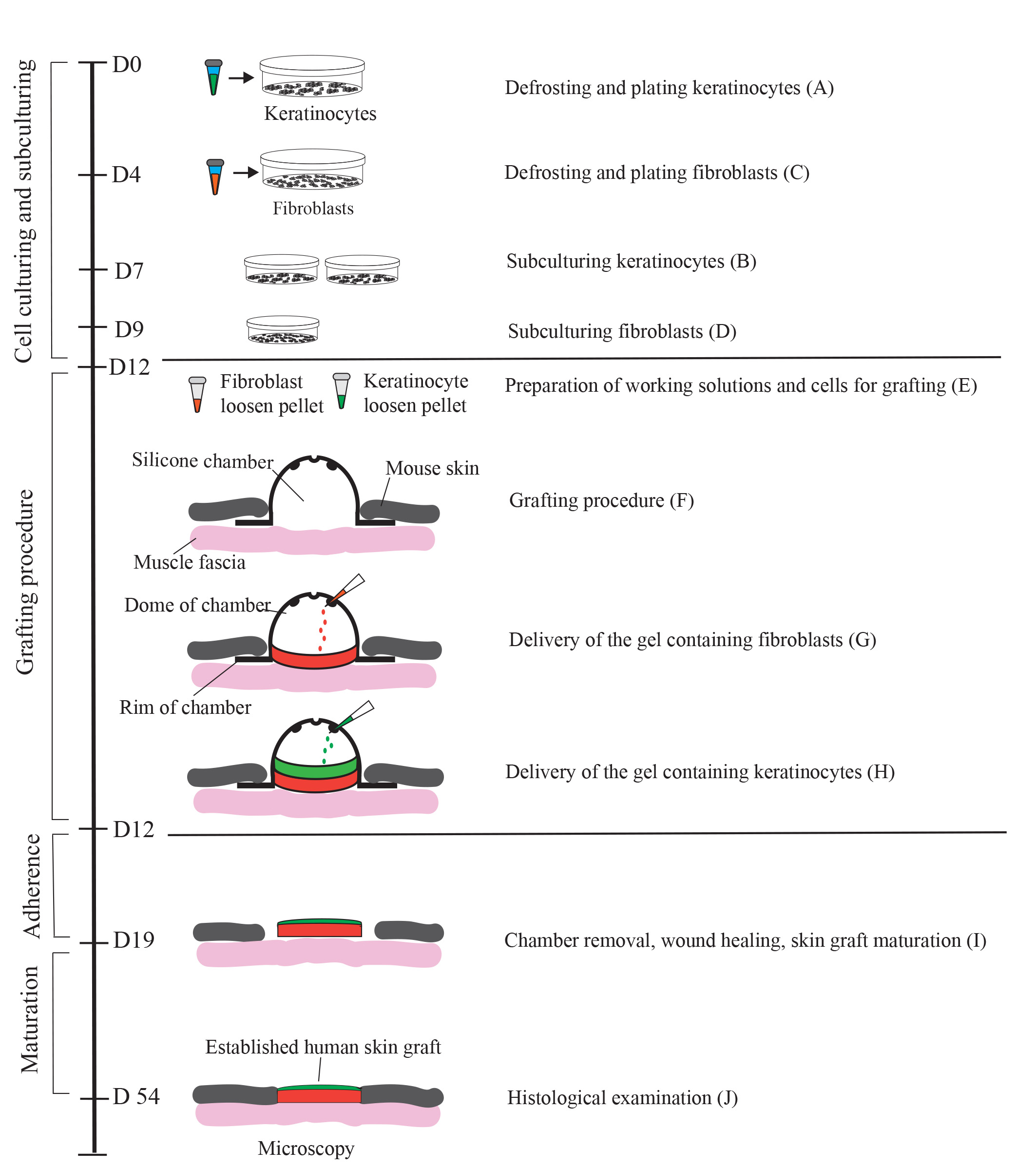
Generation of a human skin equivalent on an immunodeficient mouse using a fibrin-based grafting system. A schematic of the protocol is shown. Cultured keratinocytes and fibroblasts resuspended in a fibrin-based gel are delivered as layers into a silicon chamber inserted underneath the skin of an immunocompromised mouse. First, a fibrin gel containing encapsulated fibroblasts (up to 2 × 106 per 1.4 cm2 wound) is delivered into the chamber and allowed to solidify for 15 minutes. Second, a fibrin gel containing 1.0–2.0 × 106 keratinocytes is applied on top of the fibroblast layer. On day 7 post-grafting, the chamber is removed, and the wound with the graft is allowed to heal for 4–5 weeks. During healing, a scab forms and eventually falls off. By day 54, the graft is fully established.
Background
Progress in mouse genetics has provided a powerful research tool for elucidating the mechanisms of epidermal stem cell commitment and skin homeostasis. However, given the dramatic difference in skin structure between humans and mice, one must be cautious in applying the insights gained in studying mouse skin to human skin, especially when investigating skin diseases. For example, the inbred genetic background of mice can influence the phenotype resulting from disease-associated mutations. Humans are of mixed genetic background, and this complexity results in phenotypical variations and a different penetrance of genetically defined diseases, such as epidermolysis bullosa, a group of rare inherited skin blistering diseases. For this reason, developing complex in vivo human xenograft models is critical to advance our understanding of human skin conditions and developing new therapeutic interventions.
Several human xenograft models have been previously published, including a skin flap assay (Qiao et al., 2008), human skin transplantation-like approaches for grafting dermal-epidermal equivalents onto opened wounds of recipient mice (Escamez et al., 2004; Martinez-Santamaria et al., 2012; Yanez et al., 2015; Jorgensen et al., 2020), and chamber grafting assay (Wang et al., 2000; Diette et al., 2020). While promising, all these assays suffer from several limitations. They include (1) the complexity of the procedure that requires the generation of 3D skin equivalents in cell culture conditions before the transplantation onto a mouse, and (2) the limited availability and proliferative capacity of primary human cells, especially cells derived from patients with rare inherited skin diseases. A chamber grafting assay (Wang et al., 2000; Diette et al., 2020) provides a simplified procedure for grafting primary human skin cells since the assay promotes the self-assembly of fibroblasts and keratinocytes in an in vivo environment without the need to produce skin equivalents in a dish. However, successful production of human skin equivalents in the grafting assay still requires at least 5 × 106 primary keratinocytes per 1.4 cm2 of the wound area, which is a significant number when the availability of primary patient cells is limited. In addition, since this assay relies on the ability of keratinocytes and fibroblasts to self-assemble and form the dermis and epidermis, this limits the ability to incorporate other cell types that normally reside in the skin into an appropriate skin layer. Therefore, we aimed to modify the grafting chamber assay by decreasing the number of primary skin cells necessary for transplantation and mimicking the natural three-dimensional structure of the human skin in vivo to make a more viable in vivo model to study human skin conditions.
To achieve this, we modified the delivery of human keratinocytes and fibroblasts into the grafting chamber by first suspending these cells in a fibrin-based hydrogel. We then performed the sequential transplantation of fibroblasts followed by keratinocytes to recapitulate two major layers of the skin, the dermis and the epidermis, in a controlled manner. Due to its simplicity, this method can be easily optimized by incorporating additional cell types of different origins and ensuring that they are targeted explicitly into the dermis or the epidermis. In addition to grafting primary keratinocytes and fibroblasts, this system can be appropriate for testing the functionality of skin cells derived from induced pluripotent stem cells. The system is also applicable for testing novel skin transplantation systems for potential clinical applications, including cell harvesting and application techniques for treating cutaneous skin defects and pigmentation disorders.
Materials and reagents
Biological materials
Nude mice as recipients for grafting: homozygous nude Foxn1nu, formerly Hfh11nu, females, 6–8 weeks (Jackson Laboratory, Nu/J, #002019; https://www.jax.org/strain/002019). Alternatively, the homozygous NOD-Prkdcscid mice can be recipients for grafting (Jackson Laboratory, NOD.Cg-Prkdcscid/J, # 001303). Both nude and NOD-SCID mouse strains show efficient engraftment of human cells. However, NOD-SCID mice require depilation on the back before surgery and often before graft harvest. Therefore, careful consideration should be given to selecting the appropriate mouse strain for immunological studies to ensure compatibility with the experimental requirements and to minimize any potential confounding factors related to hair removal (Waldron-Lynch et al., 2012; Cristobal et al., 2021).
Note: Before preparing cell cultures, ensure that all mice are healthy and fully acclimated to the facility for at least a week. Ensure that the weight of the mice is above 21 g.
Fibroblasts, primary, neonatal (ATCC, catalog number: PCS-201-010)
Human epidermal keratinocytes, primary, neonatal (HEKn) (ThermoFisher, catalog number: C0015)
Reagents
DMEM/F12 (1:1) (ThermoFisher, catalog number 11320-033)
Pooled human AB serum derived (Innovative Research, ISERAB)
MEM-NEAA (ThermoFisher, catalog number: 11140050
GlutaMax (ThermoFisher, catalog number: 35050061)
2-Mercaptoethanol (ThermoFisher, catalog number: 21985023)
L-Ascorbic acid (Sigma-Aldrich, catalog number: A4544)
Hydrocortisone (Sigma-Aldrich, catalog number: H0888)
Antibiotic/Antimycotic (ThermoFisher, catalog number: 15240062)
Human bFGF (ThermoFisher, catalog number: PHG0263)
Human EGF (ThermoFisher, catalog number: PHG0313)
EpiLife medium (ThermoFisher, catalog number:MEPI500CA)
EpiLife defined growth supplement (EDGS) (ThermoFisher, catalog number: S0125)
Bovine collagen solution, type I, 3 mg/mL (Advanced BioMatrix, catalog number 5005).
Fetal bovine serum, qualified, one shot, raw (ThermoFisher, catalog number: A31605-01)
Aprotinin (Sigma-Aldrich, catalog number: A6279-10ML)
Fibrinogen from human plasma (Sigma-Aldrich, catalog number: F3879-1G)
Thrombin from human plasma (Sigma-Aldrich, catalog number: T4393-100UN)
DPBS without calcium and magnesium (ThermoFisher, catalog number: 14190-144)
0.25% Trypsin-EDTA (ThermoFisher, catalog number: 25200-056)
Accutase (StemCell, catalog number: 7920)
CRYO defined, animal component free freezing medium, 2× (ZenBio, catalog number: CNT-CRYO-50)
CryoStor CS10, 1× (StemCell, catalog number: 100-1061)
Anti-mouse keratin (K)1 antibody, Rabbit, 1:500 dilution (BioLegend, catalog number: 905602)
Anti-mouse/human K14 antibody, Chicken, 1:2,000 dilution (BioLegend, catalog number: 906004)
Anti-human Loricrin antibody, Rabbit, 1:2,000 dilution (Abcam, catalog number: 176322)
Anti-human Vimentin antibody, Mouse, 1:500 dilution (Abcam, catalog number: 16700)
Anti-Rabbit-488 secondary antibody (Invitrogen, catalog number: A11008)
Anti-Chicken-594 secondary antibody (Invitrogen, catalog number: A11042)
Anti Mouse-594 secondary antibody (Invitrogen, catalog number: 11032)
Formalin solution, neutral buffered, 10% (Sigma-Aldrich, catalog number: HT501128)
Solutions
Stock solutions
Thrombin stock solution (25 U/mL): made from powder (see Recipes)
Fibrinogen stock solution (40 mg/mL): made from powder (see Recipes)
Bovine collagen solution, type I (3 mg/mL): ready to use
Working solutions
Complete FEM medium (fibroblasts culturing) (see Recipes)
Complete EpiLife medium (keratinocytes culturing) (see Recipes)
Collagen solution (coating for keratinocyte culture) (see Recipes)
Fibrin gel containing fibroblasts (see Recipes)
Fibrin gel containing keratinocytes (see Recipes)
Recipes
Recipes for stock solutions
Thrombin stock solution
Working under aseptic conditions in a biosafety cabinet, reconstitute thrombin in sterile DPBS (without Ca2+ and Mg2+) at a concentration of 25 U/mL. Make an aliquot and freeze at -20 °C. The day before the surgery, defrost the thrombin solution overnight at 2–8 °C. Keep thrombin on ice before applying it for hydrogel preparation in the grafting procedure. The solution should be used promptly; however, it can be refrigerated at 2–8 °C for up to three days.
Fibrinogen stock solution
Working under aseptic conditions in a biosafety cabinet, reconstitute fibrinogen in sterile DPBS (without Ca2+ and Mg2+) at a concentration of 40 mg/mL stepwise. First, lay a thin layer of fibrinogen powder on top of warm (37 °C) DPBS and let it soak for 5 min at 37 °C; repeat the soaking procedure with the rest of the fibrinogen. Second, put the tube with the fibrinogen-saline solution in a water bath at 37 °C for 30–60 min and agitate the tube gently every 10 min. Ensure that there are no clumps of dry fibrinogen on the tube walls. Third, leave the tube on the shaker at 2–8 °C overnight to complete the preparation of the fibrinogen stock solution. If a significant number of undissolved clumps are left, repeat the 30-min incubation at 37 °C. Do not exceed 1 h of incubation at 37 °C. The fibrinogen-saline solution must not be vortexed. Fibrinogen should be filter-sterilized using a 0.22 μm syringe filter. Do not use vacuum filtration since this will lead to the breakdown of the molecule during the filtration. Make 50–100 µL aliquots and freeze at -20 °C.
Note: Alternatively, bovine fibrinogen and thrombin can be used.
Recipes for working solutions
Complete FEM medium (Table 1)
Prepare FEM medium by supplementing DMEM/F12 with 5% human serum, 0.5× MEM-NEAA, 0.5× GlutaMAXTM supplement, 55 µM GibcoTM 2-mercaptoethanol, 1× HyClone Antibiotic/Antimycotic, 50 µg/mL ascorbic acid, and 1 µg/mL hydrocortisone. Perform 0.22 µm sterile filtration and store at 4 °C for up to one month. Before application, add human bFGF to a final concentration of 12 ng/mL and human EGF to a final concentration of 5 ng/mL.
Table 1. Composition of complete FEM medium
Reagent Stock concentration Final concentration Amount DMEM/F12 (1:1) 92.763 mL Human serum 100% 5% 5 mL MEM-NEAA 100× 0.5× 0.5 mL GlutaMax Supplement 100× 0.5× 0.5 mL 2-Mercaptoethanol 55 mM 55 µM 0.1 mL Ascorbic acid 50 mg/mL 50 µg/mL 0.1 mL Hydrocortisone 5 mg/mL 1 µg/mL 0.02 mL Antibiotic/Antimycotic 100× 1× 1 mL 1. Sterilize using a 0.22 µm filter; 2. Add bFGF and EGF as shown below AFTER filtration Reagent Stock concentration Final concentration Amount bFGF 100 µg/mL 12 ng/mL 0.012 mL EGF 100 µg/mL 5 ng/mL 0.005 mL Total n/a n/a 0.005 mL Complete EpiLife medium (Table 2)
Prepare complete EpiLife medium by adding EpiLife Defined Growth Supplement (EDGS) following the manufacturer’s instruction. Add antibiotic/antimycotic.
Table 2. Composition of complete EpiLife Medium
Reagent Stock concentration Final concentration Amount EpiLife basal medium n/a n/a 500 mL EDGS 100× 1× 5 mL Antibiotic/Antimycotic 100× 1× 5 mL Total n/a n/a 510 mL Collagen solution
To prepare the collagen working solution for the coating of the tissue culture dishes, dilute the 3 mg/mL bovine collagen stock solution to a final working concentration of 30 µg/mL in 1× DPBS. Use fresh.
Fibrin gel containing fibroblasts (Table 3)
To prepare the fibrin gel containing fibroblasts, first make working Solution 1. To make working Solution 1, supplement DMEM/F12 1:1 with 1× Antibiotic/Antimycotic, 1% raw FBS, and aprotinin. Mix components in an Eppendorf tube and keep the tube on ice until ready to graft. Just before grafting, mix Solution 1 with fibroblasts and then add fibrinogen and thrombin. Mix components and use for grafting.
Critical: Do not add fibroblasts, fibrinogen, and thrombin into Solution 1 until ready to graft*.
Table 3. Composition of the gel containing fibroblasts
Reagent Stock concentration Final concentration Amount Solution 1 DMEM/F12 (1:1) n/a n/a 291.7 µL Antibiotic/Antimycotic 100× 1× 4 µL Raw FBS 100% 1% 4.1 µL Aprotinin (3-7 TIU/mg protein) n/a 23.5 µL Just before grafting, add fibroblasts, fibrinogen, and thrombin as shown below. Reagent Stock concentration Final concentration Amount Fibroblasts* n/a 2 × 106 50 µL Fibrinogen* 40 mg/mL 1.73 mg/mL 17.3 µL Thrombin* 25 U/mL 0.59 U/mL 9.4 µL Total n/a n/a 400 µL Fibrin gel containing keratinocytes (Table 4)
To prepare the fibrin gel containing keratinocytes, first make working Solution 2. To make a working Solution 2, supplement EpiLife medium with 1× Antibiotic/Antimycotic and aprotinin. Mix components in an Eppendorf tube and keep the tube on ice until ready to graft. Just before grafting, mix Solution 2 with keratinocytes and then add fibrinogen and thrombin. Mix components and use for grafting.
Critical: Do not add keratinocytes, fibrinogen, and thrombin into Solution 2 until ready to graft**.
Table 4. Composition of the gel containing keratinocytes
Reagent Stock concentration Final concentration Amount Solution 2 EpiLife n/a n/a 295.8 µL Antibiotic/Antimycotic 100× 1× 4 µL Aprotinin (3-7 TIU/mg protein) n/a 23.5 µL Just before grafting, add keratinocytes, fibrinogen, and thrombin as shown below. Reagent Stock concentration Final concentration Amount Keratinocytes** n/a 2 × 106 50 µL Fibrinogen** 40 mg/mL 1.73 mg/mL 17.3 µL Thrombin** 25 U/mL 0.59 U/mL 9.4 µL Total n/a n/a 400 µL
Note: For a large-scale experiment, the working hydrogel solutions for the dermal and epidermal layers can be prepared as a combined mix to facilitate the generation of multiple grafts simultaneously. To maximize efficiency, 2–4 grafts can be performed in a single experimental setup. Optional: recalculate the volumes for Solution 1 and Solution 2 based on the number of grafts.
Equipment
Silicone chambers [produced by Qure Medical, 1810 Renaissance Boulevard, Sturtevant, WI, 53177, (262) 417–1307, http://www.qure-med.com, according to the following dimensions: 12 mm inner diameter (ID), 20 mm outer diameter (OD), 10 mm tall (Part# 12–24, upper chamber)]. Chambers are made of Momentive LSR 2650, two-component liquid silicone rubber for injection molding processes (Figure 2A).
Note: The chambers are reusable and autoclavable. Manually punch three holes in the dome of the chamber using a 2 mm biopsy punch to allow for gas exchange during the time the chamber is in place under the skin.
Nikon Eclipse 90i + Photometrics Cool SNAP HQ2+ DS Fi1 (Nikon, model: Ti2-E)
Nikon eclipse Ti-U inverted microscope (Nikon, model: Ti-U)
CO2 incubator (ThermoFisher, model: HERA cell VIOS 160)
Straight forceps (VWR, catalog number: 82027-398)
Surgical scissors (VWR, catalog number: 82027-584)
Curved forceps (VWR, catalog number: 89259-946)
Pipettes P1000 (Gilson, catalog number: F144059M)
Pipettes P200 (Gilson, catalog number: F144058M)
Pipettes P20 (Gilson, catalog number: F144056M)
Sterile filtration system (Corning, catalog number: 431097)
-80 °C freezer (ThermoFisher, model: TSX Series with V-Drive)
Heating pad-T/Pump Professional (Gaymar, catalog number: TP700)
Heating pad-blanket (Cincinnati Sub-Zero, model: 274)
Liquid bandage New skin (Amazon, catalog number: 851409007011)
Software and datasets
NIS-elements Nikon (https://www.nikoninstruments.com/Products/Software)
Microsoft Excel (Microsoft, https://products.office.com/en-us/excel)
GraphPad Prism (GraphPad, https://www.graphpad.com/scientific-software/prism/)
Adobe Illustrator (https://www.adobe.com/products/illustrator/free-trial-download.html)
Procedure
The generation of complete grafts in this protocol spans 54 days and involves several distinct steps, as depicted in Graphical overview. In Step 1, lasting from Days 1 to 12, cryopreserved keratinocytes and fibroblasts are thawed, cultivated, and subcultured. Step 2, which occurs on Day 12, entails the surgical insertion of a silicon chamber and the establishment of cell-seeded fibrin hydrogels. In Step 3, which takes place on Day 19, the silicon chamber is removed to promote the adherence of the graft to the surrounding tissue. Finally, in Step 4, spanning Days 19–54, the graft establishes and undergoes maturation.
The described protocol outlines the process for conducting a single surgery.
Defrosting and plating keratinocytes
Coat two 10 cm tissue culture dishes with collagen.
Dilute bovine collagen stock solution (3 mg/mL) to a final working concentration of 30 µg/mL in 1× DPBS. For each 10 cm dish, resuspend 60 µL of the collagen solution in 6 mL of 1× DPBS in a 15 mL conical tube. Pipette to mix.
Add 6 mL of diluted collagen solution to each 10 cm (~60 cm2) tissue culture dish. Rock back and forth to ensure complete coverage.
Place into the 37 °C/5% CO2 incubator for 1 h.
Prepare complete EpiLife medium (see Recipes).
Prewarm a 25 mL aliquot of complete EpiLife medium to room temperature (RT).
Aspirate non-polymerized collagen from the 10 cm dishes prepared in step A1. Immediately add 9 mL of complete EpiLife medium and transfer the dishes into a 37 °C/5% CO2 tissue culture incubator to equilibrate for 30 min.
Pipette 5 mL of EpiLife into a 15 mL conical tube. Set aside in a biosafety cabinet.
Remove a cryovial that contains approximately 5 × 105 frozen keratinocytes from liquid nitrogen storage and immediately place it into a 37 °C water bath. Thaw quickly (approximately 1–2 min), avoiding contamination by keeping the cap above the water level.
Note: The thawing time of frozen cells depends significantly on the volume of the cells being thawed. After one minute of thawing, gently shake the vial and quickly observe the contents. If most of the cell suspension has melted, you can proceed with the next step.
Spray the entire vial with 70% ethanol, wipe, and proceed under strict aseptic conditions in a biosafety cabinet.
Transfer thawed keratinocytes into the conical tube prepared in step A5.
Centrifuge the cells at 200× g for 5 min at RT.
Gently resuspend the pellet in 1 mL of complete EpiLife medium and transfer 500 µL of the cell suspension with 2.5 × 105 keratinocytes into each 10 cm dish with the pre-equilibrated EpiLife medium prepared in step A4, to reach a proper/required plating density of 4 × 103 cells/cm2.
Gently and thoroughly disperse the cells by alternating between an up/down and then left/right motion. Repeat these motions two more times to ensure even distribution. Do not swirl the plate to mix. Once the cells are evenly dispersed, place the plate into a 37 °C/5% CO2 tissue culture incubator.
The next day, check cell attachment under a microscope.
Change the complete EpiLife medium every other day until cells reach approximately 60% confluency, then change daily until keratinocyte culture reaches 70%–80% confluency, at which point the cells must be passaged.
Note: If plated correctly at the density of 2.5–4 × 103/cm2, the culture should be ready for subculturing in 5–7 days.
Keratinocytes must be subcultured to perform better during grafting (see below). The rest of the cells can be frozen for future experiments.
Subculturing keratinocytes
Note: Two 10 cm dishes with keratinocytes at 80% confluency yield approximately 2.4 × 106 cells (see Figure 1A). Since 2.0 × 106 keratinocytes are needed per surgery, two 10 cm dishes are sufficient to obtain this number of cells.
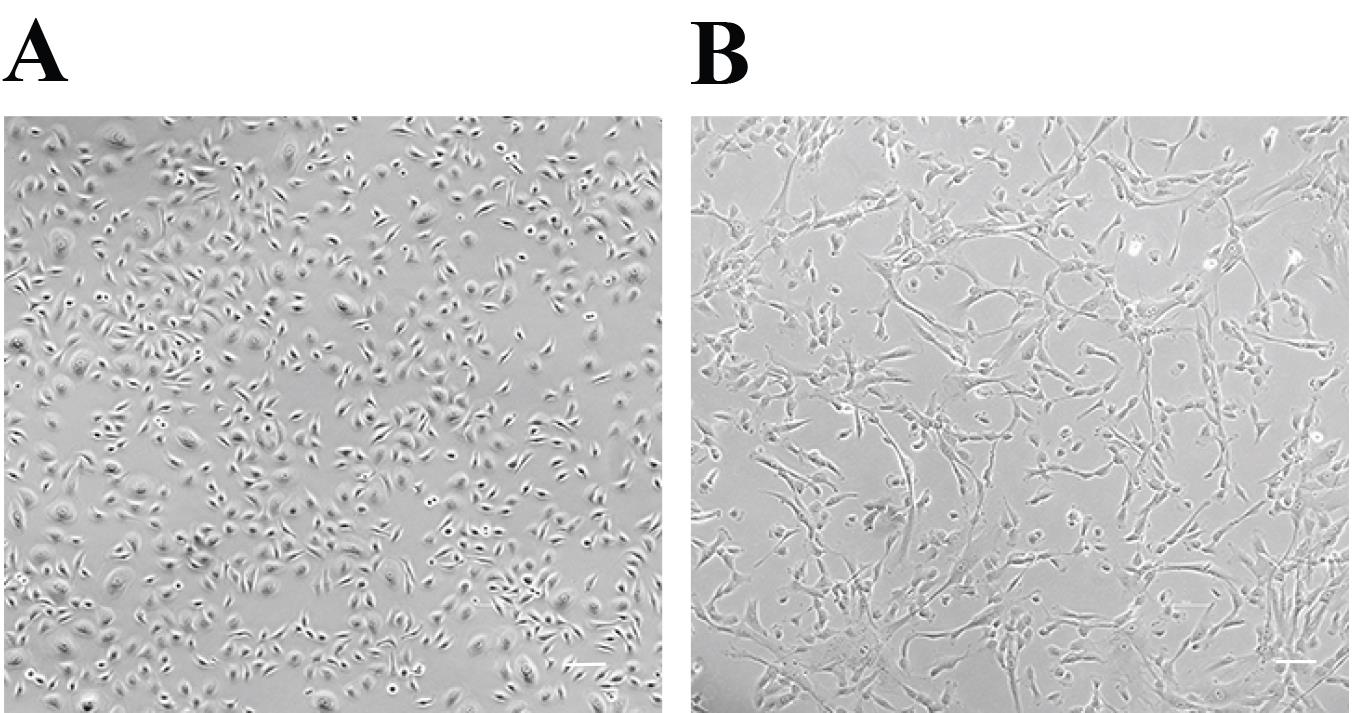
Figure 1. Representative picture of keratinocyte and fibroblast cultures ready to harvest for grafting. Keratinocytes (A) and fibroblasts (B) are shown at the appropriate confluency for grafting. Scale bar, 50 µm.Allow Accutase and complete EpiLife medium to equilibrate to RT.
Prepare two 10 cm tissue culture dishes and coat them with bovine collagen following the procedure described in step A1. Place into a 37 °C/5% CO2 tissue culture incubator for 1 h.
After 1 h of incubation, aspirate non-polymerized collagen from the 10 cm dishes prepared above and immediately add 9 mL of complete EpiLife medium. Transfer the dishes into a 37 °C/5% CO2 tissue culture incubator to equilibrate for 30 min.
Take the dish with keratinocytes that require passaging from the tissue culture incubator and aspirate spent media without disturbing the cell monolayer.
Add 6 mL of DPBS, gently rocking the dish to wash the cells. Aspirate DPBS.
Add 3 mL of Accutase and incubate at 37°C for 6–9 min.
Note: Assess the cells after 5–6 min of incubation. Many cells will start detaching or adopting a round shape morphology during this time. If more than 30% of the cells are still attached, gently tap the dish and return it to the incubator for an additional 2–3 min. After 9 min of incubation, nearly all cells should detach. It is crucial to avoid prolonged cell exposure to Accutase. Do not exceed 9 min.
Critical: If keratinocytes detach completely within only 3–4 min, this indicates that the keratinocytes have started differentiating. In this case, they should not be used for grafting; start again with earlier passage keratinocytes.
After incubation, tap the dish gently and add 3 mL of complete EpiLife medium to neutralize Accutase. Proceed to the next step immediately.
Observe under the microscope. Most cells should be floating in suspension. If not, use a wide cell scraper to collect all partially attached cells, especially from the edges of the dish.
Tilt the dish slightly and use a 10 mL serological pipette to gently pipette the keratinocytes one to two times, ensuring all cells are washed from the surface of the dish. Take care to avoid forming bubbles during this process.
Transfer the dissociated cells into a new, sterile 15 mL conical tube. Add 6 mL of DPBS to rinse away Accutase.
Centrifuge the cells at 200× g for 5 min at RT.
Aspirate supernatant. Resuspend the cell pellet in 1–5 mL of fresh EpiLife medium and count cells using a hemocytometer.
Transfer 2.5–4.0 × 103 keratinocytes per cm2 into each collagen-coated 10 cm dish with pre-equilibrated EpiLife medium prepared in step B3.
Gently and thoroughly disperse the cells by alternating between an up/down and then left/right motion. Repeat these motions two more times to ensure even distribution. Do not swirl the plate to mix. Once the cells are evenly dispersed, place the plates into a 37 °C/5% CO2 tissue culture incubator and leave the plates undisturbed for at least 24 h to allow the cells to attach to the surface of the dish.
The next day, examine cell attachment under a microscope.
Change complete EpiLife medium every other day until the keratinocyte culture reaches approximately 60% confluency. Once it reaches this density, change the medium daily until the culture reaches 70%–80% confluency. At this point, the cells are ready to be passaged.
Note: If keratinocytes are plated at a density of 2.5–4 × 103 cells/cm2, the culture should be ready for subculturing in 4–5 days (see Figure 1A).
Defrosting and plating fibroblasts
Prepare complete FEM medium as described above (see Recipes).
Prewarm a 25 mL aliquot of complete FEM medium to RT.
Add 9 mL of complete FEM medium into two 10 cm dishes and transfer them into a 37 °C/5% CO2 tissue culture incubator to pre-equilibrate for 30 min.
Pipette 5 mL of complete FEM medium into a 15 mL conical tube. Set aside in a biosafety cabinet.
Remove the cryovial containing approximately 5 × 105 frozen fibroblasts from liquid nitrogen storage and immediately place it into a 37 °C water bath. Thaw quickly (approximately 1–2 min), taking care to avoid contamination by keeping the cap above the water level.
Note: Thawing time depends on the volume of frozen cells. After one minute, shake the vial and quickly observe the contents. If most of the cell suspension has melted, you can proceed with the next step.
Spray the entire vial with 70% ethanol, wipe, and proceed under strict aseptic conditions in a biosafety cabinet.
Transfer thawed fibroblasts into a conical tube with 5 mL of complete FEM medium prepared in step C4.
Centrifuge the cells at 200× g for 5 min at RT.
Gently resuspend the pellet in 1 mL of complete FEM medium and transfer 500 µL of the cell suspension with 2.5 × 105 fibroblasts into each 10 cm dish with the pre-equilibrated FEM medium prepared in step C3, to reach a proper/required plating density of 4 × 103 cells/ cm2.
Gently and thoroughly disperse the cells by alternating between an up/down and then left/right motion. Repeat these motions two more times to ensure an even distribution. Do not swirl the plate to mix. Once the cells are evenly dispersed, place the plate into a tissue culture incubator and leave it undisturbed for at least 24 h to allow the cells to attach to the dish.
The next day, examine cell attachment under a microscope.
Change complete FEM medium every other day until the cells reach approximately 80% confluency. Once they reach this density, passage the cells.
Note: If fibroblasts are cultured in FBS-containing medium, they should be transitioned into FEM medium (see Recipes, Table 1). Culturing fibroblasts in complete FEM medium for four days is sufficient for a successful transition, which will result in changes in fibroblast phenotype and accelerated growth of these cells, as shown in Figure 1B.
Fibroblasts must be subcultured to perform better during grafting (see below). The rest of the cells can be frozen for future experiments.
Subculturing fibroblasts
Note: One 10 cm dish with fibroblasts at 85% confluency yields approximately 2.5 × 106 cells (see Figure 1B). Since 2.0 × 106 fibroblasts are needed per surgery, one 10 cm dish is sufficient to obtain this number of cells.
Prepare one 10 cm tissue culture dish and allow Trypsin-EDTA and complete FEM medium to equilibrate to RT.
Add 9 mL of complete FEM medium to the dish and transfer it into a 37 °C/5% CO2 tissue culture incubator to equilibrate for 30 min.
Take the dish with fibroblasts that require passaging from the tissue culture incubator and aspirate spent media without disturbing the cell monolayer.
Add 10 mL of DPBS, gently rocking the dish to wash the cells. Aspirate DPBS.
Add 3 mL of Trypsin-EDTA and incubate at 37 °C in a tissue culture incubator for 2–3 min.
After incubation, tap the dish gently and add 6 mL of complete FEM medium to neutralize Trypsin-EDTA.
Observe the cells under a microscope. Most cells should be floating in suspension. Alternatively, use a wide cell scraper to collect all partially attached cells, especially from the edges of the dish.
Tilt the dish and gently pipette with a 10 mL serological pipette one to two times to wash all fibroblasts from the surface of the dish. Avoid forming bubbles.
Transfer the dissociated cells into a new, sterile 15 mL conical tube. Add 3 mL of DPBS to dilute Trypsin-EDTA.
Centrifuge cells at 200× g for 5 min at RT.
Aspirate supernatant. Resuspend the cell pellet in 1–5 mL of fresh complete FEM medium and count cells using a hemocytometer.
Transfer 2.5 × 103 fibroblasts per cm2 to the dish with pre-equilibrated FEM medium prepared in step D2.
Gently and thoroughly disperse the cells by alternating between an up/down and then left/right motion. Repeat these motions two more times to ensure even distribution. Do not swirl the plate. Once the cells are evenly dispersed, transfer the plate into a 37 °C/5% CO2 tissue culture incubator and leave the plate undisturbed for at least 24 h to allow the cells to attach to the dish.
The next day, check cell attachment under a microscope.
Change the complete FEM medium every other day until cells reach approximately 70%–80% confluency (Figure 1B).
Note: If fibroblasts are plated at the density of 2.5 × 103/cm2, the culture should be ready for subculturing in 3 days.
Preparation of working solutions and cells for grafting (for one graft)
Defrost thrombin stock solution (25 U/mL) overnight on ice at 4 °C.
Defrost the fibrinogen aliquot in a fridge for approximately 1 h before use.
On the day of the surgery, prepare the working Solution 1 and Solution 2 according to Tables 3 and 4 (see Recipes for working solutions). Immediately place all components on ice while preparing cells and mice for surgery.
Collect expanded keratinocytes and fibroblasts from section B and section D, respectively.
Note: It is important that both keratinocytes and fibroblasts are of good quality and within 75% and 85% confluency, respectively, on the day of surgery (Figure 1A–1B). The use of differentiated or senescent cells will result in graft failure. Collect cells immediately before the surgery.
Collect 2.0 × 106 fibroblasts as described in steps D1–D10, count cells using a hemocytometer, and centrifuge the cells to obtain a pellet. Resuspend the pellet in 50 µL of complete FEM medium and put the tube with resuspended fibroblasts on ice. Promptly use for surgery to preserve cell viability.
Collect 2.0 × 106 keratinocytes as described in steps B1–B11, count cells using a hemocytometer, and centrifuge the cells to obtain a pellet. Resuspend the pellet in 50 µL of complete EpiLife medium and put the tube with resuspended keratinocytes on ice. Promptly use for surgery to preserve cell viability.
(Optional) If frozen-then-thawed keratinocytes and fibroblasts are grafted, follow the procedure below (described for one graft):
Thaw 2.0 × 106 keratinocytes and 2.0 × 106 fibroblasts as described in steps A6–A9 and C5–C8, respectively. Count the cells before centrifuging to ensure that the correct number of viable cells are used.
Carefully resuspend the pellet containing thawed fibroblasts in 50 µL of complete FEM medium and the pellet containing thawed keratinocytes in 50 µL of complete EpiLife medium. Keep the resuspended cells on ice and promptly use them for surgery to preserve cell viability.
Note: It is crucial to freeze and thaw cells under optimal conditions to ensure cell viability and functionality. See the supplementary information below.
Place the following on ice to transport to the animal facility for grafting. All volumes shown below are for one graft:
Solution 1 323.3 µL
Suspension of fibroblasts 50 µL
Solution 2 323.3 µL
Suspension of keratinocytes 50 µL
Fibrinogen (40 mg/mL stock) 50 µL
Thrombin (25 U/mL stock) 25 µL
Grafting procedure
Follow the protocol for the insertion of a silicone chamber into a nude mouse as outlined in Diette et al. (2020) or refer to Figure 2B–2D and Video 1. Briefly:
 Video 1. Procedure for chamber insertion
Video 1. Procedure for chamber insertionAdminister anesthesia to the mice via an intraperitoneal injection with prepared anesthetic solution in adherence to animal protocol.
Wipe the back skin of the anesthetized mouse with an alcohol swab and let the skin dry.
Use forceps to pinch and elevate the skin above the flat area of the back. Cut a piece of the skin with curved scissors to create a round incision, approximately 1 cm in size (Figure 2B).
Apply a 200 µL drop of sterile DPBS with antibiotics onto the opened muscle fascia to provide lubrication. Gently release a rim of the skin surrounding the incision (approximately 5 mm) by lifting the edges with forceps. Simultaneously, glide along the rim of the incision with a 1 mL tip, separating the skin from the underlying muscle fascia.
Using straight forceps, pull up the edge of the skin near the incision, creating an elongated slit-like opening between the skin and muscle fascia. Simultaneously, grasp a silicone chamber with curvy forceps and slightly squeeze the dome of the chamber.
Slide one side of the chamber’s rim beneath the skin on one side of the slit (Figure 2C).
With forceps, gently pull the skin over the chamber’s rim on the other side of the slit simultaneously releasing the curvy forceps. Release both forceps and secure the entire lower rim of the chamber beneath the skin by ensuring that the rim is flat under the skin (Figure 2D).
After the chamber has been successfully inserted, transfer the mouse to a clean cage placed on a heating pad without a wire feeder.
Proceed promptly with the cell transplantation procedure as described in section G.
Critical: Ensure that the wound bed inside the implanted chamber does not dry during the procedure.
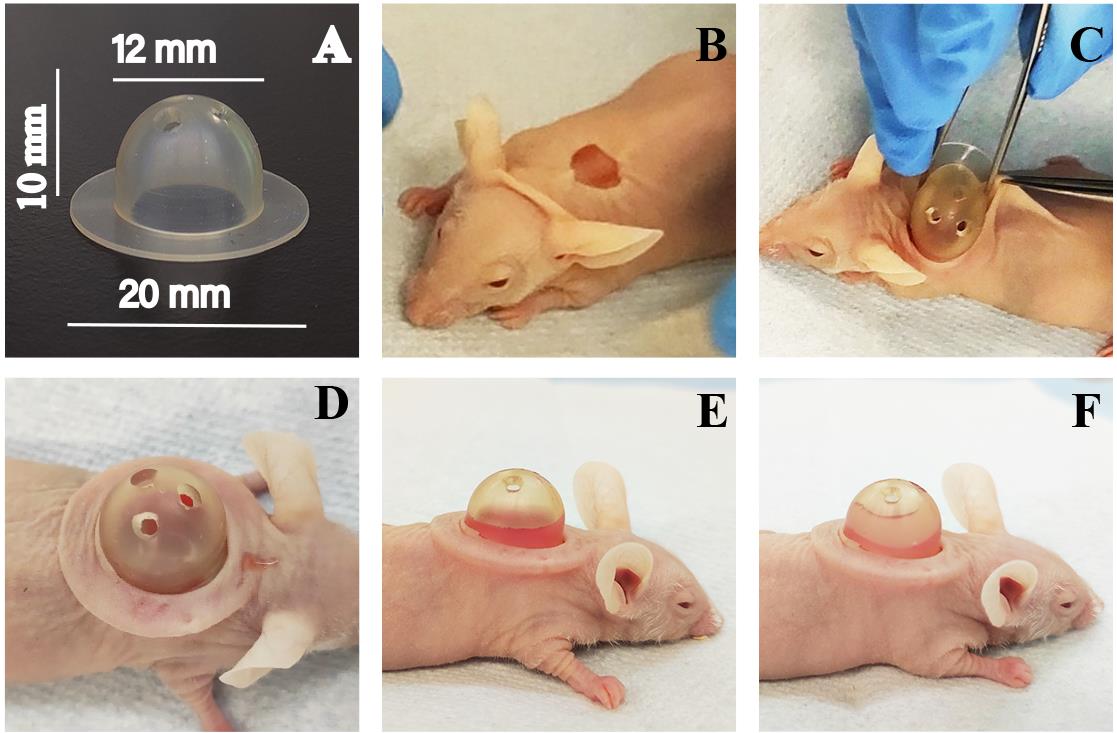
Figure 2. Visual representation of the grafting procedure. (A) Dimensions of the silicone chamber are shown. (B) The mouse skin is cut to create a hole with a circumference of approximately 1 cm for chamber implantation. The chamber is then inserted into the wound (C–D), and a fibroblast-comprised fibrin hydrogel is delivered to form the dermal layer (E). Following the polymerization of the dermal layer, a keratinocyte-comprised fibrin hydrogel is delivered on top (F).
Delivery of the gel containing fibroblasts
Keep all tubes with working solutions, fibrinogen, thrombin, and cell suspensions on ice before use. Spray the vials with 70% ethanol, wipe them, and proceed under strict aseptic conditions in a biosafety cabinet or a clean procedure room in an animal facility.
Critical: To maintain the integrity and viability of the samples, it is imperative to perform all steps expeditiously, ensuring that reagents and materials are kept chilled on ice throughout the procedure.
Using a pipette, transfer the entire volume of cold working Solution 1 (323.3 µL) into the tube containing the suspended fibroblasts prepared in step E4a (or E5 if thawed cells are used). Gently mix the contents, taking care to avoid the formation of bubbles.
Immediately before transferring into an implanted chamber, add 9.4 µL of 25 U/mL thrombin and 17.3 µL of 40 mg/mL fibrinogen to the cell suspension from step G1. Resuspend gently by pipetting 2–3 times, avoiding bubbles (see Table 3).
Immediately transfer the final mix (400 µL) into the chamber using a narrow-tip 1 mL pipette.
Gently shake the mouse to facilitate the distribution of the hydrogel solution throughout the wound bed.
Note: With secure and proper chamber placement, the fibrin gel should be slightly visible in the lower portion of the silicone chamber (Figure 2E).
Place the mouse in the cage on a warming pad and allow it to remain there for a period of 15 min. This timeframe allows the fibrin hydrogel to undergo polymerization, resulting in the formation of a dermal layer. It is important to provide sufficient time for the hydrogel to solidify and establish the desired structural integrity.
Delivery of the gel containing keratinocytes
While the dermal layer solidifies, prepare the epidermal fibrin gel containing keratinocytes.
Using a pipette, transfer the entire volume of cold working Solution 2 (323.3 µL) into the tube containing the keratinocyte suspension prepared in steps E4b (or E5 if thawed cells are used). Gently mix the content, taking care to avoid the formation of bubbles.
Immediately before transplanting into an implanted chamber with the dermal layer, add 9.4 µL of 25 U/mL thrombin and 17.3 µL of 40 mg/mL fibrinogen to the cell suspension. Resuspend gently by pipetting 2–3 times, avoiding bubbles (Table 4).
Immediately transfer the final mixture (400 µL) into the chamber on top of the solidified fibrin gel containing fibroblasts.
Note: Avoid pulling or tilting the silicon chamber when transferring the mixture. It is important to handle the chamber carefully to prevent any disruption or displacement of the grafted material.
Gently shake the mouse to ensure even distribution of the hydrogel-containing keratinocytes over the solidified hydrogel containing fibroblasts previously transferred into the silicone chamber.
Note: With secure and proper chamber placement, the hydrogels should be visible and fill one-half to three-fourths of the silicone chamber (Figure 2F).
Place the mouse in the cage on a warming pad and allow it to remain there for a period of 15–40 min to let the fibrin gel polymerize to create the epidermal layer.
Critical: Ensure that the mouse remains calm for approximately 15 min to allow each layer to polymerize. Additionally, after applying both fibrin layers, keep the mouse undisturbed for at least 1 h. Consequently, the most optimal duration for anesthesia is 2 h.
Keep the chamber on mice for seven days.
Note: During the first 10 days post-surgery, check the mice regularly and provide them with moist chow daily. This helps to ensure the mice are hydrated and supports their post-operative recovery.
Chamber removal, wound healing, and skin graft maturation
On day 7 post-surgery (Day 19), remove the chamber from grafted mice as described in Diette et al. (2020) or refer to Video 2. Briefly:
Video 2. Procedure for chamber removalAdminister anesthesia to the mice via an intraperitoneal injection with prepared anesthetic solution in adherence to animal protocol.Administer anesthesia to the mice via an intraperitoneal injection with prepared anesthetic solution in adherence to animal protocol.
Wipe the skin around the chamber and the dome of the chamber with an alcohol swab.
Carefully loosen the skin around the chamber with sterile forceps.
With a clean, gloved finger, gently squeeze the dome of the silicone chamber and carefully pull one side from under the skin. Carefully release one edge of the rim using forceps.
Ensure that the graft is not attached to the inner part of the chamber and the forming skin graft stays in the wound.
Notes:
If the edge of the graft and connective tissues around it remain attached to the silicone chamber, run another pair of forceps along the silicone lip of the chamber to free the graft.
The resulting graft should appear as an off-white plug with no yellow areas, which indicates minimal tissue decay (Figure 3A–3D). The wound surface can exhibit varying levels of dryness, which does not significantly affect graft formation.
Apply New skin liquid bandage onto the mouse skin around the open wound to prevent mouse skin contraction.
Place the mouse into a clean cage (without a wire feeder) that has been resting on a heating pad for at least 1 h. After the mouse recovers from anesthesia, remove the heating pad and monitor the mouse for the duration of the healing (approximately six weeks after surgery).
Note: The scab that forms after chamber removal will naturally detach within 2–2.5 weeks.
Harvest the graft for histological analysis or live in vivo studies six weeks after surgery (day 54 of the protocol).
Note: The size of the human skin grafts after chamber removal is constrained by the chamber dimensions and ranges from 1.0 to 1.5 cm in diameter (Figure 3A–3D). Following healing, the grafts typically measure between 0.4 and 1.2 cm in diameter (Figure 3E–3H). This size is suitable for various applications, such as assessing skin cell functionality, modeling the phenotype of skin disorders, investigating wound healing stages, studying aging, and evaluating skin barrier function. The graft can be analyzed earlier, at five weeks post-surgery, if necessary.
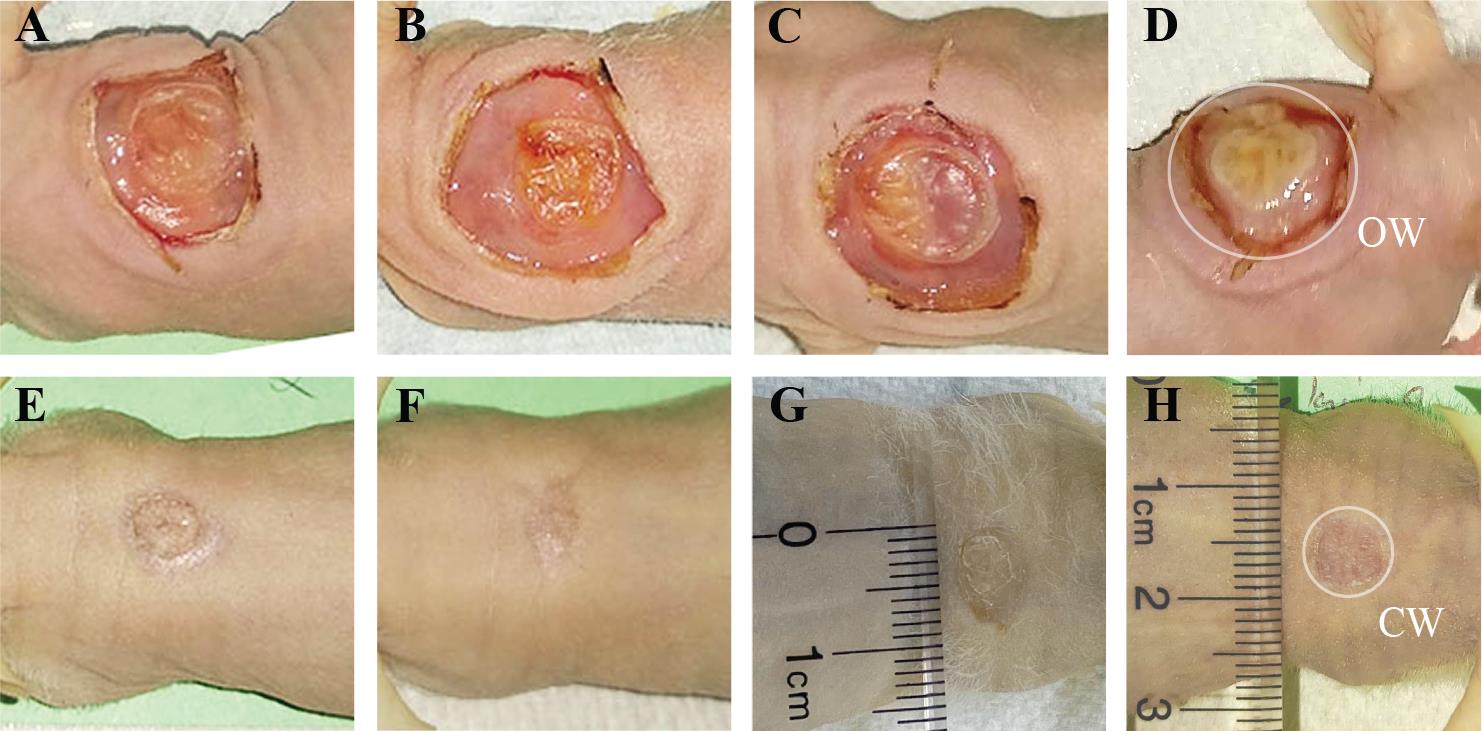
Figure 3. Representative images of grafts at different stages of the healing process. The fibrin-based grafting was performed with (A, E) 2.0 × 106 freshly cultured keratinocytes together with 2.0 × 106 of freshly cultured fibroblasts; (B, F) 1.0 × 106 freshly cultured keratinocytes together with 1.0 × 106 freshly cultured fibroblasts, and (C, G) 2 × 106 frozen-then-thawed keratinocytes together with 2 × 106 frozen-then- thawed fibroblasts. As a control, grafts generated using a classical grafting chamber assay with 5.0 × 106 freshly cultured keratinocytes and 5.0 × 106 freshly cultured fibroblasts are shown (D, H). Representative images of forming grafts on the day of chamber removal (day 7) are shown on top and corresponding fully established grafts at 5 weeks post-grafting are shown on the bottom. OW–opened wound after chamber removal, and CW–closed wound at the day of harvest of the skin graft.
Histological examination of human skin xenografts
Euthanize the mouse on the day of harvesting (Day 54) in accordance with the approved protocol by the Institutional Animal Care and Use Committee (IACUC).
Use a permanent marker to mark the borders of the graft and carefully cut the mouse skin around the graft, maintaining a 5 mm distance from the edge of the graft.
Gently peel off the skin and transfer it to Whatman paper to ensure that it lays flat.
Fix sections with skin grafts in a buffered solution of 10% Formalin for 24 h and then transfer them into 70% ethanol.
Fixed tissue can be paraffin-embedded and sectioned for further histological analysis. If histological analysis is performed, prepare 7 μm sections that include both the center and edge portions of the graft.
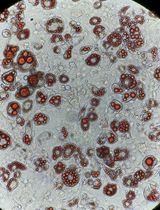
Confirm the presence of the human skin graft through a conventional histological analysis of tissue sections stained with hematoxylin and eosin (H&E) (Yanez et al., 2015). In this staining method, the epidermis shows up as a thin, compact multilayer structure on the top of the skin, exhibiting a dark pink color with a stratum corneum overlay (Figure 4A). The dermis appears as a light-pink, substantial layer beneath the epidermis, featuring nuclei stained in purple. Notably, a human graft typically displays a thicker stratum corneum and epidermis compared to adjacent mouse skin (Figure 4A, 4D, 4G, and 4J).
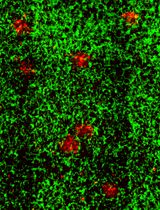
Conduct immunohistochemical analysis following the protocol outlined in Yanez et al. (2015). To label the epidermis, use the anti-K14 antibody (BioLegend 906004) at a 1:2000 dilution. Co-staining with the anti-mouse K1 antibody (BioLegend 905602) at a 1:500 dilution aids in delineating human graft borders (refer to Figure 4B, 4E, 4H, and 4K, where K14 is represented in red and K1 is in green). For visualizing the human dermis, employ a human specific anti-Vimentin antibody (ab16700) at a 1:500 dilution. When co-stained with the anti-human Loricrin antibody (ab176322) at a 1:2000 dilution, the human Vimentin staining provides insights into the proper localization of the human dermis beneath the human epidermis (see Figure 4C, 4F, 4I, and 4L, with Vimentin represented in red and Loricrin in green). Use corresponding secondary antibodies at a 1:300 dilution, as listed in the reagents section. Capture images using a Nikon Eclipse 90i microscope equipped with a Photometrics Cool SNAP HQ2+ DS Fi1 Nikon confocal system or another comparable microscope system.
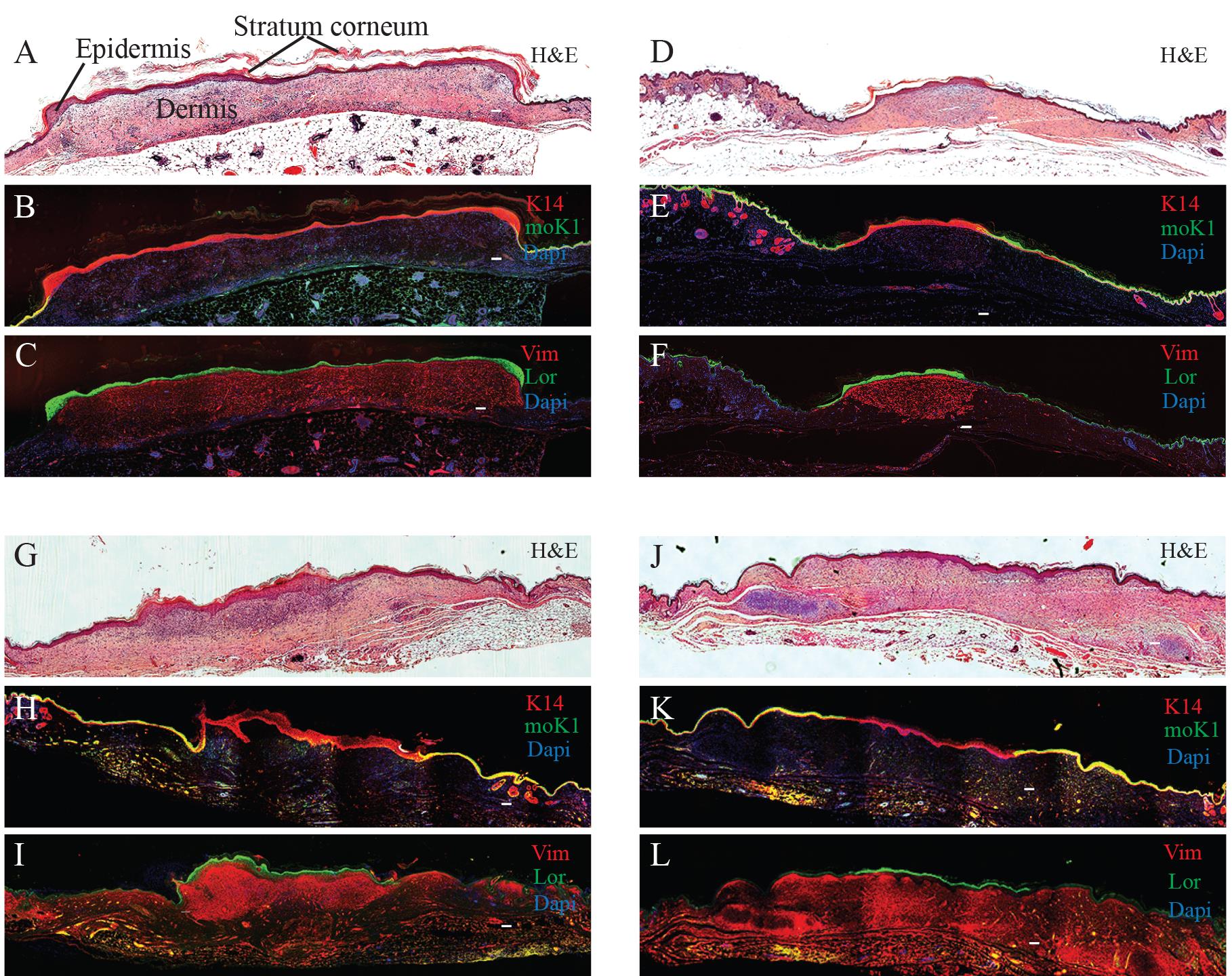
Figure 4. Histological examination of human skin xenografts. Representative images of fibrin-based grafts generated with 2.0 × 106 freshly cultured keratinocytes together with 2.0 × 106 freshly cultured fibroblasts (A–C), 1.0 × 106 freshly cultured keratinocytes together with 1.0 × 106 freshly cultured fibroblasts (D–F), as well as with 2.0 × 106 frozen-then-thawed keratinocytes together with 2.0 × 106 frozen-then-thawed fibroblasts (G–I) are shown. As a control, grafts generated using a classical grafting chamber assay with 5.0 × 106 freshly cultured keratinocytes and 5.0 × 106 freshly cultured fibroblasts were also analyzed (J–L). H & E staining of the grafts (A, D, G, J). Immunofluorescence staining using mouse-specific Keratin (K)1 (moK1; green) and human/mouse-specific K14 (red). Note that the moK1 antibody stains only the mouse epidermis (B, E, H, K). Immunofluorescence staining using human-specific Loricrin (Lor; green) detects the human epidermis, while human-specific Vimentin (Vim, red) detects the human dermis (C, F, I, L). Nuclei stained with DAPI (blue). Scale bar, 100 μm.
Data analysis
To assess the quality of the grafts, several parameters can be assessed such as (1) the length of the developed human skin graft and (2) the thickness of the epidermis and dermis using histological analysis and immunofluorescence staining, as well as (3) the wound contraction. To compare the means of graft measurements between the grafted groups, conduct an ANOVA test (GraphPad Prism).
Graft measurements
Wound contraction
Using a digital camera and a sterile ruler calibrated in centimeters placed laterally underneath the graft, capture an image of the opened wound (OW) on Day 19 after chamber removal and the closed wound (CW) on Day 54 before graft harvesting (Figure 3D and 3H).
Utilizing the Measure tool in Adobe Illustrator, ascertain the number of pixels equivalent to 1 centimeter on the ruler within the photograph. Establish a pixel-to-centimeter conversion.
Determine the number of pixels representing the diameter of the OW and the CW.
Recompute the wound diameters into centimeters, using the pixel-to-centimeter conversion established earlier.
Divide the calculated wound diameter in two to find the radius of the wound (r).
Recalculate the wound area (cm2) following:
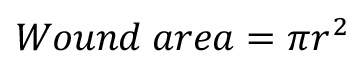
Define the percentage of wound contractions as follows:
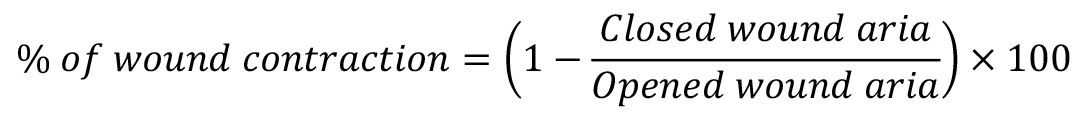
Skin layers measurement
Use NIS-elements Nikon Software to merge colors and set the 100 µm scale bar (Figure 4).
Utilize the Measure tool in Adobe Illustrator to measure the size of the human graft and the thickness of the epidermis and dermis based on immunohistochemical staining of skin layers.
Validation of protocol
To establish this grafting method, a total of 28 grafting procedures were performed. Eight grafting procedures were performed with 2 × 106 freshly cultured keratinocytes together with 2 × 106 freshly cultured fibroblasts with a 100% success rate. When the number of cultured cells decreased, the success rate of grafting decreased to 80%. Specifically, five grafting procedures were performed with 1 × 106 freshly cultured keratinocytes together with 1 × 106 freshly cultured fibroblasts, and four grafts successfully formed. Importantly, a typical success rate of a classical chamber grafting assay that utilizes 5 × 106 of freshly cultured keratinocytes together with 5 × 106 of freshly cultured fibroblasts also fluctuates between 60% and 80% (Wang et al., 2000; Diette et al., 2020). Therefore, our grafting approach allows for more consistent human skin graft formation with fewer cell numbers compared to other methods of skin grafting.
Interestingly, when 2 × 106 frozen-then-thawed keratinocytes together with 2 × 106 frozen-then-thawed fibroblasts were used for grafting, our approach resulted in a 72% success rate (8 successful engraftments out of 11 grafts), providing feasibility data for using frozen-then-thawed skin cells in skin transplantation. Additionally, as a control, four classical skin grafting chamber assays were also performed.
The procedure described in this manuscript is currently being used in our laboratory in at least three different projects and is reproducible among multiple laboratory members.
The quality of the human graft, particularly the size and thickness of the epidermis and dermis, are important factors that influence graft persistence and usefulness in downstream applications. We plotted different measurements of the grafts generated under different conditions and found significant differences in the parameters of the resulting grafts (Figure 5A–5D). Four grafts from each group were analyzed in week 6 after surgery.
First, we analyzed the contraction of the wound area during graft healing. The extent of mouse skin contraction can significantly impact the size of resulting human xenografts once they have healed. Our experimental data revealed that grafts created with 2.0 × 106 freshly cultured keratinocytes together with 2.0 × 106 freshly cultured fibroblasts exhibited wound contraction of 52.8% ± 11.2%, while grafts generated with 1.0 × 106 cultured keratinocytes together with 1.0 × 106 freshly cultured fibroblasts demonstrated a contraction of 88.7% ± 3.7%. The utilization of 2 × 106 frozen-then-thawed keratinocytes together with 2 × 106 frozen-then-thawed fibroblasts resulted in the contraction of 62% ± 5.2%. In comparison, the control graft produced using the classical chamber assay performed with 5.0 × 106 cultured keratinocytes together with 5.0 × 106 cultured fibroblasts exhibited a contraction rate of 60.9% ± 4.7% (Figure 5A). The accuracy of graft size measurements was affirmed through concurrent histological analysis, which showed that the grafts generated with 2.0 × 106 freshly cultured keratinocytes together with 2.0 × 106 freshly cultured fibroblasts exhibited the largest surface area (Figure 4A–4C and Figure 5B).
The reported contraction rates of our grafts were in alignment with other reports that utilized other hydrogel-based methods (Del Rio et al., 2002; Llames et al., 2004; Kalyanaraman and Boyce, 2009; Martinez-Santamaria et al., 2012; Yanez et al., 2015; Jorgensen et al., 2020). For instance, Kalyanaraman and Boyce (2009) reported a contraction rate of approximately 64%–70% in the grafts generated with a 4 cm2 fibroblast-populated collagen-glycosaminoglycan sponge as the dermal component (Kalyanaraman and Boyce, 2009). Another study demonstrated a 50% contraction rate by week 3 when a 6.25 cm wound was covered with a three-layered fibrin-based bio-printed skin (Jorgensen et al., 2020). Similarly, 3D-printed collagen-based grafts exhibited a 62.5% contraction rate by week 6 when applied to a 2.5 cm wound (Yanez et al., 2015).
The thickness of the graft is crucial as it affects the degree of shrinkage within the wound bed over time and influences the diffusion of nutrients from the wound bed during the initial healing stages. We measured the thickness of the dermis and epidermis in our grafts based on histological analysis. The average dermal layer thickness within our grafts ranged from 314.2 to 621.4 µm, which was on the lower end of dermal thickness in the native human skin, with no significant differences between grafting variants (Figure 5C). The thickness of the epidermis ranged between 34.2 and 92.3 µm, which was also on the lower end of the epidermal thickness of native human skin (Lintzeri et al., 2022). The only deviation from this number was observed in grafts generated with 2 × 106 frozen-then-thawed keratinocytes together with 2 × 106 frozen-then-thawed fibroblasts. These grafts developed a thicker epidermis, which ranged from 128 to 140 µm (Figure 5D). The differences in the parameters of the grafts generated with frozen-then-thawed cells vs. those produced with freshly cultured cells probably result from functional alterations during the freezing and thawing processes.
The observed differences in graft characteristics highlight the importance of cell numbers and processing methods in achieving desired outcomes in skin grafting procedures.
In summary, grafts generated with 2.0 × 106 freshly cultured keratinocytes and 2.0 × 106 freshly cultured fibroblasts were reproducible and of superior quality. Therefore, this composition emerges as the most optimal variant within the grafting protocol delineated in this study. Grafts made with frozen-then-thawed keratinocytes and fibroblasts can also serve as an appropriate alternative if needed.
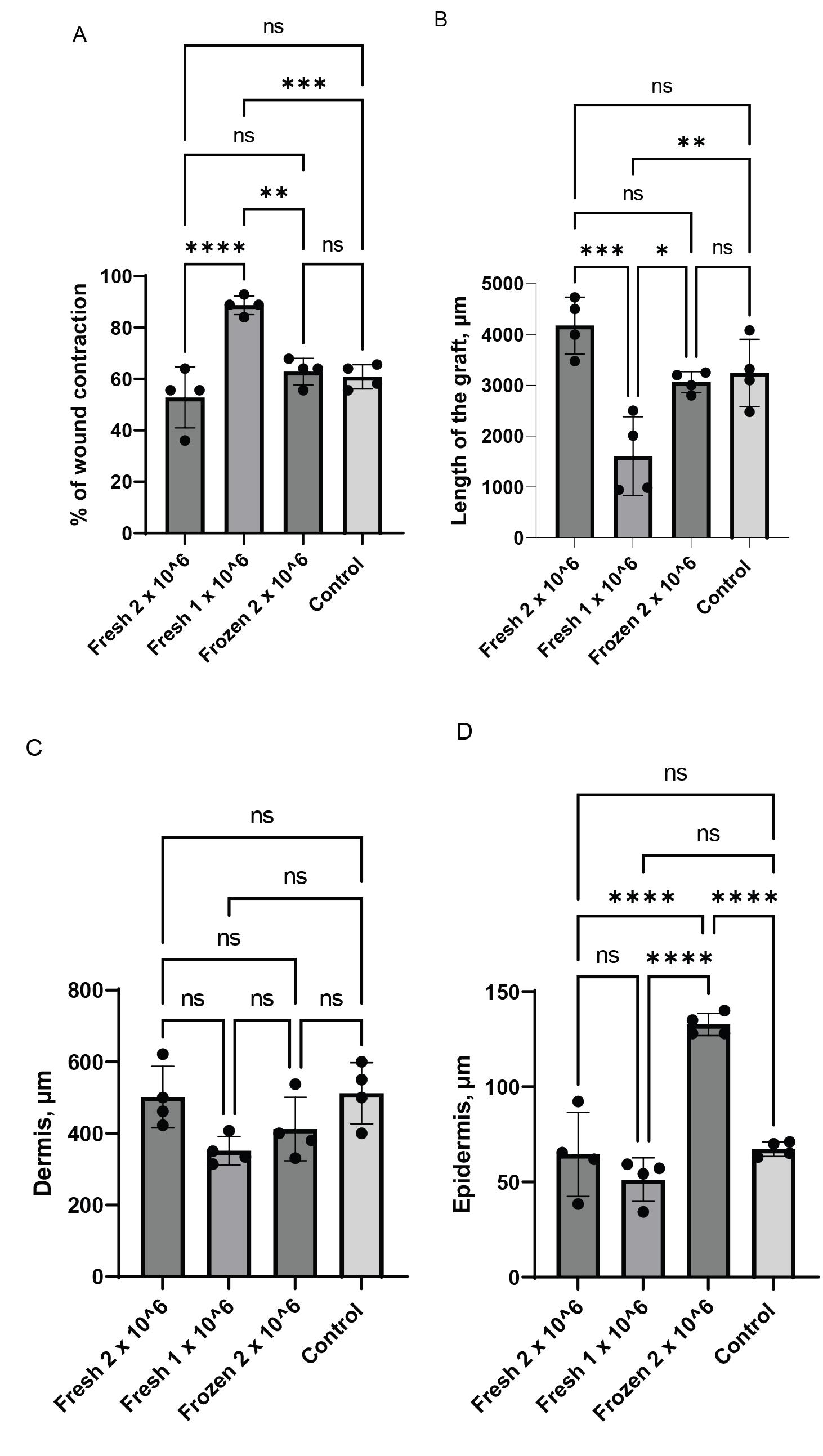
Figure 5. Digital planimetry. Individual plots for a wound contraction rate are shown in A. The length of grafts (B), the thickness of the dermis (C), and the thickness of the epidermis (D) were determined by analyzing immunofluorescence images of grafts developed at 6 weeks after surgery (****p < 0.0001; ***p < 0.002; **p < 0.0094; *p < 0.02).
General notes and troubleshooting
General notes
For the first time, our study reports the successful layer-by-layer engraftment of skin cells in vivo. It shows the feasibility of transplanting layered skin without the need to perform 3D skin bioprinting or generate complex 3D skin equivalents in a dish, both labor intensive and requiring specialized equipment (Cooper et al., 1993; Jorgensen et al., 2020). Therefore, with further modifications, the provided method may serve as a foundation for new clinical protocols for skin transplantation. Our protocol is expected to yield superior outcomes compared to grafting approaches involving keratinocyte sheets, semi-split, or fragile full-thickness grafts.
Our protocol is specifically optimized for the use of human neonatal keratinocytes and fibroblasts on passage two. However, it can be tailored to skin cells collected at later passages (up to passage four for keratinocytes and up to passage six for fibroblasts) and of different origins (data not shown).
Our protocol also allows for the successful engraftment of frozen-then-thawed keratinocytes and fibroblasts (Figures 3C and 3G). This is the first report that shows the successful generation of in vivo full-thickness skin grafts using frozen-then-thawed skin cells without prior cell culture expansion. Since the transportation and storage of frozen cells are more accessible than in vitro–produced skin equivalents, the feasibility of transplanting frozen skin cells may dramatically simplify skin transplantation in burn patients and patients with chronic wounds.
The human skin grafts generated with this protocol persist for at least three months after grafting onto immunocompromised mice.
Human fibrin-based hydrogels are commonly used as a matrix for tissue bioengineering and skin transplantation and are FDA-approved. To ensure the clinical applicability and success of our method, we use highly pure and cell culture–tested human plasma-derived fibrinogen and thrombin in this protocol.
In our modified chamber assay, the final concentration of fibrinogen is 1.73 mg/mL and the thrombin concentration is 0.59 U/mL. These concentrations are significantly lower than the recommended ranges for clinical and research applications, where fibrinogen concentrations typically range from 5 to 80 mg/mL and thrombin concentrations range from 1 to 125 U/mL. However, the concentrations used in our formulation produce soft gels that solidify within 15 min. The soft gel structure has relatively large pore sizes that facilitate cell migration within the gel. In addition, the presence of abundant surrounding medium promotes cell viability. The final stiffness of the gel does not affect the graft size, and the gel effectively covers the entire wound area. Therefore, these conditions are likely to enhance cell viability, support cell division and migration, and facilitate cellular rearrangement in a favorable environment.
A potential limitation of our proposed method is the time required to form hydrogel-encapsulated cells. Compared to the standard chamber assay, creating dermal and epidermal layers in our approach results in a 15-min delay in transferring cells from tissue culture dishes to the wound surface. However, this delay does not affect cell viability, and our method results in less contraction of the grafts (52.8%) compared to the standard chamber assay (60.1%)
Troubleshooting
Graft failure. The quality of keratinocytes is critical for successful engraftment. Preventing keratinocytes from reaching a confluency level higher than 75% is essential. Exceeding this threshold can lead to terminal differentiation of the cells, impeding their effective engraftment. Similarly, fibroblasts should not be allowed to reach 85% confluency before using them for surgery, as they should be mitotically active at the time of grafting. Make at least three biological repeats.
The number of fibroblasts is insufficient for surgery. The dermal component can be successfully generated with fewer than 1.0 × 106 fibroblasts without compromising the reproducibility of the method (data not shown).
The number of keratinocytes is insufficient for surgery. Our protocol can be performed with as low as 0.5 × 106 cultured keratinocytes and 0.5 × 106 cultured fibroblasts. However, the success rate of these grafts is lower, approximately 40% (data not shown).
Frozen cells do not engraft as efficiently as freshly cultured cells. It is crucial to freeze and thaw cells properly for their successful engraftment. Our protocol uses Cryo50 (ZenBio) for cryopreserving keratinocytes and CryoStor CS10 (StemCell Technologies) for fibroblasts. The freezing and thawing procedures are conducted according to the manufacturer’s recommendations, which are further described in Supplementary Materials.
The fibrin gel adheres to the silicon chamber during the grafting procedure. During the grafting procedure, one potential challenge is the adhesion of the fibrin gel to the silicon chamber's walls. To prevent this, ensure that the chamber is clean and free of debris or moisture before introducing the fibrin gel. Avoid overloading the chamber and follow the protocol’s recommended volumes of cell-loaded gels. Overfilling the chamber can lead to excess gel contacting the walls, increasing the likelihood of gel adhesion. Following the protocol diligently and using the specified volumes of gel solutions will contribute to the overall success of the procedure and minimize any issues related to the adherence of the gel to the chamber walls.
Acknowledgments
We are grateful for funding support from the following: the National Institutes of Health (R01AR059947, R01AR078551 and U01AR075932), the Department of Defense (DOD) (W81XWH-18-1-0706), the Epidermolysis Bullosa (EB) Research Partnership, the EB Medical Research Foundation, the Cure EB Charity, the Dystrophic Epidermolysis Bullosa Research Association (DEBRA) International, the Gates Frontiers Fund, and AVITA Medical. This protocol is partly based on the work by Diette, N., Kogut, I. and Bilousova, G. (2020). Generation of a Full-Thickness Human Skin Equivalent on an Immunodeficient Mouse. Methods Mol Biol 2109: 169–183.
Competing interests
DRR has an equity interest in AVITA Medical. KB is an employee of AVITA Medical, and AH was an employee at the time the research was conducted. AVITA Medical may potentially benefit from the research findings presented here. Other authors do not have any potential conflicts of interest to declare.
Ethical considerations
All animals were housed and handled in accordance with the Institutional Animal Care and Use Committee (IACUC) of The University of Colorado Anschutz Medical Campus. All studies were performed with the approval of the IACUC committee.
References
- Cooper, M. L., Andree, C., Hansbrough, J. F., Zapata-Sirvent, R. L. and Spielvogel, R. L. (1993). Direct comparison of a cultured composite skin substitute containing human keratinocytes and fibroblasts to an epidermal sheet graft containing human keratinocytes on athymic mice. J Invest Dermatol 101(6): 811–819.
- Cristobal, L., Asunsolo, A., Sanchez, J., Ortega, M. A., Alvarez-Mon, M., Garcia-Honduvilla, N., Bujan, J. and Maldonado, A. A. (2021). Mouse Models for Human Skin Transplantation: A Systematic Review. Cells Tissues Organs 210(4): 250–259.
- Del Rio, M., Larcher, F., Serrano, F., Meana, A., Munoz, M., Garcia, M., Munoz, E., Martin, C., Bernad, A. and Jorcano, J. L. (2002). A preclinical model for the analysis of genetically modified human skin in vivo. Hum Gene Ther 13(8): 959–968.
- Diette, N., Kogut, I. and Bilousova, G. (2020). Generation of a Full-Thickness Human Skin Equivalent on an Immunodeficient Mouse. Methods Mol Biol 2109: 169–183.
- Escamez, M. J., Garcia, M., Larcher, F., Meana, A., Munoz, E., Jorcano, J. L. and Del Rio, M. (2004). An in vivo model of wound healing in genetically modified skin-humanized mice. J Invest Dermatol 123(6): 1182–1191.
- Jorgensen, A. M., Varkey, M., Gorkun, A., Clouse, C., Xu, L., Chou, Z., Murphy, S. V., Molnar, J., Lee, S. J., Yoo, J. J., et al. (2020). Bioprinted Skin Recapitulates Normal Collagen Remodeling in Full-Thickness Wounds. Tissue Eng Part A 26(9-10): 512–526.
- Kalyanaraman, B. and Boyce, S. T. (2009). Wound healing on athymic mice with engineered skin substitutes fabricated with keratinocytes harvested from an automated bioreactor. J Surg Res 152(2): 296–302.
- Lintzeri, D. A., Karimian, N., Blume-Peytavi, U. and Kottner, J. (2022). Epidermal thickness in healthy humans: a systematic review and meta-analysis. J Eur Acad Dermatol Venereol 36(8): 1191–1200.
- Llames, S. G., Del Rio, M., Larcher, F., Garcia, E., Garcia, M., Escamez, M. J., Jorcano, J. L., Holguin, P. and Meana, A. (2004). Human plasma as a dermal scaffold for the generation of a completely autologous bioengineered skin. Transplantation 77(3): 350–355.
- Martinez-Santamaria, L., Guerrero-Aspizua, S. and Del Rio, M. (2012). [Skin bioengineering: preclinical and clinical applications]. Actas Dermosifiliogr 103(1): 5–11.
- Qiao, J., Philips, E. and Teumer, J. (2008). A graft model for hair development. Exp Dermatol 17(6): 512–518.
- Waldron-Lynch, F., Deng, S., Preston-Hurlburt, P., Henegariu, O. and Herold, K. C. (2012). Analysis of human biologics with a mouse skin transplant model in humanized mice. Am J Transplant 12(10): 2652–2662.
- Wang, C. K., Nelson, C. F., Brinkman, A. M., Miller, A. C. and Hoeffler, W. K. (2000). Spontaneous cell sorting of fibroblasts and keratinocytes creates an organotypic human skin equivalent. J Invest Dermatol 114(4): 674–680.
- Yanez, M., Rincon, J., Dones, A., De Maria, C., Gonzales, R. and Boland, T. (2015). In vivo assessment of printed microvasculature in a bilayer skin graft to treat full-thickness wounds. Tissue Eng Part A 21(1–2): 224–233.
Supplementary information
The following supporting information can be downloaded here:
- Supplementary information
Article Information
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Pavlova, M., Balaiya, V., Flores, J. C., Ferreyros, M., Bush, K., Hopkin, A., Kogut, I., Roop, D. R. and Bilousova, G. (2024). The Development of an Advanced Model for Multilayer Human Skin Reconstruction In Vivo. Bio-protocol 14(2): e4919. DOI: 10.21769/BioProtoc.4919.
Category
Biological Engineering
Cell Biology > Cell Transplantation > Xenograft
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.