- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Phylogenetic Inference of Homologous/Orthologous Genes among Distantly Related Plants

Published: Vol 13, Iss 23, Dec 5, 2023 DOI: 10.21769/BioProtoc.4893 Views: 2290
Reviewed by: Xin QiaoYao XiaoYe XuAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2023
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
The recent surge in plant genomic and transcriptomic data has laid a foundation for reconstructing evolutionary scenarios and inferring potential functions of key genes related to plants’ development and stress responses. The classical scheme for identifying homologous genes is sequence similarity–based searching, under the crucial assumption that homologous sequences are more similar to each other than they are to any other non-homologous sequences. Advances in plant phylogenomics and computational algorithms have enabled us to systemically identify homologs/orthologs and reconstruct their evolutionary histories among distantly related lineages. Here, we present a comprehensive pipeline for homologous sequences identification, phylogenetic relationship inference, and potential functional profiling of genes in plants.
Key features
• Identification of orthologs using large-scale genomic and transcriptomic data.
• This protocol is generalized for analyzing the evolution of plant genes.
Keywords: HomologBackground
Evolution of plant genes is inextricably coupled with various evolutionary events, including endosymbiotic events, whole-genome duplication/triplication (WGD/T), gene loss, and horizontal gene transfer (Zhang et al., 2022). Archaeplastida, including green plants (Viridiplantae), glaucophytes (Glaucophyta), and red algae (Rhodophyta), originate anciently and most of them have experienced multiple WGD/T events, resulting in dramatic changes in copy numbers and complicated evolutionary trajectories of their homologous genes (Qiao et al., 2019). Homologs, orthologs, and paralogs are important concepts for the evolutionary classification of genes, being prevalent in recent comparative genomic studies. Homologs are genes sharing a common origin; orthologs and paralogs are two types of homologous genes, which separately evolved via speciation and gene duplication (Thornton and DeSalle, 2000; Koonin, 2005). Homologous genes generally have a relatively higher degree of sequence similarity than non-homologous genes. Sequence similarity–based searching and phylogenetic analyses are useful tools for identifying homologous sequences of genes and reconstructing their evolutionary routes.
Although the definition of homology/orthology has nothing to do with biological functions, there are major functional connotations (Koonin, 2005). Homologous/orthologous genes among different plants typically perform similar or equivalent functions, which is theoretically plausible and empirically supported. Thus, for a newly identified gene in non-model plants, identifying its homologs/orthologs in model plants or crops that have well-documented functional annotations is very useful to assign its possible functions. Phylogenetic analyses can reconstruct the evolutionary trajectories of homologs/orthologs among various species, which can facilitate the understanding of the molecular mechanisms underpinning its biological functions. Here, taking the acetyltransferase like protein HOOKLESS1 (HLS1) as an example (Lehman et al., 1996; Li et al., 2004), we provide a detailed procedure for homologs/orthologs identification using large-scale genomic and transcriptomic data of distantly related plants. This protocol includes generalized steps and parameters for evolutionary analyses of plant genes, and some of these steps and parameters can be customized based on the genes of interest.
Equipment
Server with a 64-bit Linux-based operating system (Ubuntu 18.04.6 LTS): 512 GB RAM and Intel Xeon (R) Gold 6238 CPU
Desktop with a Windows 10 operating system: Intel Core i5-8300H CPU and 8 GB RAM
Software and datasets
Software and databases used in this protocol are as follows:
Miniconda3-py39_4.12.0-Linux-x86_64 (https://mirrors.tuna.tsinghua.edu.cn/anaconda/miniconda/Miniconda3-py39_4.12.0-Linux-x86_64.sh)
TBtools v1.120 (Chen et al., 2020)
Diamond v2.1.7.161 (Buchfink et al., 2015)
MAFFT v7.453 (Katoh and Standley, 2013)
trimAL v1.4.rev15 (Capella-Gutiérrez et al., 2009)
IQ-TREE v2.2.2.6 (Minh et al., 2020)
InterProScan 5.63-95.0 (Jones et al., 2014)
1KP dataset (One Thousand Plant Transcriptomes Initiative, 2019)
MEME 5.5.3 (Bailey and Elkan, 1994)
iTOL (Interactive Tree Of Life) (Letunic and Bork, 2021)
Jalview v2.11.2.0 (Waterhouse et al., 2009)
Procedure
We show a detailed procedure for homologs/orthologs identification with large-scale genomic data (Figure 1).
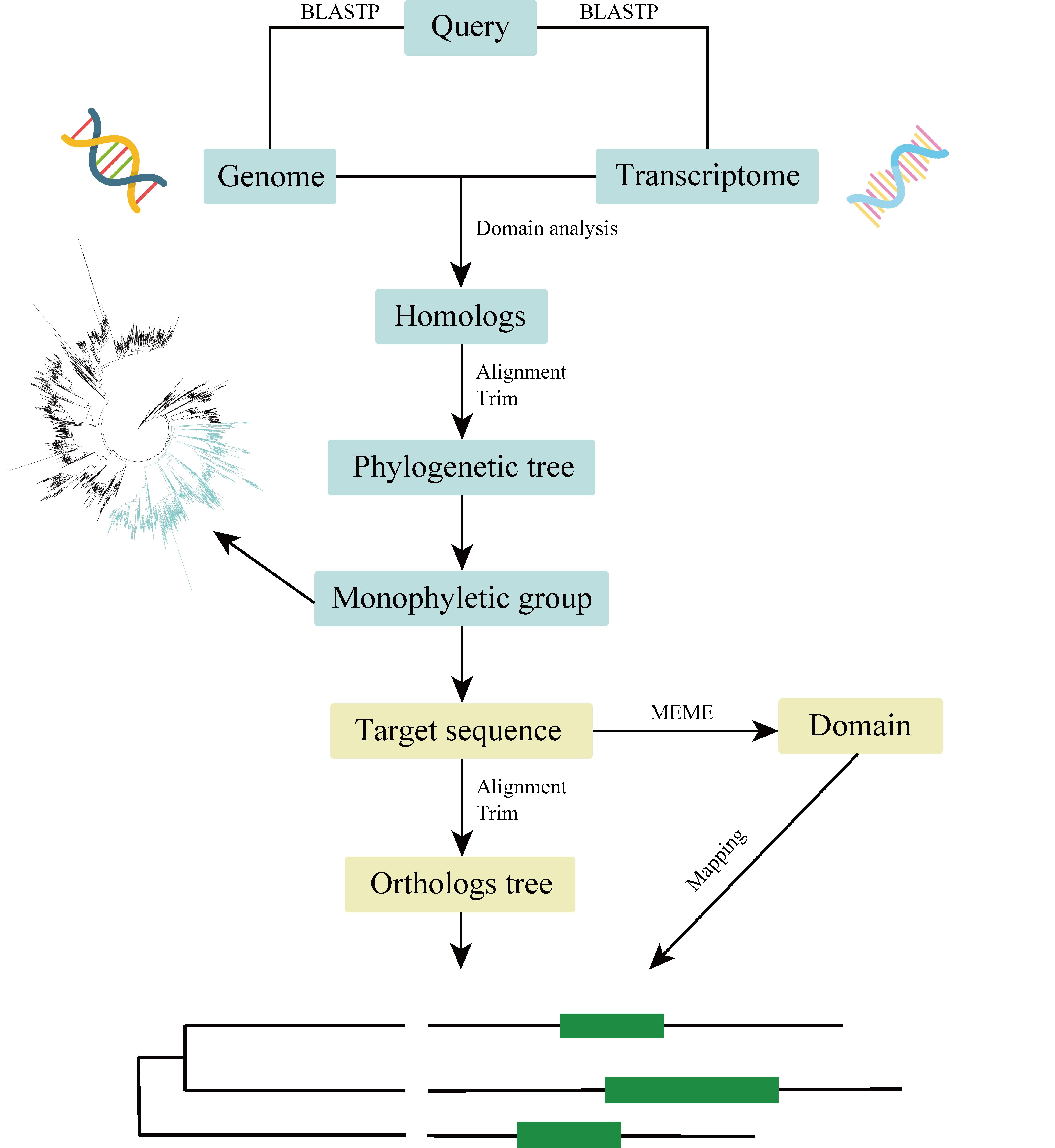
Figure 1. Pipeline for homologs/orthologs identification with large-scale genomes and transcriptomes
Software installation
Miniconda3-py39_4.12.0-Linux-x86_64
Miniconda3 is a package manager for downloading and installing bioinformatics software. It can be downloaded and installed in the server by the following commands:
wget https://mirrors.tuna.tsinghua.edu.cn/anaconda/miniconda/Miniconda3-py39_4.12.0-Linux-x86_64.sh
bash Miniconda3-py39_4.12.0-Linux-x86_64.sh
Diamond v2.1.7.161
Diamond is a fast protein aligner for protein sequences that can be downloaded and installed using the following commands:
wget https://github.com/bbuchfink/diamond/releases/download/v2.1.7/diamond-linux64.tar.gz
tar -zxvf diamond-linux64.tar.gz
Add the path of Diamond to the environment variable.
MAFFT v7.453
MAFFT is a package for sequence alignment that can be installed by conda:
conda install mafft
trimAL v1.4.rev15
TrimAL is a tool for automated alignment trimming.
wget https://github.com/inab/trimal/archive/refs/tags/v1.4.1.tar.gzhttps://github.com/inab/trimal/archive/refs/tags/v1.4.1.tar.gz
tar -zxvf trimal-1.4.1.tar.gz
cd ./trimal-1.4.1/source/
make
Add the current directory to the environment variable after compilation.
IQ-TREE v2.2.2.6
Q-TREE 2 is a widely used tool for maximum-likelihood phylogeny inference.
wget https://github.com/iqtree/iqtree2/releases/download/v2.2.2.6/iqtree-2.2.2.6-Linux.tar.gz
tar -zxvf iqtree-2.2.2.6-Linux.tar.gz
Add the ./iqtree-2.2.2.6-Linux/bin to the environment variable.
InterProScan 5.63-95.0
InterProScan is a protein function annotation software. It can be download and installed following the instruction: InterProScan documentation-interproscan-docs documentation (https://interproscan-docs.readthedocs.io/en/latest/).
TBtools v1.120
TBtools is an integrated tool for bioinformatic analysis and may be downloaded from https://github.com/CJ-Chen/TBtools/releases/download/1.123/TBtools_windows-x64_1_123.exe.
Jalview v2.11.2.0
Jalview a free cross-platform program for multiple sequence alignment editing, visualization, and analysis. This software may be downloaded from https://www.jalview.org/.
Genome and transcriptome download and processing
Plant genomes download.
A total of 39 streptophytes (land plants and charophytes), 54 chlorophytes, 9 rhodophytes, and 1 glaucophyte were selected, covering all main clades of Archaeplastida (Table S1). The protein sequences or coding sequences (CDS) and GFF annotation files were downloaded. The used transcriptomes data of algae are available at the 1KP website (https://db.cngb.org/onekp/).
Removing redundant transcripts and short genes.
Based on the GFF annotation files of each genome, the redundant transcripts and short genes are removed by TBtools: (a) the longest transcript of each gene is retained to remove redundancy resulting from alternative splicing variations (detailed steps are briefly displayed in Video 1); (b) protein sequences length of genes is calculated by TBtools, and sequences shorter than 50 amino acids are manually filtered.
 Video 1. Removing the redundant transcripts by TBtools
Video 1. Removing the redundant transcripts by TBtools
Identifying orthologous genes within large-scale genomic/transcriptomic data
Identifying candidate homologs from genomes.
In order to identify candidate homologs of Arabidopsis thaliana HLS1 (AtHLS1), we conducted similarity searches with relative stringency threshold (E value < 1 × 10-5) against the protein sequences of plant genomes. For genomic data, the relative stringency threshold (E value < 1 × 10-5) is used, and the reasons include: (1) gene annotation of genomic data is based on multiple evidences (such as gene database and transcriptomic data), which ensured the completeness and quality of gene sequences; (2) the genomic data in this study is mainly derived from the Phytozome (https://phytozome-next.jgi.doe.gov/) and has high qualities. We provide brief descriptions of files used in the command line in Table S2.
diamond makedb --in all_genome_sequences.fa -d all_genome_sequences
diamond blastp --db all_genome_sequences.dmnd --query AtHLS1.fa --out genome_out.result --outfmt 6 --sensitive -e 1e-5 --block-size 1.0 --index-chunks 1
Identifying candidate homologs from transcriptomes.
We used a relatively relaxed threshold to search candidate homologs from plant transcriptomes (E value < 1 × 10-2). The reason includes the intrinsic incompleteness of transcriptomes resulting from alternative splicing and premature termination. The detailed steps are briefly displayed in Video 2.
Video 2. BLASTp for AtHLS1 at the 1KP websiteFiltering sequences with blast-hits.
Candidate homologs from both genomes and transcriptomes are integrated into a single fasta file (ID_sequences.fa), and InterProScan database is used to filter homologous sequences without a conserved functional domain: N-acetyltransferase (PF00583). Consolidate the homologous sequences with “N-acetyltransferase” domain into a new fasta file (PF00583_ID_sequences.fa) for further phylogenetic analyses.
Interproscan.sh -i ID_sequences.fa -f tsv -appl Pfam -o interproscan_ID_sequences.txt
Orthologs inference with phylogenetic analyses
Alignment and trimming of homologous sequences.
Homologous sequences are aligned by MAFFT using the following command:
mafft PF00583_ID_sequences.fa > mafft_out.fa
Note: An alignment of up to ~200 sequences × ~2,000 sites is suitable for an accurate option (L-INS-i), and an alignment of <~30,000 sequences is suitable for fast option (FFT-NS-2).
TrimAl is a widely used tool for automated alignment trimming in large-scale phylogenetic analyses. Before trimming, we perform manual inspection for sequence alignment using Jalview and exclude one sequence (in red rectangle) for its error alignment (Figure 2). We use a relatively relaxed threshold (‘-gt=0.13’: retaining columns that have at least 13% gap-free sites for keeping as much informative sites of conserved domains as possible) for trimming multiple sequence alignment (MSA). The trimmed MSA is further visually inspected to (1) filter the obvious ambiguously aligned regions and (2) retain the regions of functional domains, ensuring a greater proportion of reliably phylogenetically informative sites.
trimal -in mafft_out.fa -out mafftout_0.13.fas -gt 0.13

Figure 2. Multiple sequence alignment of AtHLS1 and its homologs. We empirically focused on the alignment region of conserved domains and found an obvious sequence that is poorly aligned (no amino acid residue was aligned).Constructing a phylogenetic tree of homologous proteins.
The maximum likelihood phylogenetic tree of homologous proteins is inferred using IQ-TREE 2, and the best-fitting model is determined by ModelFinder. Branch supports are evaluated by the ultrafast bootstrap (UFBoot) approach and SH approximate likelihood ratio test (SH-aLRT test) with 1,000 replicates.
iqtree2 -s mafftout_0.13.fas -st AA -m MF
iqtree2 -s mafftout_0.13.fas -m Q.plant+I+R6 -alrt 1000 -bb 1000 –bnni -pre 0.13_result
Confirmation of orthologous sequences using function domains/motifs.
The conserved functional domains/motifs are analyzed by MEME suite. The parameter “number of motifs expected to be found” is set to ten (a common setting for this parameter) and other parameters are as default. Corresponding functional domains and motifs of each orthologous sequence are mapped to the phylogenetic tree using iTOL (Video 3). Two conserved residues (L327 and E346 in AtHLS1) are checked and highlighted in the MSA. Both the conserved functional domains/motifs and residues are used for the confirmation of orthologous sequence (Figure 3).
Video 3. Mapping the domain/motifs to the phylogenetic tree
Figure 3. Phylogenetic tree, two conserved residues, and conserved motifs of plant HLS1 homologs
Acknowledgments
This work is supported by the Natural Science Fund for Colleges and Universities in Jiangsu Province of China (21KJB180015). This protocol was adapted from our recent work (Wang et al., 2023). We thank the editor and anonymous reviewers for their helpful suggestions.
Competing interests
The authors declare that there are no conflicts of interest or competing interests.
References
Bailey, T. L. and Elkan, C. (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2: 28–36.
- Buchfink, B., Xie, C. and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12(1): 59–60.
- Capella-Gutiérrez, S., Silla-Martínez, J. M. and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25(15): 1972–1973.
- Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y. and Xia, R. (2020). TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 13(8): 1194–1202.
- Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., McWilliam, H., Maslen, J., Mitchell, A., Nuka, G., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30(9): 1236–1240.
- Katoh, K. and Standley, D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 30(4): 772–780.
- Koonin, E. V. (2005). Orthologs, Paralogs, and Evolutionary Genomics. Annu. Rev. Genet. 39(1): 309–338.
- Lehman, A., Black, R. and Ecker, J. R. (1996). HOOKLESS1, an Ethylene Response Gene, Is Required for Differential Cell Elongation in the Arabidopsis Hypocotyl. Cell 85(2): 183–194.
- Letunic, I. and Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49: W293–W296.
- Li, H., Johnson, P., Stepanova, A., Alonso, J. M. and Ecker, J. R. (2004). Convergence of Signaling Pathways in the Control of Differential Cell Growth in Arabidopsis. Dev. Cell 7(2): 193–204.
- Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A. and Lanfear, R. (2020). IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 37(5): 1530–1534.
- One Thousand Plant Transcriptomes Initiative (2019). One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574(7780): 679–685.
- Qiao, X., Li, Q., Yin, H., Qi, K., Li, L., Wang, R., Zhang, S. and Paterson, A. H. (2019). Gene duplication and evolution in recurring polyploidization–diploidization cycles in plants. Genome Biol. 20(1): e1186/s13059-019-1650-2.
- Thornton, J. W. and DeSalle, R. (2000). Gene Family Evolution and Homology: Genomics Meets Phylogenetics. Annu. Rev. Genomics Hum. Genet. 1(1): 41–73.
- Wang, Q., Sun, J., Wang, R., Zhang, Z., Liu, N., Jin, H., Zhong, B. and Zhu, Z. (2023). The origin, evolution and functional divergence of HOOKLESS1 in plants. Commun. Biol. 6(1): e1038/s42003-023-04849-4.
- Waterhouse, A. M., Procter, J. B., Martin, D. M. A., Clamp, M. and Barton, G. J. (2009). Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25(9): 1189–1191.
- Zhang, Z., Ma, X., Liu, Y., Yang, L., Shi, X., Wang, H., Diao, R. and Zhong, B. (2022). Origin and evolution of green plants in the light of key evolutionary events. J. Integr. Plant Biol. 64(2): 516–535.
Supplementary information
The following supporting information can be downloaded here:
- Table S1. Sources of the genomic data used in this protocol
- Table S2. Brief description of files used in the command line
Article Information
Copyright
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Xu, Z., Sun, W., Zhu, Z., Zhong, B. and Zhang, Z. (2023). Phylogenetic Inference of Homologous/Orthologous Genes among Distantly Related Plants. Bio-protocol 13(23): e4893. DOI: 10.21769/BioProtoc.4893.
Category
Systems Biology > Genomics > Phylogenetics
Bioinformatics and Computational Biology
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.