- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Post-translational Modification–enhanced Pull-down Method to Study Degron Domains and the Associated Protein Degradation Complexes
Published: Vol 13, Iss 18, Sep 20, 2023 DOI: 10.21769/BioProtoc.4816 Views: 2746
Reviewed by: Ralph Thomas BoettcherQingliang ShenAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2023

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






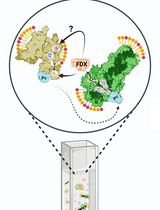
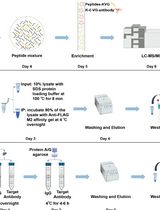
Abstract
The identification and characterization of the ubiquitin E-ligase complexes involved in specific proteins’ degradation via the ubiquitin-proteasome system (UPS) can be challenging and require biochemical purification processes and in vitro reconstitution assays. Likewise, evaluating the effect of parallel phosphorylation and ubiquitination events occurring in vivo at dual phospho/ubiquitin-regulated motifs (called Phospho-Degrons or pDegrons) driving UPS degradation of the targeted protein has remained elusive. Indeed, the functional study of such E1-E2-E3 complexes acting on a protein-specific level requires previously or otherwise acquired knowledge of the nature of such degradation complex components. Furthermore, the molecular basis of the interaction between an E3 ligase and its pDegron binding motif on a target protein would require individually optimized in vitro kinase and ubiquitination assays. Here, we describe a novel enzymatically enhanced pull-down method to functionally streamline the discovery and functional validation of the ubiquitin E-ligase components interacting with a phospho-degron containing protein domain and/or sub-domain. The protocol combines key features of a protein kinase and ubiquitination in vitro assay by including them in a pull-down step exerted by a known or putative pDegron-tagged peptide using the cell extracts as a source of enzymatically active post-translational modification (PTM) modifying/binding native proteins. The same method allows studying specific stimuli or treatments towards the recruitment of the molecular degradation complex at the target protein’s phospho-degron site, reflecting in vivo–initiated events further enhanced through the assay design. In order to take full advantage of the method over traditional protein–protein interaction methods, we propose to use this PTM-enhanced (PTMe) pull down both towards the degradation complex discovery/ID phase as well as for the functional pDegron recruitment validation phase, which is the one described in the present protocol both graphically and in a stepwise fashion for reproduceable results.
Key features
• Suitable to study UPS-regulated (a) cytosolic and/or nuclear proteins, (b) intracellular region of transmembrane proteins, and (c) protein sub-domains bearing a known/putative pDegron motif.
• Requires a biotin-tagged recombinant version of the target protein and/or sub-domain.
• Allows the qualitative and quantitative analysis of endogenous ubiquitin (Ub) E-ligases recruitment to a known or putative pDegron bearing protein/sub-domain.
• Allows simultaneous testing of various treatments and/or conditions affecting the phosphorylative and/or ubiquitylation status of the studied pDegron bearing protein/sub-domain and the recruited factors.
Graphical overview

Background
Regulation of protein degradation is particularly relevant for the cell to control the expression levels of regulatory proteins along with the underlying biological processes. The major known cellular proteostasis mechanism is mediated by the ubiquitin-proteasome system (UPS) and uses a dedicated set of widespread enzymes called ubiquitin E-ligases. Their role is to flag specific proteins by adding a ubiquitin moiety to specific lysine residues that will then be further identified, targeted, and complexed by E-ligases (typically initiated by E3, followed by E2 and E1 types) and ultimately degraded by the UPS (reviewed in [1]). The essential amino-acid sequence allowing ubiquitin-targeted protein degradation has been called Degrons [2–4]. Degrons in which phosphorylation residue(s) play either a permissive or inhibitory role towards degradation are best defined as phospho-(regulated) Degrons and are ubiquitously found in regulatory proteins involved in growth and developmental processes [4,5]. Phospho-Degrons in which the alternative phosphorylation vs. ubiquitylation requirement establishes a biological switch are also referred as phospho-inhibited degrons [6]. Phospho-Degron motifs can be predicted by the parallel presence of in vivo PTM-modified Ser/Thr/Tyr amino-acid residues within a relative short distance from an in vivo modified lysine [7–9]. At present, the post-translational modification (PTM) status of putative Phospho-Degrons can be exerted by independent assays looking either at the ubiquitylation (in vitro ubiquitination assay) or phosphorylation status (through in vitro kinase assays). We have designed an integrated strategy and method to enhance specificity towards both (a) identifying a putative phospho-degron and simultaneously (b) functionally confirm the ID of the ubiquitin E1,2,3-ligases recruited by the phospho-degron-bearing protein/domain. Central to the described method is the synthesis of a biotin-tagged protein domain/subdomain or peptide bearing the native ubiquitin-binding motif amino acidic sequence for the putative phospho-Degron. This recombinant biotin-tagged protein domain/peptide is used as the bait component of the PTM-enhanced (PTMe) pull-down method. The method relies on the ex vivo–enhanced phosphorylation and ubiquitylation (PTMs) events induced in vivo. This bears increased advantages over performing separate co-immunoprecipitations (co-IP)/far westerns and individual kinase or ubiquitylation assays in immunoprecipitated proteins, as shown in Figure 1. The method can indeed be used prior to further refining individual protein interactions within a protein degradation complex, requiring deletion and single-point recombinant mutants, therefore sparing lengthy preparatory work for the expression and purification of the individual recombinant proteins before having clarified their functional involvement in the degradation complex. PTMe pull-down conditions can also be used for proteomics profiling in the degradation complex discovery phase, but the LC-Ms/Ms approach may not be available to all laboratories. We have successfully used this method towards the characterization of a growth factor–regulated phospho-Degron that we initially identified in an angiogenic/metastatic membrane receptor kinase of the Ephrin family, over-expressed in malignant mesothelioma cells [6]. We have adopted the described protocol in combination with immune-enriched proteomics towards further identifying the protein complex for the degradation of the angiogenic kinase EphB4 [6], whose inhibition by an IGF-II autocrine signal determines its over-expression in a malignant mesothelioma cell line [5].
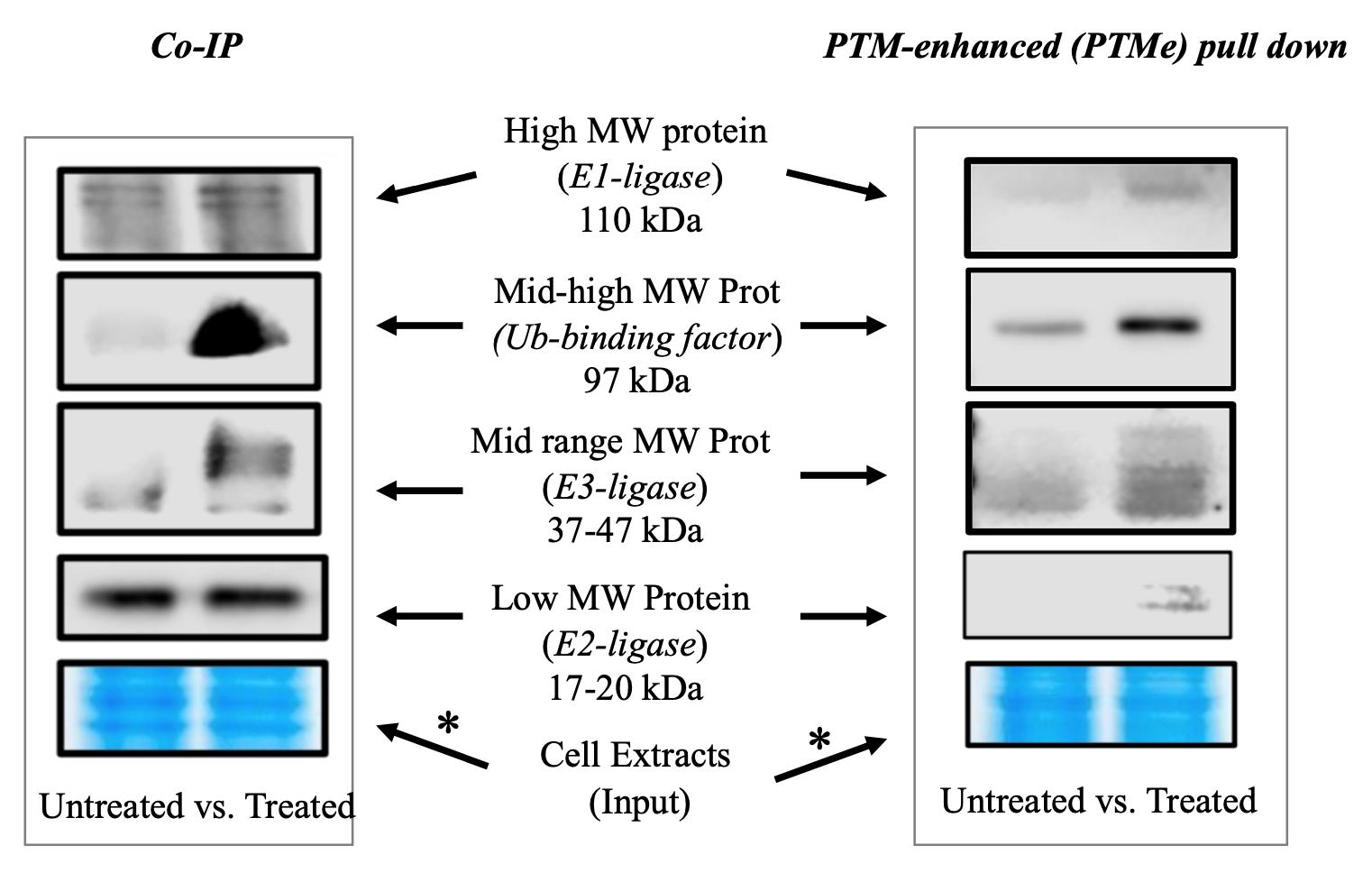
Figure 1. Immuno-blotting detection branch of the post-translational modification–enhanced (PTMe) pull-down method and comparison with traditional co-immune precipitation (co-IP) on the same cellular extract batch from treated vs. untreated cells. An aliquot of the material used in each condition was run on a gel and used for Coomassie stain. The asterisk-marked arrows refer to the complex components for which the method can provide results not observed with traditional co-IP as specified under background. The right-side image is part of the study by Scalia et al. (2023) [6], in which the method was first used. *Please note that two different areas of the same Coomassie gel are shown for comparing the post-normalization protein amount of the same cell extract batches used for both the displayed co-IP and PTMe pull-down experiments.
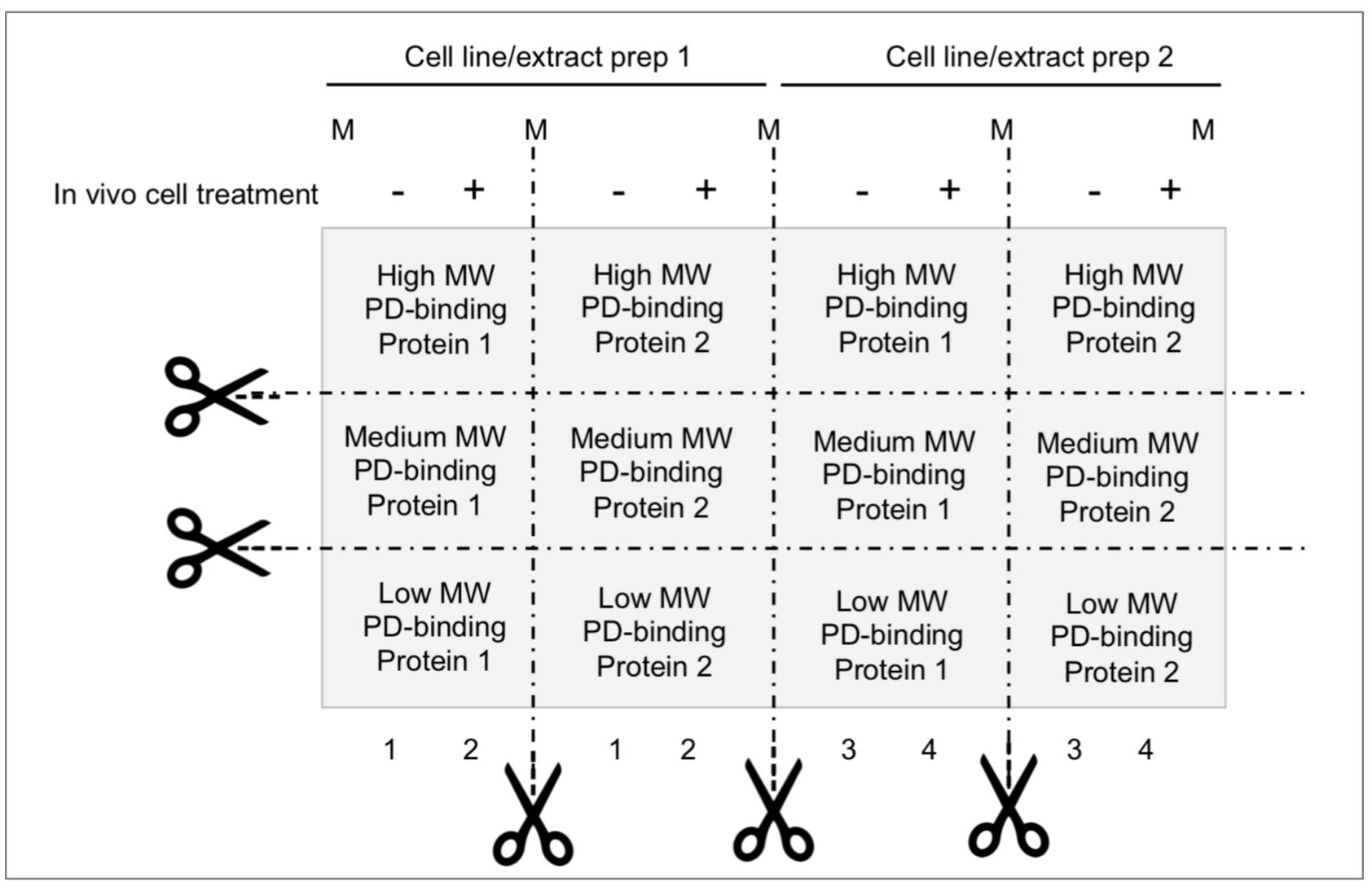
Figure 2. Membrane partition suggested for parallel analysis of ubiquitin (Ub)-E-ligases detection/pDegron recruitment study using post-translational modification–enhanced (PTMe) pull-down assay. Membrane partition is suggested given the relevance of studying same-cell extract batches deriving from individual in vivo treatment conditions and underlying PTMe pulled-down complexes from individual experiment. Proper control suggested between conditions is a Coomassie stain of equal amounts of (protein-normalized) cell extract input resolved by SDS-PAGE.
Materials and reagents
Materials include disposable sterile items like tubing and cell culture supplies obtained from Thermo Inc. (Waltham, MA, USA). Reagents, along with the working concentrations used, have been conveyed in Table 1.
Table 1. Reagents
| Name | Reagent Specs | Working conc. | Provider | Product# |
|---|---|---|---|---|
| Putative PD-synthetic peptide (N/C-biotin tagged) | Assay substrate/bait HPLC-grade | 50 μg/ pull-down reaction (rxn) | New England Peptides (MA) | custom synthesis |
| Ubiquitin, human synthetic | Ub-moiety donor | 1 μg/rxn | Boston Biochemical | U-100H |
| Tris-HCl | pH buffer | 10 mM | Sigma-Aldrich | 77-96-1 |
| PBS | pH buffer | 1× to final rxn vol. | Sigma-Aldrich | D1408 |
| ATP | phosphate moiety donor | 4 mM | Sigma-Aldrich | A1852 |
| DTT | reducing agent | 4 mM | Sigma-Aldrich | D9779 |
| MnCl2 | prot kinase cofactor (Mn++) | 2 mM | Sigma-Aldrich | 1375137 |
| MgCl2 | prot kinase cofactor (Mg++) | 2 mM | Sigma-Aldrich | 1374248 |
| NP40 | Rxn/pull-down facilitator | 0.05% | Sigma-Aldrich | 9036-19-5 |
| D5/W5 inhibitors cocktail | Protease inhibitors | See Recipe 2 | ||
| Kaleidescope prestained protein markers | broad range visible protein markers | 6 µL/lane | Bio-Rad | 1610324 |
| WB-Master Protein Standard | Chemiluminescence Sensitive Markers | 10 µL/lane | Genscript | M00521 |
| SuperSignalTM West Femto Chemiluminescent Substrate | Chemiluminescence Ultrasensitive Reagents system | 0.5 mL/reagent/ Membrane strip | Fisher | 34095 |
| Antigen (/Ab Clone) | Animal source/ clonality | Working concentration | Provider | Clone /product# |
| Ubiquitin (P4D1) | mouse monoclonal | 1:1,000 | Santa Cruz | P4D1/sc8017 |
| Anti-mouse IgG-HRP | chicken monoclonal | 1:2,000 | Santa Cruz | sc2954 |
| Anti-IgGk-LC-HRP | Mouse monoclonal | 1:1,000 | Santa Cruz | sc156102 |
| Streptavidin mag beads | n/a | 2 μL/mL | Genscript | L00936 |
Recipes
The key component of the PTMe pull-down method stands in the homonymous reaction/incubation mixture described below.
PTMe pull-down reaction mixture
PTMe Reaction (rxn) Stock conc. Volume per rxn Final conc. N-Biotin pDegron-peptide coupled w/STP-magnetic beads (see text) n/a 50 μg-STP bound/rxn Tris-HCl, pH 7.5 80 μL 10 mM DTT, 100 mM 8 μL 4 mM ATP, 100 mM 8 μL 4 mM MnCl2,100 mM 4 μL 2 mM MgCl2, 100 mM 4 μL 2 mM Ubiquitin, human 1 µg/µL 1 μL 1 μg Cell extracts (2.5–3.5 µg/µL) Up to 95 μL 0.2–0.3 mg PBS, 0.05% NP40 +D5/W5 protease inhibitors mix
To 200 μL (as needed) 1× Protease and related activities inhibitors stocks
Water soluble inhibitors (W5)
Inhibitor [Stock] µL stock to produce [working]
for 5 mL pull-down buffer[Working] NaVO3 0.2 M 25 µL 1 mM Benzamide 0.25 M 40 µL 2 mM β-glycerophosphate 1 M 50 µL 10 mM NaF 1 M 50 µL 10 mM DTT 1 M 5 µL 1 mM * Sub-total volume (165 µL) - Mol Grade H2O to 10× 483.5 µL n/a PBS, 0.05% NP40 n/a 4,500 µL 1× DMSO soluble inhibitors stock (D5)
Inhibitor [Stock] µL stock to produce [working]
5 mL pull-down buffer
[Working] Pepstatin 10 mg/mL 5 µL 10 µg/mL Leupeptin 10 mg/mL 5 µL 10 µg/mL Aprotonin 10 mg/mL 5 µL 10 µg/mL Okadaic acid 100 µM 5 µL 100 nM PMSF 0.5 M 5 µL 0.5 mM Sub-total volume (25 µL) - DMSO for 10× 487.5 µL n/a PBS, 0.05% NP40 n/a 4,500 µL 1× It is advisable to prepare a larger volume of 10× W5 and D5 stocks, make 0.5 mL aliquots, and save them for single fast thawing (water bath, room temperature). Keep at -20 °C for three months or at -80 °C for six months.
*DTT is added separately to the final buffer containing water-soluble and DMSO-soluble protease inhibitors, to produce a buffer containing 1 mM DTT.
PBST is the antibody probing buffer and is prepared starting by PBS at 1× dilution in sterile deionized H2O with Tween 20 detergent at 0.001% (w/w).
Note: All reagents were purchased from Sigma-Aldrich, St. Louis, MO, USA
Equipment
Cell culture facility (SterilGard III tissue culture hood; CO2 Auto Zero tissue culture incubator, Thermo Scientific, Waltham, MA or comparable models)
4 °C refrigerator or same temperature cold room
-20 °C and -80 °C freezers
Benchtop microcentrifuge supporting 14,000× g spins (e.g., Eppendorf 5418 model or comparable)
Gentle orbital rotation instrument (Boekel Orbital rotator, Feasterville, PA or comparable)
Thermo block with heating capability of 95 °C (Thermo Digital Block Dry Heater, Thermo Scientific, Waltham MA or comparable)
Protein electrophoresis/western blot equipment (NOVEX XCell II and NOVEX TransBlot system, Thermo Scientific, Waltham MA or comparable)
Gel image multi-signal detection system (Odissey XF imaging system, LICOR, Lincoln, NE or comparable). The suggested equipment brands are only indicative and not considered as limiting the outcome of the described protocol
Procedure
The protocol has been conveyed in three sequential workflows that can take place on sequential days, with the exception of Workflow B that must be continued until proteins transfer to PVDF membrane. Day three can start from the post-overnight membrane blocking step in workflow C. The author acknowledges that the optimization of the present protocol may be favored by previous investigator’s hands-on experience with protein immunoprecipitation and protein kinase or other PTM-based in vitro assay. Therefore, in order to speed up the optimization of the present method, it is advised getting familiar with the mentioned procedures prior to dive straight into the integrated approach described herein.
Benchwork considerations (Figure 3)

Figure 3. Stepwise procedure, workflow AAn N-/C-terminally tagged protein domain bearing a pDegron or a corresponding Biotin-conjugated synthetic peptide is used as a substrate/bait (New England Peptides, Gardner, MA) in a modified pull-down/ubiquitination/phosphorylation in vitro assay designed to enhance the specific post-translational modifications (ubiquitination and phosphorylation) induced by the ubiquitin-ligase/phosphorylation native kinase enzymatic activities in the cell extracts from either treated cells or those obtained from parallel untreated cells.
The pDegron protein/peptide bait can be either synthetized by a commercial provider or generated through recombinant DNA cloning and protein purification methods, depending on the length of the domain and on the laboratory cost/time benefit evaluations. The pDegron synthetic peptide was reconstituted in sterile molecular biology-grade water as per manufacturers’ recommendation at a 1 mg/mL concentration, aliquoted in sterile microfuge tubes, and stored at -20 °C until use. We did not find signs of degradation or loss within six months but have not tested longer storage times. The purified phospho-Degron-tagged peptide is therefore used as a bait for the in vitro protein complex capture assay in the presence of pre-treated cell lysates as a source of the native enzymatic factors targeting the degron peptide (namely, protein kinases or PKs and Ub-ligases), along with defined reconstituted inorganic components aimed to enhancing both protein kinase and ubiquitylation activities triggered by the in vivo treatment during the in vitro pull-down incubation step. In the case of the pDegron characterized by the authors through the described method, its putative presence in the studied protein kinase (PK) was first demonstrated by looking at the differential effect of a cancer-secreted growth factor signal on the (Cyclohexymide-sensitive) protein levels on the over-expressed PK [5]. The same GF signal was then specifically correlated to the phosphorylation and ubiquitination status of the C-terminal region of the PK, allowing the definition of the pDegron as a phospho-inhibited type. This means that the pDegron is active and leads to the protein UPS-mediated degradation upon de-phosphorylation. Therefore, the recruitment of degradation factors (namely the E3-E2-E1-binding ubiquitin ligases) to the pDegron (peptide mimicking the C-tail region of the PK) was enhanced through a comparative experimental design (as shown in Figure 1), which included untreated cell extracts (with intact GF signal, inhibited pDegron, and over-expressed target protein degradation rescue status) vs. GF-signal-deprived cell extracts (with dephosphorylated pDegron and recruited degradation factors) [6].
Benchwork considerations (Figure 4)
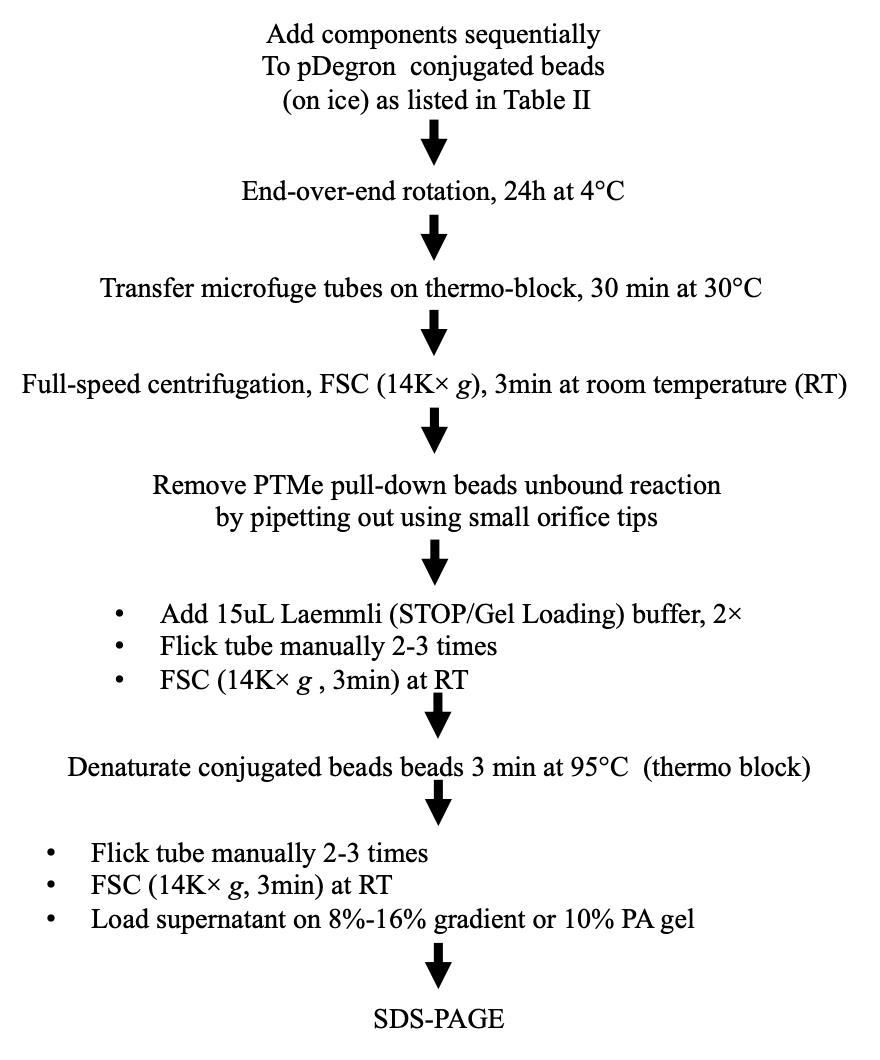
Figure 4. Stepwise procedure, workflow BFor protocol validation and during the discovery phase, two cell pellets (for untreated and treated conditions, respectively) were pooled from 4× 150 mm plates at semi-confluency and used to generate the two cell extract batches (with several frozen aliquots generated to avoid multiple freezing/thawing cycles) displayed in Figure 1. Specifically, a single preparation (of treated and untreated cells) was used towards performing, at different times, (a) co-IP, (b) PTMe pull down, and (c) Ms/Ms profiling from a single biological source. The traditional co-IP method demanded the highest starting protein amount (we used up to 0.5–0.7 mg of cell extract per IP condition for the results shown in Figure 1), followed by the PTMe pull down (0.2–0.3 mg/condition); the most sensitive was the immuno-enriched LC-Ms/Ms (0.05–0.150 mg/condition).
Although we obtained good results on the PTMe pull down with less than 100 μg whole cell extracts for the detection of binding proteins through mass spectrometry, we suggest increasing the protein amount to the maximum usable volume of normalized protein extracts for the pull-down reaction step towards immuno-blot detection (0.2–0.3 mg of enzymatically active protein extracts). Enzymatically active cell extracts were obtained by (1) adding the described protease inhibitors (Recipe 2) to the lysis buffer, (2) performing all cell-free steps on ice, (3) adopting fast freezing (liquid nitrogen immersion) and slow thawing (on ice) of the tested cell extracts, and (4) avoiding multiple freezing/thawing cycles.
First, combine 50 μg of Nt-Biotin-tagged synthetic pDegron-bearing peptide with 6.25 μL of Streptavidin (STP) magnetic beads (Genscript, Piscataway NJ) for 2 h at 4 °C with rotation, followed by a PBS 0.05% NP40 wash. Then, use the STP beads–coupled N-Biotin peptide bait to assemble a 200 μL mixture containing 0.75 mg whole-cancer cell extracts (enzymatically active) along with 10 mM Tris pH 7.5, 4 mM ATP, 2 mM MnCl2, 2 mM MgCl2, 1 μg human recombinant Ubiquitin, and protease/phosphatase inhibitors at the final concentrations summarized in Recipe 2.
In order to determine the optimal amount of starting cell extracts needed for a successful result, an aliquot of the pulled down product was used in parallel with 2–5 μL of total cell extracts for SDS-PAGE followed by gel staining with either Coomassie blue or Silver Stain. Protein–protein interactions along with cell extracts–induced phosphorylation/ubiquitination post-transcriptional modifications were allowed to occur by incubating the mixture for 24–48 h at 4 °C with gentle rotation.
At the end of the pull-down incubation, use the microfuge tubes containing pDegron beads–bound proteins either (a) without wash for LC-Ms/Ms analysis or (b) transferred to a thermo block pre-equilibrated at 30 °C for further incubation, enhancing the cell extracts endogenous protein kinase and ubiquitination events. Alternatively, transfer an aliquot (e.g., 1/3 volume) of the PTMe-complexed beads into a microfuge tube and wash twice with 1 mL of ice-cold PBS to stop the reaction; use the beads for LC-Ms/Ms (as displayed in the graphic overview). The need for PTMe to detect native proteins bound to our pDegron through mass spectrometry was mitigated by the ex vivo treatment of our cells to increase the specific proteins interaction to our pDegron [6]. Nonetheless, in those cases where no biologic background is available about the regulation of the putative pDegron domain, PTMe for both the proteomic step and the immuno-blot detection may become the preferred approach.
After incubation in step B3, add Stop/SDS Gel loading (Laemmli) buffer and DTT (1 mM final concentration) to the beads and perform denaturation at 95 °C for 3 min. Following brief vortexing and full-speed centrifugation, load the supernatant sample on a polyacrylamide gel (fixed 10%, 12%, or 8%–16% gradient) for SDS-PAGE.
Although the workflow ends with protein separation on SDS-PAGE for conceptual simplicity, for practical reasons it must continue to protein transfer to the suggested solid membrane (PVDF), in order for the protocol to be suspended as suggested in workflow C (Figure 5).
Benchwork considerations (Figure 5)
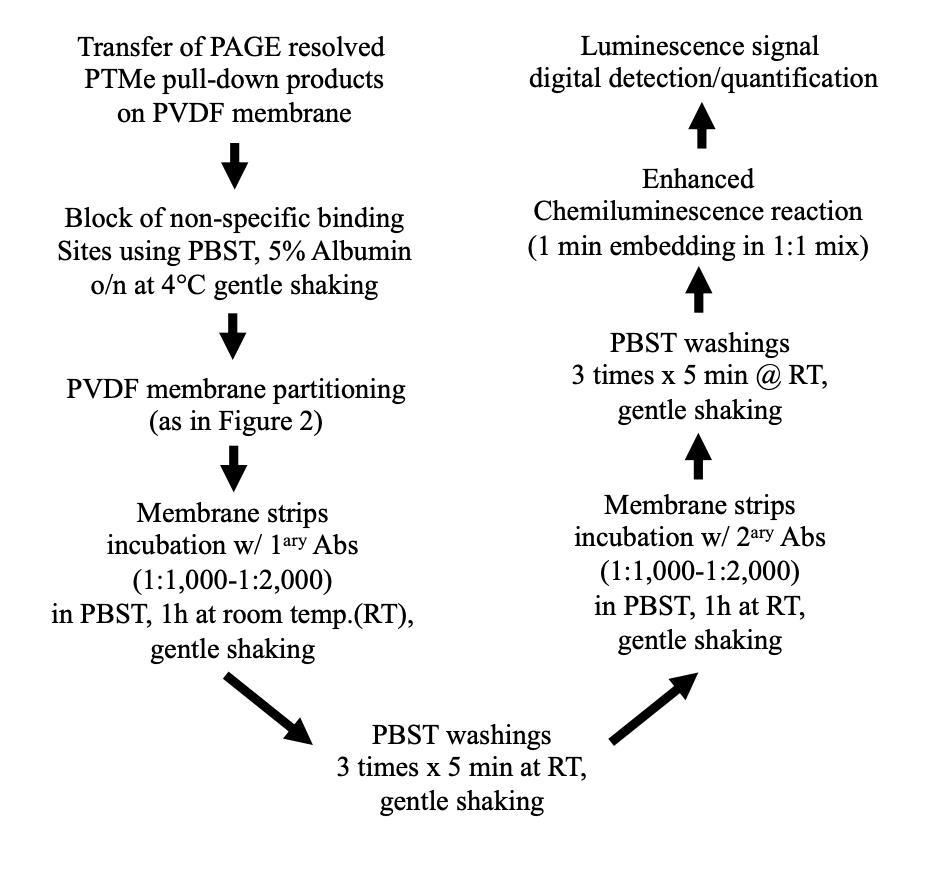
Figure 5. Stepwise procedure, workflow CTransfer resolved proteins onto PVDF (which requires pre-activation with soaking for 5 s in organic solvent, as suggested by the manufacturer, followed by equilibration in PBST buffer for 2–5 min).
Block non-specific binding sites through incubation in PBST 5% BSA overnight at 4 °C.
To obtain the most from each cell extract preparation and single experiment, cut membrane strips from the post gel-to-membrane transfer solid support (PDVF) as shown in Figure 2 and perform MW-specific immunoblotting on each strip immediately after step C3. Membrane partition and differential p-Degron binding components immune detection of the same extracts (rate limiting or PTMe) require former establishment of the identity and MW of the protein complex components, through either the suggested enriched proteomic approach or other proteomic workflows.
The use of enhanced luminescence signal (we used SuperSignal West Femto kit from Pierce) for detection of femto-molar amounts of PVDF-transferred protein turned out to be a key factor for the results of the described method (see under troubleshooting tips, Table IV).
The PTMe pull-down method described herein was optimized using a tagged peptide of the studied protein bearing a previously identified phospho-Degron motif [5].
Upon denaturation of the pull-down complex bound to the studied (tagged) phospho-Degron peptide, proceed to either a proteomic study or a co-IP assay, by resolving the pull-down endogenous proteins by SDS-PAGE and by detecting the resolved factors by western blotting.
In case of using the PTMe pull down towards protein complex components ID by LC-Ms/Ms profiling (not part of the present protocol), we suggest adding an ex vivo or in vitro reversible cross-linking step, as described in Scalia et al. (2023) [6].
Validation of protocol
Given the biological complexity driving the formation of degradation complexes and the context-specific variables involved, the direct comparison of traditional co-IP with pull down using the same cell extracts is a mandatory strategy we suggest for protocol validation. The case-specific advantages observed will drive the choice. As shown in Figure 1, PTMe pull down provided greater specificity for the treatment-induced recruitment of all the degradation complex components, while co-IP was accurate only for two of the pDegron binding components tested (identified by previous proteomic screening), which included the complete set of Ub-E-ligases along with a key chaperone/unfoldase co-factor involved in the underlying process. The advantage of validating all components biologically responsible for the degradation of the studied protein in a single experiment using a single cellular extract/preparation/treatment per se justifies the implementation of the proposed PTMe pull-down method over other protocols.
General notes and troubleshooting
The described protocol is best suited for proteins and sub-domains with predicted pDegron function. This can be accomplished through preliminary sub-domain comparisons with existing protein domains known to be ubiquitylated in vivo (the best is pre-tested by western blot for K48 lysine ubiquitylation using available commercial antibodies). Troubleshooting has been conveyed in Table 2 below.
Table 2. PTMe pull-down method troubleshooting
| Problem | Potential technical issues | Solution |
|
| Include protease inhibitors in your cell extract/lysis buffer (see Recipe 2). |
| Low amount or expression of target protein(s) of interest in lysates | Confirm presence of expected or proband protein bands at specific MW range by either silver stain or Coomassie blue method. | |
| Increase the amount of pull-down proteins by: | |
| Potential consumption of ECL reagent in the PVDF membrane from other proteins than the one studied | Cut and perform western blot (WB) of single strip of transfer membrane corresponding to the protein of interest as in Figure 2. | |
| Amount of pull-down protein factors lower than detection limit of chemiluminescent detection system used | Use femto-molar grade WB luminescence detection kit (when using HRP-conjugated secondary Ab) or use IR-labeled secondary Ab. |
Acknowledgments
No public funding was used for this study and the study relative to the described protocol.
Method first reported in Scalia et al. (2023) [6]; discovery and design of the pDegron domain used for the present protocol development: Scalia et al. (2019) [5].
Competing interests
The authors declare no competing interests.
References
- Hershko, A. and Ciechanover, A. (1992). The ubiquitin system for protein degradation. Annu. Rev. Biochem. 61(1): 761–807. doi: 10.1146/annurev.bi.61.070192.003553
- Van Roey, K., Uyar, B., Weatheritt, R. J., Dinkel, H., Seiler, M., Budd, A., Gibson, T. J. and Davey, N. E. (2014). Short Linear Motifs: Ubiquitous and Functionally Diverse Protein Interaction Modules Directing Cell Regulation. Chem. Rev. 114(13): 6733–6778. doi: 10.1021/cr400585q
- Davey, N. E. and Morgan, D. O. (2016). Building a Regulatory Network with Short Linear Sequence Motifs: Lessons from the Degrons of the Anaphase-Promoting Complex. Mol. Cell 64(1): 12–23. doi: 10.1016/j.molcel.2016.09.006
- Hunter, T. (2007). The Age of Crosstalk: Phosphorylation, Ubiquitination, and Beyond. Mol. Cell 28(5): 730–738. doi: 10.1016/j.molcel.2007.11.019
- Scalia, P., Pandini, G., Carnevale, V., Giordano, A. and Williams, S. J. (2019). Identification of a novel EphB4 phosphodegron regulated by the autocrine IGFII/IRA axis in malignant mesothelioma. Oncogene 38(31): 5987–6001. doi: 10.1038/s41388-019-0854-y
- Scalia, P., Merali, C., Barrero, C., Suma, A., Carnevale, V., Merali, S. and Williams, S. J. (2023). Novel Isoform DTX3c Associates with UBE2N-UBA1 and Cdc48/p97 as Part of the EphB4 Degradation Complex Regulated by the Autocrine IGF-II/IRA Signal in Malignant Mesothelioma. Int. J. Mol. Sci. 24(8): 7380. doi: 10.3390/ijms24087380
- Tang, X., Orlicky, S., Liu, Q., Willems, A., Sicheri, F. and Tyers, M. (2005). Genome‐Wide Surveys for Phosphorylation‐Dependent Substrates of SCF Ubiquitin Ligases. Meth. Enzymol.: 433–458. doi: 10.1016/s0076-6879(05)99030-7
- Hao, B., Oehlmann, S., Sowa, M. E., Harper, J. W. and Pavletich, N. P. (2007). Structure of a Fbw7-Skp1-Cyclin E Complex: Multisite-Phosphorylated Substrate Recognition by SCF Ubiquitin Ligases. Mol. Cell 26(1): 131–143. doi: 10.1016/j.molcel.2007.02.022
- Nash, P., Tang, X., Orlicky, S., Chen, Q., Gertler, F. B., Mendenhall, M. D., Sicheri, F., Pawson, T. and Tyers, M. (2001). Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature 414(6863): 514–521. doi: 10.1038/35107009
Article Information
Copyright
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Scalia, P. and Williams, S. J. (2023). A Post-translational Modification–enhanced Pull-down Method to Study Degron Domains and the Associated Protein Degradation Complexes. Bio-protocol 13(18): e4816. DOI: 10.21769/BioProtoc.4816.
Category
Molecular Biology > Protein > Ubiquitinylation
Molecular Biology > Protein > Protein-protein interaction
Molecular Biology > Protein > Targeted degradation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.