- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Purification of Long Non-coding RNAs on Replication Forks Using iROND (Isolate RNAs on Nascent DNA)
Published: Vol 13, Iss 21, Nov 5, 2023 DOI: 10.21769/BioProtoc.4869 Views: 2263
Reviewed by: Emilia KrypotouThirupugal GovindarajanZheng Zachory WeiDr. Amit K. Tripathi
Original research article
The authors used this protocol in:

Jan 2023

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Fork stability is key to genome DNA duplication and genetic integrity. Long non-coding RNAs (LncRNAs) may play vital roles in fork stabilization and chromatin remodeling. Existing techniques such as NCC-RNA sequencing are useful to identify LncRNAs on nascent chromatin DNA. However, there is still a lack of methods for LncRNAs purification directly from replicative forks, hindering a deep understanding of the functions of LncRNAs in fork regulation. Here, we provide a step-by-step protocol named iROND (isolate RNAs on nascent DNA). iROND was developed and modified from iPOND, a well-known method for purifying fork-associated proteins. iROND relies on click chemistry reaction of 5'-ethynyl-2'-deoxyuridine (EdU)-labeled forks and biotin. After streptavidin pull down, fork-associated LncRNAs and proteins are purified simultaneously. iROND is compatible with downstream RNA sequencing, qPCR confirmation, and immunoblotting. Integrated with functional methods such as RNA fluorescent in situ hybridization (RNA FISH) and DNA fiber assay, it is feasible to screen fork-binding LncRNAs in defined cell lines and explore their functions. In summary, we provide a purification pipeline of fork-associated LncRNAs. iROND is also useful for studying other types of fork-associated non-coding RNAs.
Key features
• Purify long non-coding RNAs (LncRNAs) directly from replication forks.
• Connects to RNA sequencing for screening easily.
• Allows testing various genotoxic stress responses.
• Provides LncRNA candidate list for downstream functional research.
Graphical overview
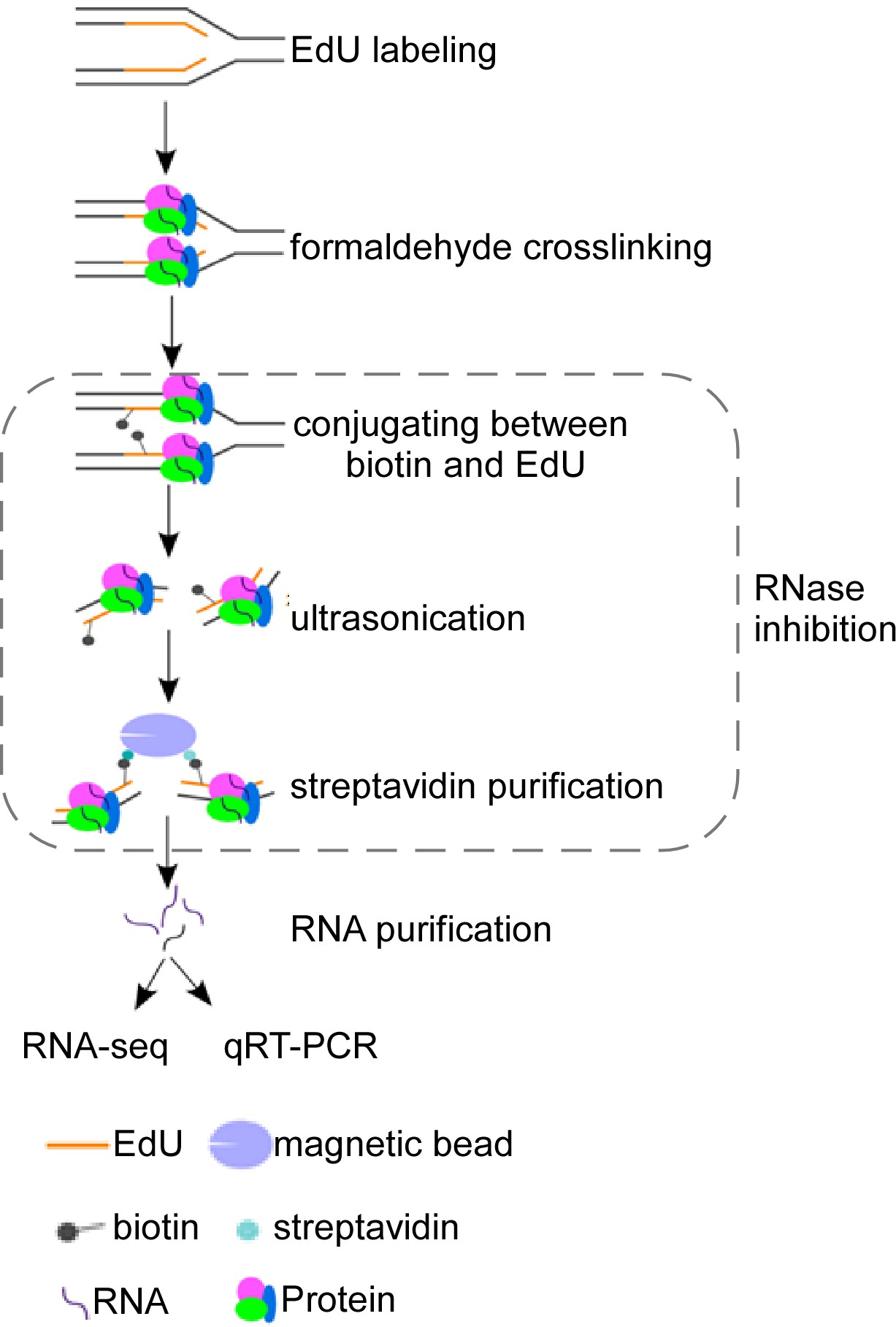
Schematic overview of isolate RNAs on nascent DNA (iROND) protocol. Cells were pulse-labeled with 5'-ethynyl-2'-deoxyuridine (EdU) for 10 min before paraformaldehyde fixation. EdU-positive forks were ligated with biotin through Click-IT chemistry reaction. Genomic DNA was ultrasonically cracked and crosslinked with streptavidin for pulling down. Both RNA and protein components were purified. RNA components were used for downstream RNA sequencing and qPCR validation. Protein components were used for immunoblotting to evaluate binding dynamics of fork-associated proteins such as helicase, topoisomerase, and DNA polymerases.
Background
DNA replication and DNA repair are central topics in genome stability research. Currently, a vast majority of studies focuses on protein components intensively. Through the investigation of core factors such as ATM, ATR, CHK1, CHK2, DNA-PK, and P53, as well as their associated signaling pathways, the fundamental regulatory mechanisms in DNA replication and DNA repair have been revealed (Jackson and Bartek, 2009). For example, using isolate proteins on nascent DNA (iPOND) technology, numerous replication fork-associated protein components have been identified, including DNA polymerases, helicases, and epigenetic modifiers (Sirbu et al., 2012). The initiation, elongation, and termination of steady-state replication forks are strictly controlled to ensure the integrity of the genome replication process (Dungrawala and Cortez, 2015). In order to cope with genotoxic drug-induced fork collapse or stalling, endogenous DNA replication stress response mechanisms can ensure DNA replication progression and reduce the accumulation of DNA breaks (Berti et al., 2020). In terms of DNA repair, numerous specific techniques have been developed. For instance, GFP-based reporter systems were used to evaluate DNA repairing efficiency (Pierce et al., 1999). Single-cell comet assay is used broadly to examine general DNA damage levels (Collins, 2004). Overall, the involvement of protein components in genome stability maintenance is presently well understood.
Besides protein components, a few recent studies raised the potential importance of RNA components in DNA repair and replication, facilitated by indirect identification techniques. For example, the NCC-RNA-seq technique, based on the principle of biotin affinity purification, has been used to identify multiple long and short RNA molecules associated with nascent chromosomes (Gylling et al., 2020). The functions of these RNAs in chromosome reshaping and maturation warrant further investigation. Additionally, using RNA affinity purification coupled with mass spectrometry, the long non-coding RNA (LncRNA) NORAD plays a critical role in genome stability through interaction with PUMILIO protein (Elguindy and Mendell, 2021). Comparative transcriptomics is an effective approach for discovering functional non-coding RNAs. In our previous studies, we identified a novel LncRNA, DISCN, by comparing the expression profiles of LncRNAs in mouse embryonic stem cells (mESCs) with differentiated cells. DISCN exhibits specific high expression in embryonic and neural stem cells. DISCN forms a functional complex with the nucleolar protein nucleolin and the DNA single-strand binding protein RPA, through which it keeps an RPA protein pool to ensure the efficiency of DNA replication stress response and DNA repair (Wang et al., 2021). These studies support the notion that non-coding RNAs form a new regulatory layer in genomic stability system. However, limited by direct purification techniques, the roles of non-coding RNAs in DNA replication or DNA repair are still not understood.
We have recently developed a novel method called iROND (isolate RNAs on nascent DNA). Using iROND, we identified a specific fork-binding LncRNA Lnc956, in the embryonic stem cell system. Lnc956 forms a stable nucleic acid–protein complex with TRIM28 and HSP90B1, facilitating the integration of the CMG helicase at replication fork sites and ensuring replication fork stability in mESCs (Zhang et al., 2023). Here, we provide a step-by-step protocol for iROND. iROND is applicable in different types of rapidly proliferating cell lines and compatible with downstream functional validation systems such as RNA fluorescence in situ hybridization (RNA-FISH) and DNA fiber assays.
Materials and reagents
Biological materials
Mouse embryonic stem cells (derived from C57Bl/6J blastocysts)
Mouse NIH3T3 cells (kindly provided by Dr. Bingyu Mao at Kunming Institute of Zoology, Chinese Academy of Sciences)
Materials
35 mm cell culture dish (Corning, catalog number: 430165)
60 mm cell culture dish (NEST, catalog number: 705001)
100 mm culture dish (NEST, catalog number: 704001)
150 mm culture dish (Corning, catalog number: 430599)
200 μL PCR tube (Axygen, catalog number: AXYPCR02LC)
1.5 mL centrifugal tube (Axygen, catalog number: AXYMCT150C)
15 mL centrifugal tube (NEST, catalog number: 601052)
50 mL centrifugal tube (NEST, catalog number: 602002)
0.22 μm filter (Millipore, catalog number: SLGP033RB)
Reagents
5-ethynyl-2′-deoxyuridine (EdU) (Life Technology, catalog number: A10044)
Thymidine (Sigma-Aldrich, catalog number: T1895)
37% (w/v) formaldehyde solution (Sigma-Aldrich, catalog number: F1635); caution: toxic
Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9888)
Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P3911)
Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: P5655)
Sodium phosphate dibasic (Na2HPO4) (Sigma-Aldrich, catalog number: S5136)
Glycine (Sigma-Aldrich, catalog number: G7126)
Triton X-100 (Sigma-Aldrich, catalog number: X100)
BSA (Sigma-Aldrich, catalog number: V900933)
Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, catalog number: D8418)
Copper (II) sulfate pentahydrate (CuSO4·5H2O) (Sigma-Aldrich, catalog number: 209198)
(+) Sodium L-ascorbate (Sigma-Aldrich, catalog number: A4034)
Biotin azide (Invitrogen, catalog number: B10184)
SDS (Sangon Biotech, catalog number: A600485)
Tris (Sangon Biotech, catalog number: A610195)
Glycerol (Sangon Biotech, catalog number: A600232)
Bromophenol blue (Sangon Biotech, catalog number: A602230)
EDTA (Sigma-Aldrich, catalog number: E9884)
Agarose (Merck, catalog number: A4718)
Dithiothreitol (DTT) (Merck, catalog number: D8255)
Aprotinin (Merck, catalog number: A6279)
Leupeptin (Merck, catalog number: L2884)
Streptavidin agarose (Novagen, catalog number: 69203-3)
RNase inhibitor (Thermo Fisher, catalog number: EO0381)
Diethyl pyrocarbonate (DEPC) (Sigma-Aldrich, catalog number: D5758)
Protease K (Thermo Fisher, catalog number: 4333793)
Chloroform (Sigma-Aldrich, catalog number: 366911)
Isopropyl alcohol (Sigma-Aldrich, catalog number: W292912)
Ammonium acetate (Sigma-Aldrich, catalog number: A7262)
Alcohol (Sigma-Aldrich, catalog number: AX0442)
Acetic acid (Sigma-Aldrich, catalog number: 695092)
TRIzol (Thermo Fisher, catalog number: 15596026)
Hydroxyurea (HU) (Sigma-Aldrich, catalog number: H8627)
EdU solution (see Recipes)
DEPC-H2O (see Recipes)
Thymidine solution (see Recipes)
Chase medium (see Recipes)
1× PBS solution (see Recipes)
1% Formaldehyde solution (see Recipes)
1.25 M Glycine solution (see Recipes)
Permeabilization buffer (see Recipes)
Wash buffer (see Recipes)
Biotin-azide solution (see Recipes)
100 mM CuSO4 solution (see Recipes)
Sodium L-ascorbate solution (see Recipes)
Lysis buffer (pH 8.0) (see Recipes)
Salt wash buffer (see Recipes)
2× SDS Laemmli sample buffer (2× SB) (see Recipes)
50× Tris acetate-EDTA (TAE) buffer (see Recipes)
EdU solution
Reagent (use for storing) Final concentration Quantity EdU (50 mg) 10 mM 25.2 mg DMSO n/a 10 mL Total n/a 10 mL Reagent (use for cell labeling) Final concentration Quantity EdU (10 mM) 10 μM 1 μL mESCs medium (see Step A1) n/a 999 μL Total n/a 1 mL DEPC-H2O
Reagent Final concentration Quantity DEPC n/a 1 mL H2O n/a 999 mL Total n/a 1,000 mL Thymidine solution
Reagent (use for storing) Final concentration Quantity Thymidine (1 g) 10 mM 24.2 mg DEPC-H2O n/a 10 mL Total n/a 10 mL Chase medium
Reagent (use for cell labeling) Final concentration Quantity Thymidine (10 mM) 10 μM 1 μL mESCs Medium n/a 999 μL Total n/a 1 mL 1× PBS solution
Reagent Final concentration Quantity NaCl 137 mM 8 g KCl 3 mM 0.2 g Na2HPO4 8 mM 1.15 g KH2PO4 2 mM 0.24 g DEPC-H2O n/a 1 L Total n/a 1 L 1% Formaldehyde solution
Reagent Final concentration Quantity Formaldehyde (37%) 1% 0.27 mL DEPC-PBS n/a 9.73 mL Total n/a 10 mL 1.25 M Glycine solution
Reagen Final concentration Quantity Glycine 1.25 M 46.92 g DEPC-H2O n/a 500 mL Total n/a 500 mL Permeabilization buffer
Reagent Final concentration Quantity Triton X-100 0.25% 2.5 mL DEPC-PBS n/a 997.5 mL Total n/a 1 L Wash buffer
Reagent Final concentration Quantity BSA 0.5% 0.5 g DEPC-PBS n/a 100 mL Total n/a 100 mL Biotin-azide solution
Reagent Final concentration Quantity Biotin-azide 1 mM 1 mg DMSO n/a 1.624 mL Total n/a 1.624 mL 100 mM CuSO4 solution
Reagent Final concentration Quantity CuSO4·5H2O 100 mM 1.248 g DEPC-H2O n/a 50 mL Total n/a 50 mL Sodium L-ascorbate solution
Reagent Final concentration Quantity (+) sodium L-ascorbate 100 mM 20 mg DEPC-H2O n/a 1 mL Total n/a 1 mL Lysis buffer (pH 8.0)
Reagen Final concentration Quantity Tris 10 mM 1.21 g SDS 7 mM 2 g DEPC-H2O n/a 200 mL RNase inhibitor 10 U/μL 200 μg Aprotinin 1 μg/mL 200 μg Leupeptin 1 μg/mL 200 μg Total n/a 200 mL Salt wash buffer
Reagent Final concentration Quantity NaCl 1 M 14.625 g DEPC-H2O n/a n/a Total n/a 250 mL 2× SDS Laemmli sample buffer (2× SB)
Reagent Final concentration Quantity SDS 123 mM 0.4 g Bromophenol blue 1 mM 0.01 g Glycerol 18% 2 mL 1 M Tris (pH 6.8)
1M DTT
1 mM
1 mM
1.25 mL
1.25 mL
H2O n/a 8 mL Total n/a 12.5 mL 50× Tris acetate-EDTA (TAE) buffer
Reagent Final concentration Quantity Tris 2 M 242 g Acetic acid n/a 57.1 mL EDTA 0.1 M 37.2 g H2O n/a 942.9 mL Total n/a 1,000 mL
Equipment
Thermo Forma series II water jacketed CO2 incubator (Thermo Scientific, model: 3111)
Nikon eclipse Ti inverted microscope (Nikon, model: Ti)
Ultrasonic cell disruptor (Diagenode, Bioruptor Plus)
90 µm nylon mesh (Small Parts Inc., catalog number: B000FN0PGQ)
Roll shaker for incubation (Thermo Scientific, model: PHMT)
Real-time fluorescence quantitative PCR instrument (Bio-Rad, model: CFX96Touch)
Vortexer (VWR analog vortex mixer, catalog number: 10153-838)
4 °C refrigerator (Haier, model: HYC-1090)
-20 °C freezer (Haier, model: DWL262)
NanoDrop (Thermo Scientific, ND2000)
-80 °C freezer (Thermo Scientific, model: TSX60086VRAKLB)
Software
Microsoft Excel (Microsoft, https://products.office.com/en-us/excel) (Office Excel 2016, September 22, 2015)
GraphPad Prism (GraphPad, https://www.graphpad.com/scientific-software/prism/) (GraphPad Prism 8.3.0, October 29, 2019)
Procedure
Label cells with EdU
For the preparation of mESC medium and culture and expansion of mESCs and NIH3T3, please refer to our publication (Zhang et al., 2023). Before EdU labeling, incubate cells with 37 °C pre-warmed fresh mESCs medium for 2 h, with 25 mL of medium for each 15 cm diameter dish containing ~1.5 × 107 cells.
To perform EdU labeling, take the dishes to a biological safety cabinet, add 25 μL of EdU stock (10 mM) into 25 mL of pre-existing medium for each dish, and mix well gently to make sure that the final concentration of EdU is 10 μM. Depending on the percentage of mESCs in S phase (more than 60%), we calculated that at least 9 × 109 cells in each dish were labeled by EdU.
Put the dishes back into the incubator quickly and label for 10 min.
Note: Typically, fork-associated RNA components display a low-dose manner. To improve the productivity, using a cell line with high percentage of S-phase cells is critical to secure the RNA output. In our study, we used mESCs, as these always keep more than 60% of cells in S phase. If you need to evaluate replication stress dynamics, add genotoxic drugs such as 4 mM hydroxyurea (HU) into the dish and incubate for 2 h.
To get post-replicated chromatin samples, remove EdU-positive medium, carefully wash cells three times with 5 mL each time of pre-warmed chase medium. Chase medium, containing 10 μM thymidine, is used to compete with EdU to chase DNA replication. After 1 h incubation, the EdU-labeled DNA has been constructed to mature chromatin. So, this group is used as a chromatin control. Then, add 25 mL of pre-warmed chase medium and incubate for 1 h.
Formaldehyde crosslink and harvest cells
Once finished with EdU labeling, thymidine chase, or drugs treatment, remove culture medium quickly and add 10 mL of ice-cold PBS containing 1% formaldehyde to each dish to fix cells for 20 min at room temperature.
Caution: Formaldehyde is toxic to human health.
Critical: Do not wash cells with PBS buffer before fixation since PBS washing may cause RNA degradation.
Add 1 mL of 1.25 M glycine to each dish to quench formaldehyde.
Scrape cells and harvest into 50 mL RNase-free centrifuge tubes. Record the fixation volume.
Critical: Keep tubes on ice all the time.
Spin down at 900× g for 5 min at 4 °C.
Remove supernatant.
Use ice-cold 1× PBS buffer containing 10 U/μL RNase inhibitor to wash sediments three times with the same volume as the fixation volume from step B3.
Pause point: Samples can be quickly immersed into liquid nitrogen and stored at -80 °C for at least four weeks.
Cell permeabilization
Resuspend cells in ice-cold permeabilization buffer containing 10 U/μL RNase inhibitor. Adjust cell density to 1 × 107 cells/mL. Vortex well and incubate at room temperature for 30 min on a shaker (40 rpm).
Centrifuge at 900× g for 5 min at 4 °C.
Remove supernatant.
Wash cells with ice-cold 1× PBS containing 0.5% BSA and 10 U/μL RNase inhibitor; the volume used is equal to the permeabilization buffer in step C1.
Centrifuge at 900× g for 5 min at 4 °C and remove supernatant.
Repeat steps C4 and C5 once. Keep samples on ice for click reaction.
Click reaction
Prepare click reaction cocktail as listed in Table 1.
Table 1. Click reaction cocktail (5 mL for 1 × 108 cells)
Reagent Stock concentration Working concentration Control reaction volume (mL) Experimental reaction volume (mL) 1× PBS 4.225 4.225 Biotin-azide 1 mM 10 μM 0.05 CuSO4 100 mM 2 mM 0.1 0.1 (+) Sodium L-ascorbate 100 mM 10 mM 0.5 0.5 DMSO 0.05 RNase inhibitor 40 U/μL 1 U/μL 0.125 0.125 Total volume 5.0 5.0 Critical: The biotin-azide is sensitive to light. The reaction cocktail should be stored in the dark.
Critical: CuII is unstable in water. The reaction cocktail should be prepared fresh.
Resuspend cells into click reaction cocktail. Adjust reaction volume to reach the density of 1 × 108 cells per 5 mL.
Vortex gently and incubate for 2 h at room temperature on a shaker (40 rpm).
Critical: Keep the reaction tubes in the dark.
Centrifuge at 900× g for 5 min at 4 °C and remove supernatant.
Wash cells with ice-cold 1× PBS buffer containing 0.5% BSA and 10 U/μL RNase inhibitor. The PBS volume used is equal to the permeabilization buffer in step C1.
Centrifuge at 900× g for 5 min at 4 °C and remove supernatant.
Repeat steps D5 and D6 once.
Cell lysis and sonication
Keep lysis buffer on ice. Add leupeptin, aprotinin, and RNase inhibitor before use.
Add 100 μL of lysis buffer per 1.5 × 107 cells and resuspend well. Transfer cell suspension into a 1.5 mL RNase-free centrifuge tube.
Sonicate cells using a microtip sonicator with the following settings: pulse, 20 s; 40 s pause; 15 cycles; power: 13–16 watts.
Critical: Tubes should be immersed into an ice-cold water bath in the process of sonication to prevent overheating.
Note: After sonication, the supernatant should be clear, but not cloudy.
Centrifuge at 12,000× g for 15 min at 4 °C.
Transfer the supernatant through a 90 µm nylon mesh and collect into a new 1.5 mL RNase-free centrifuge tube; then, keep samples on ice.
Note: To examine DNA fragment size at this step, pick up 5 μL of supernatant to extract DNA. Perform electrophoresis using 1% agarose gel at 120 V for 30 min to detect DNA fragments (Figure 1).
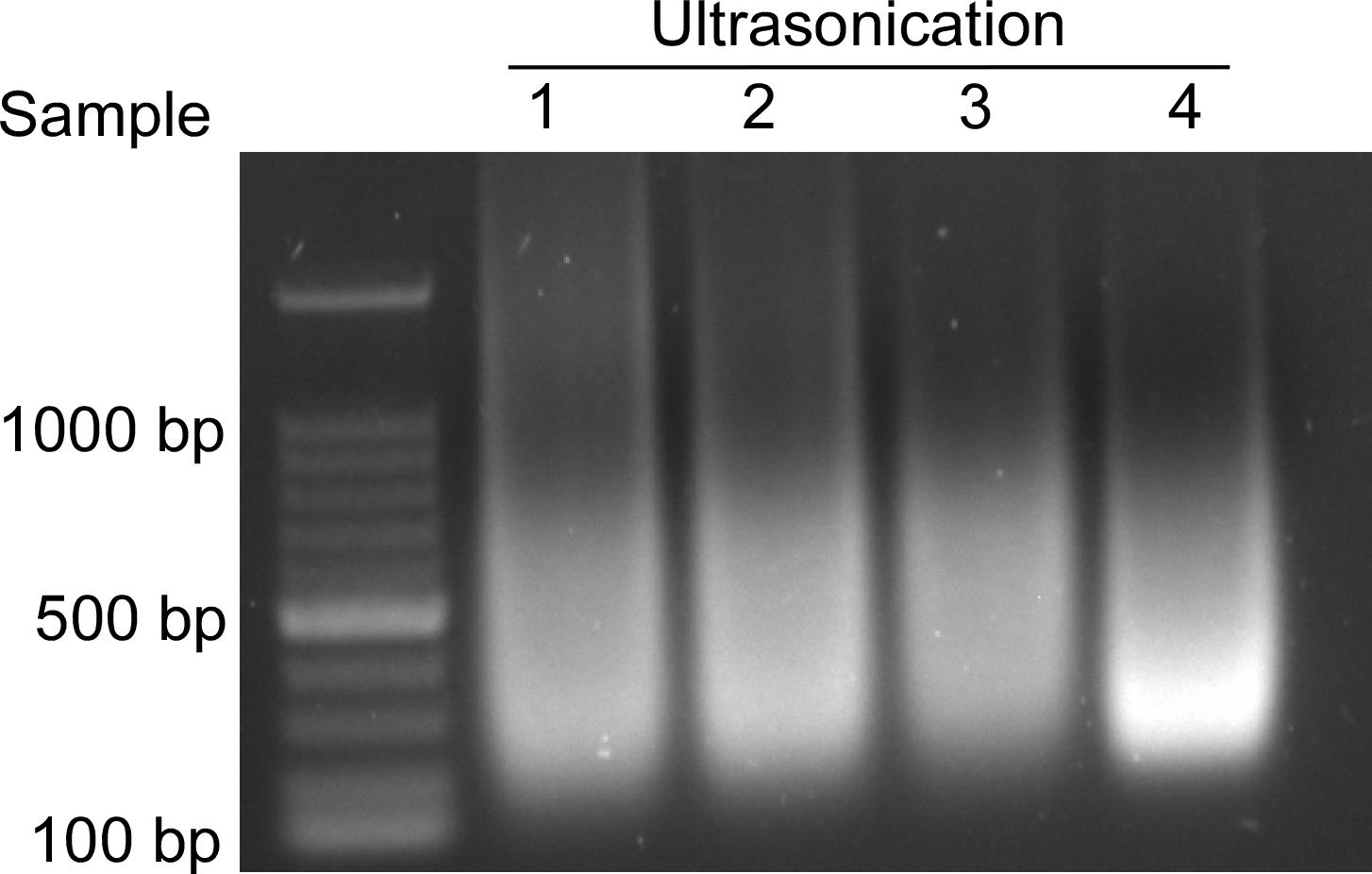
Figure 1. DNA fragments after sonication. The size of DNA fragments is mainly concentrated in the 200–1,000 base pair range.DNA extraction.
Take 5 μL from the supernatant from step E5, add protease K to the final concentration of 50–100 μg/mL, mix well, and incubate at 50 °C for 3 h.
Add an equal volume of chloroform/isopropyl alcohol (volume ratio: 24:1), mix well, centrifuge at 10,000× g for 10 min, and transfer the supernatant to the new centrifuge tube.
Add 10% volume of 10 M ammonium acetate and mix gently.
Add twice the volume of isopropyl alcohol and mix gently.
Centrifuge at 10,000× g for 10 min and discard supernatant.
Add the same volume of 75% ethanol and mix well, centrifuge at 10,000× g for 5 min, and discard the supernatant.
Air-dry pellets for 5 min and dissolve with 10 μL of double-distilled water for electrophoresis.
Dilute the samples with the same volume of ice-cold PBS to improve the efficiency of following Biotin capture. Make sure to add 1 μg/mL leupeptin, 1 μg/mL aprotinin, and 10 U/μL RNase inhibitor.
Pick up 15 μL of each sample, mix well with 15 μL of 2× SB, and boil at 95 °C for 10 min. These samples can be used as input for immunoblotting targeting replication fork proteins such as PCNA.
Critical: This step is crucial since immunoblotting of PCNA can be used to confirm the pull-down efficiency and specificity.
Pick up 15 μL of each sample, mix well with 500 μL of TRIzol, and store at -80 °C for RNA purification.
Critical: This step is crucial; samples will be used to calculate enrichment fold change in the following qPCR confirmation.
Streptavidin capture
Vortex streptavidin agarose beads well.
Calculate agarose beads volume for each sample. Typically, 100 μL of streptavidin agarose beads is used per 1 × 108 cells.
Wash streptavidin agarose beads two times with lysis buffer and one time with PBS before use, using 1 mL for each wash.
Add beads into each sample and incubate for 16 h at 4 °C on a rotator (40 rpm).
Critical: Keep the rotator in the dark.
Centrifuge at 1,800× g for 1 min at 4 °C and remove supernatant.
Wash beads three times with ice-cold lysis buffer and one time with 1 mL of salt wash buffer. For each wash, samples should be rotated for 5 min before centrifugation.
Elution of proteins and RNAs
Separate the beads of each sample into two parts equally to elution protein and RNA components, respectively.
Add 2 μL of 2× SB into one part, mix well, and boil samples at 95 °C for 25 min to elute proteins. These are ready to use for immunoblotting.
For elution of RNAs, add 500 μL of TRIzol into another part of each sample, and mix well.
Add 100 μL of chloroform into each tube, vortex tightly for 30 s, and leave on ice for 5 min.
Centrifuge at 12,000× g for 10 min at 4 °C.
Transfer the supernatant of each sample into a new RNase-free tube and record the volume.
Add an equal volume of isopropanol and leave on ice for 10 min.
Centrifuge at 12,000× g for 10 min at 4 °C. Remove the supernatant.
Wash the pellet with 75% alcohol.
Centrifuge at 12,000× g for 10 min at 4 °C. Remove the supernatant and air dry for 1 min.
Add 40 μL of RNase-free double-distilled water to dissolve the pellet.
Measure RNA concentration using a NanoDrop. Approximately 200–400 ng of RNA can be extracted from 1× 108 cells.
Note: RNA purification is critically important; the optical density (OD) 260/230 value should be between 1.5 and 2.4, and the OD260/280 value must be between 1.8 and 2.4.
Data analysis
Immunoblotting is useful to confirm the pull-down specification of fork DNA fragments. PCNA and Histone H3 can be used as a fork-binding protein marker and mature chromatin marker, respectively. Please refer to Supplementary Figure 1A in our publication (Zhang et al., 2023).
RNA samples can be used either for qPCR analysis or RNA sequencing (Figures 2 and 3). Please refer to Figure 1B, C, and D in our publication (Zhang et al., 2023).
Validation of protocol
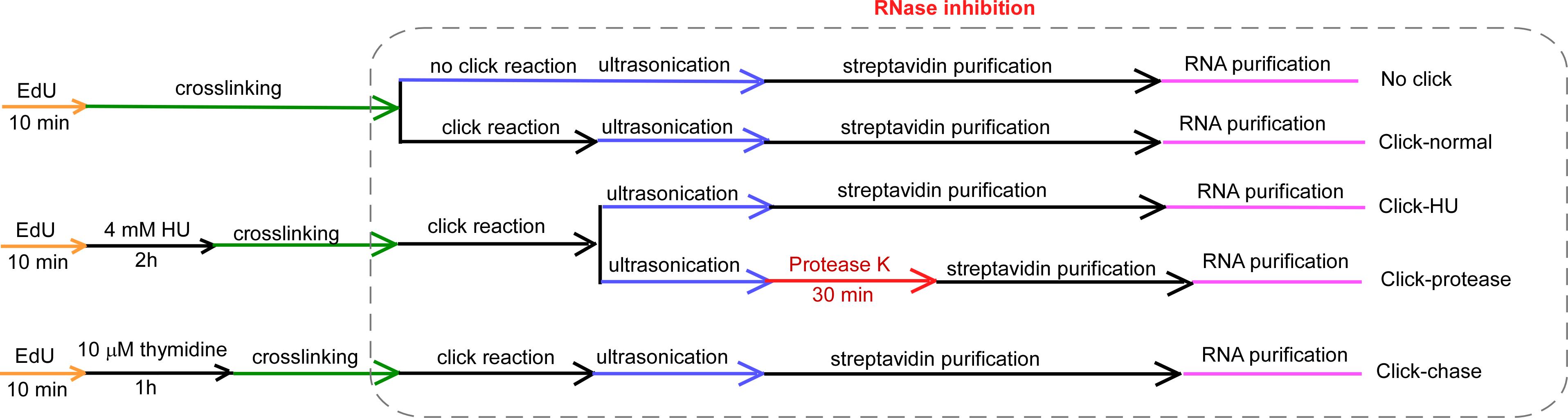
Figure 2. Design of isolate RNAs on nascent DNA (iROND)-based LncRNA screening in mouse embryonic stem cells (mESCs) and NIH3T3 cells (Zhang et al., 2023). We set up the following experimental items: 1) no click group: cells were pulse-labeled by EdU but click reaction was omitted, as a non-specific binding control. 2) Click-normal group: cells were cultured in normal conditions and standard iROND was performed, as a steady-state sample. 3) Click-HU group: cells were pulse-labeled by EdU and treated with 4 mM hydroxyurea (HU) to induce replication stress, as a genotoxic sample. 4) Click-Protease group: all procedures were the same as group 3, with extra treatment of 100 μg/mL protease K for 30 min to degrade protein components. 5) Click-chase group: cells were chase-labeled with thymidine to get a mature chromatin control.
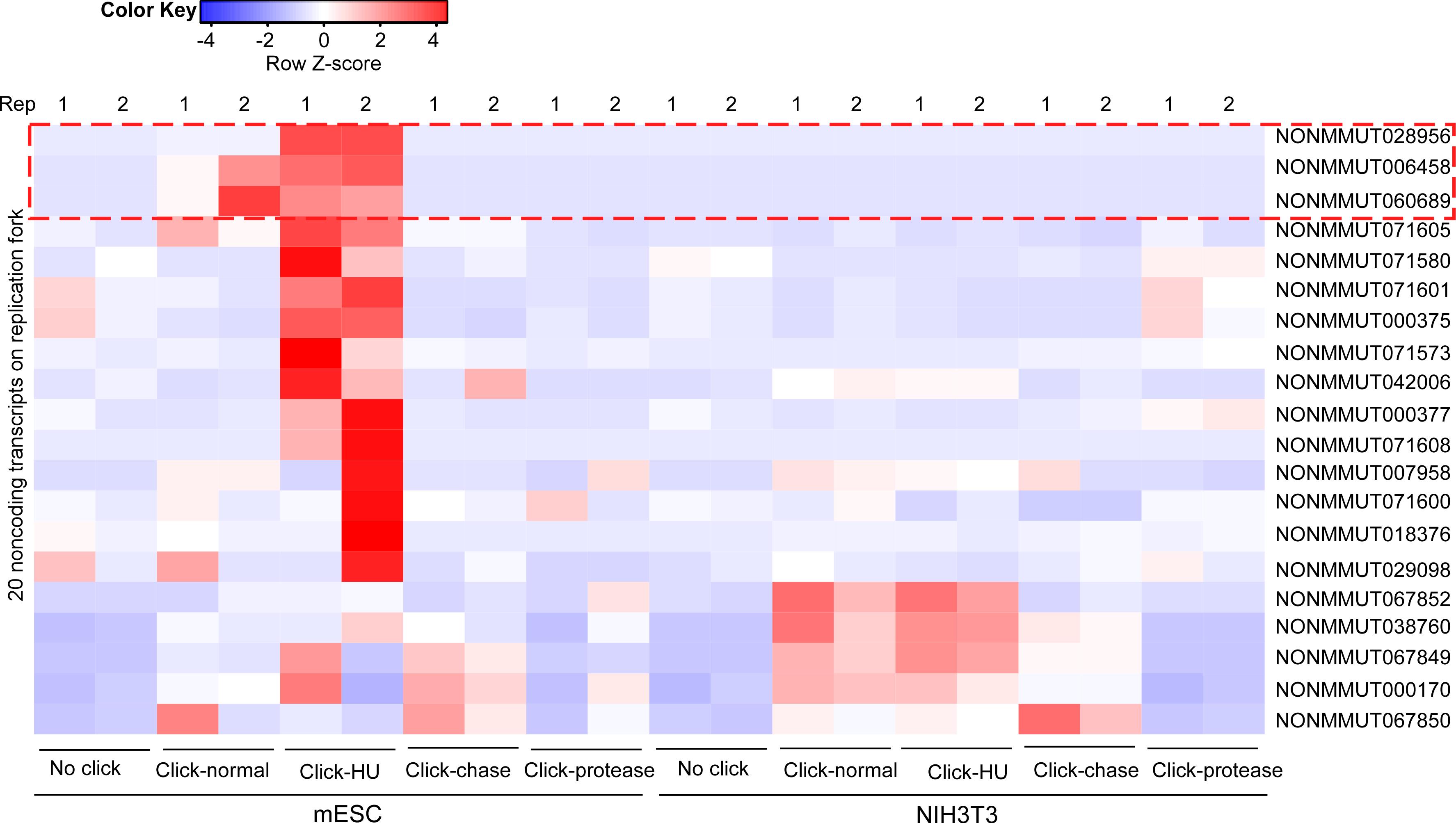
Figure 3. RNA sequencing and bioinformatic comparison of isolate RNAs on nascent DNA (iROND) samples from mouse embryonic stem cells (mESCs) and NIH3T3 cells under indicated conditions (Zhang et al., 2023). Two biological repeats were performed for each group. Notably, a set of LncRNAs were significantly enriched in mESC Click-HU group.
General notes and troubleshooting
Compared with published methods for studying LncRNAs in genomic dynamics or stability, iROND is more specific for purifying the fork-associated LncRNA subpopulation (Gylling et al., 2020; Wang et al., 2021). One limitation is that iROND needs billions of cells for each assay. The more S-phase cells, the easier performing the assay is. In our study, we used mESCs and NIH3T3 cells; both proliferate very fast. This is critical for iROND success. We assume that it might be difficult if low proliferating cell lines are used. Alternatively, synchronization into S phase before EdU labeling may be a reasonable choice.
In the steps of EdU labeling and thymidine chase, pre-warming medium is important to minimize disturbance to cells in a very short time.
In order to label replication forks as specifically as possible, EdU labeling must be done in 10 min. It is quite hard for only one person to handle many culture dishes in a very short period of time. So, cooperation of two or three people is recommended.
Acknowledgments
The work was supported by National Natural Science Foundation of China (31930027 to P.Z. and 32000422 to W.Z.), National Key Research and Developmental Program of China (2021YFA1102002), Yunnan Fundamental Research Projects (202001AT070140 and 2019FB049). The protocol is derived from the assay systems described in Zhang et al. (2023).
Competing interests
The authors have no competing interests.
References
- Berti, M., Cortez, D. and Lopes, M. (2020). The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat. Rev. Mol. Cell Biol. 21(10): 633–651.
- Collins, A. R. (2004). The Comet Assay for DNA Damage and Repair: Principles, Applications, and Limitations. Mol. Biotechnol. 26(3): 249–261.
- Dungrawala, H. and Cortez, D. (2015). Purification of Proteins on Newly Synthesized DNA Using iPOND. The Nucleus : 123–131.
- Elguindy, M. M. and Mendell, J. T. (2021). NORAD-induced Pumilio phase separation is required for genome stability. Nature 595(7866): 303–308.
- Gylling, H. M., Gonzalez-Aguilera, C., Smith, M. A., Kaczorowski, D. C., Groth, A. and Lund, A. H. (2020). Repeat RNAs associate with replication forks and post-replicative DNA. RNA 26(9): 1104–1117.
- Jackson, S. P. and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461(7267): 1071–1078.
- Pierce, A. J., Johnson, R. D., Thompson, L. H. and Jasin, M. (1999). XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes & Development 13(20): 2633–2638.
- Sirbu, B. M., Couch, F. B. and Cortez, D. (2012). Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat. Protoc. 7(3): 594–605.
- Wang, L., Li, J., Zhou, H., Zhang, W., Gao, J. and Zheng, P. (2021). A novel lncRNA Discn fine-tunes replication protein A (RPA) availability to promote genomic stability. Nat. Commun. 12(1): 5572.
- Zhang, W., Tang, M., Wang, L., Zhou, H., Gao, J., Chen, Z., Zhao, B. and Zheng, P. (2023). Lnc956 -TRIM28-HSP90B1 complex on replication forks promotes CMG helicase retention to ensure stem cell genomic stability and embryogenesis. Sci. Adv. 9(4): eadf6277.
Article Information
Copyright
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Zhang, W., Tang, M., Wang, L., Zheng, P. and Zhao, B. (2023). Purification of Long Non-coding RNAs on Replication Forks Using iROND (Isolate RNAs on Nascent DNA). Bio-protocol 13(21): e4869. DOI: 10.21769/BioProtoc.4869.
- Zhang, W., Tang, M., Wang, L., Zhou, H., Gao, J., Chen, Z., Zhao, B. and Zheng, P. (2023). Lnc956 -TRIM28-HSP90B1 complex on replication forks promotes CMG helicase retention to ensure stem cell genomic stability and embryogenesis. Sci. Adv. 9(4): eadf6277.
Category
Molecular Biology > RNA > RNA purification > Affinity purification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.