- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vivo Electroporation of Skeletal Muscle Fibers in Mice
Published: Vol 13, Iss 13, Jul 5, 2023 DOI: 10.21769/BioProtoc.4759 Views: 2015
Reviewed by: Salma MerchantAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2021
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
In vitro models are essential for investigating the molecular, biochemical, and cell-biological aspects of skeletal muscle. Still, models that utilize cell lines or embryonic cells do not fully recapitulate mature muscle fibers in vivo. Protein function is best studied in mature differentiated tissue, where biological context is maintained, but this is often difficult when reliable detection reagents, such as antibodies, are not commercially available. Exogenous expression of tagged proteins in vivo solves some of these problems, but this approach can be technically challenging because either a mouse must be engineered for each protein of interest or viral vectors are required for adequate levels of expression. While viral vectors can infect target cells following local administration, they carry the risk of genome integration that may interfere with downstream analyses. Plasmids are another accessible expression system, but they require ancillary means of cell penetration; electroporation is a simple physical method for this purpose that requires minimal training or specialized equipment. Here, we describe a method for in vivo plasmid expression in a foot muscle following electroporation.
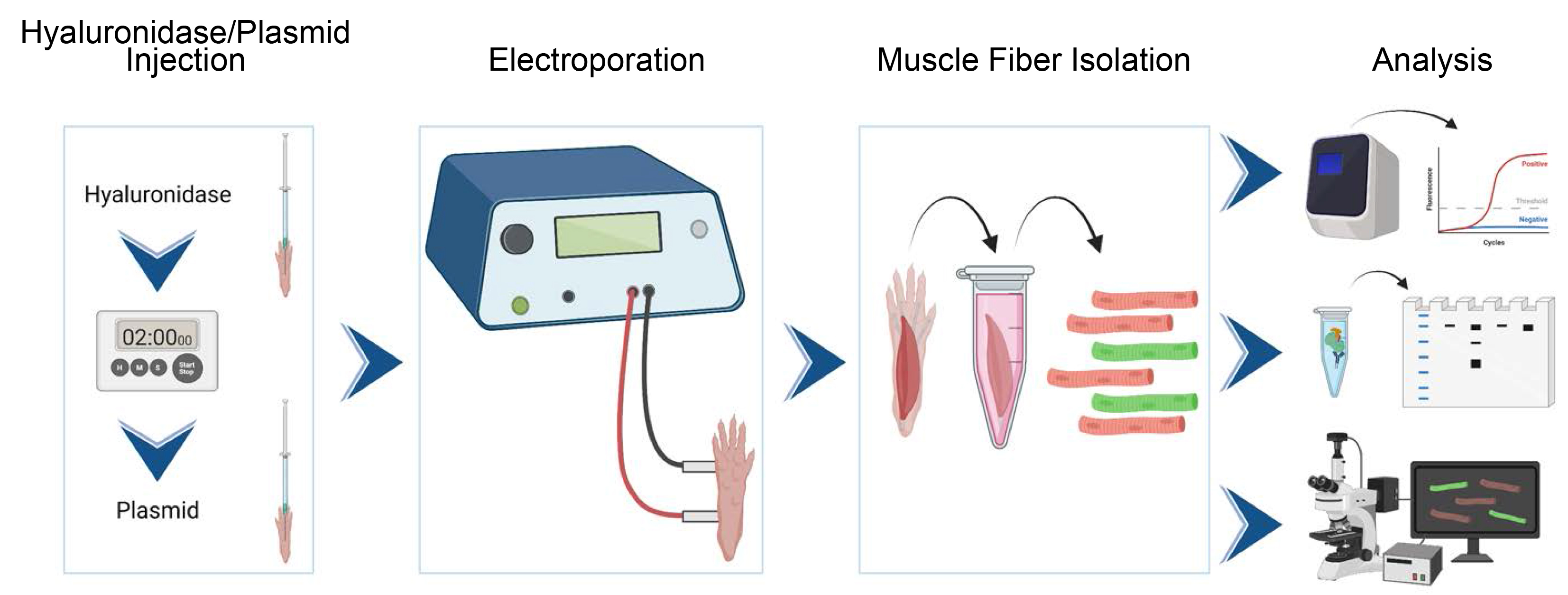
Graphical overview
Background
Skeletal muscle is the largest human organ by mass and is essential for autonomous locomotion (Janssen et al., 2000). It is also the target of several degenerative genetic diseases collectively termed muscular dystrophies and myopathies. Recent improvements in genome sequencing have rapidly expanded the list of genes and gene variants causing muscle diseases, many of which have unknown or unconfirmed functions (Lek and MacArthur, 2014; Biancalana and Laporte, 2015; Angelini et al., 2018; Fichna et al., 2018). Besides the obvious desire to understand these novel disease-causing genes for therapeutic development, an exploration of their expression patterns and function, including cellular trafficking and localization, binding partners, and responses to various environmental stressors, is required to expand the current understanding of muscle cell biology. In vitro systems are convenient for the study of new gene products, as they consist of a single or defined mixture of cell types, are usually amenable to genetic manipulation (both knockout and overexpression), and deliver results quickly relative to in vivo systems. However, mature skeletal muscle fibers differ substantially from myotubes cultured in vitro in terms of size, structure, and three-dimensional organization (Dessauge et al., 2021). For example, nuclei regularly cluster in cultured myotubes but are separated into myonuclear domains in vivo. Furthermore, muscle in vivo consists primarily of contractile units (sarcomeres), which are interwoven with highly organized mitochondrial, t-tubule, and sarcoplasmic reticular systems, but typical muscle cell culture systems do not develop well-organized sarcomeres (Denes et al., 2019). These factors combine to influence expression, localization, and behavior of proteins in skeletal muscle fibers, which can differ substantively from what is observed in vitro (Deshmukh et al., 2015).
Historically, animal models (knockout or transgenic) have been the gold standard for probing protein function in vivo but are associated with substantial production costs and long characterization times. Another disadvantage of engineered mice is that a separate mouse is required for each protein of interest, and studying the behavior of several interacting proteins becomes daunting. One would like to use common in vitro tools, like fluorescent and biochemical tags, in vivo. Labeled proteins can be expressed via vectors in skeletal muscle, provided that the vector is able to enter the cell. Adeno-associated viral vectors can freely transduce muscle following intramuscular injection but are labor intensive to produce (Gregorevic et al., 2004; Qiao et al., 2011). Other viral vectors (e.g., lentivirus or retrovirus) can transduce stem cells in vitro, which can then be transplanted into muscle, but this is technically impractical in most cases (Li et al., 2005; Ousterout et al., 2015). A simpler approach is to transfect cells in vivo with plasmid DNA. This is efficiently accomplished through electroporation, a physical method for reversibly permeabilizing the cell membrane with strong electric field. Here, we describe a method for expressing tagged proteins in skeletal muscle fibers of the flexor digitorum brevis (FDB) muscle of the foot through direct intramuscular injection of plasmid DNA followed by electroporation. While this method is theoretically applicable to most skeletal muscles, the FDB possesses several advantages: it is small, accessible, and easily dissociable to generate a suspension of single muscle fibers for downstream analyses. This protocol provides a simple system for evaluating protein functions in skeletal muscle in vivo (Demonbreun and McNally, 2015; Foltz et al., 2021) with supplies that are attainable for most research labs.
Materials and reagents
Standard laboratory bulk materials
Microfuge tubes
Pipette tips
1 mL syringe
26 G needles
0.2 μm filters
Dissecting board and pins
Dissection scissors and forceps
Additional materials and reagents
Hyaluronidase (Sigma-Aldrich, catalog number: H4272); make a 0.5 mg/mL stock in sterile H2O, aliquot, and store at -20 °C
Isoflurane: must be stored in a locked drawer and accessible only by authorized users
Hank’s balanced salt solution (HBSS) (Thermo Fisher, Gibco, catalog number: 14025092)
Collagenase A (Roche, catalog number: 10103586001); prepare a 1% (w/v) stock solution in sterile DMEM, aliquot, and store for < 6 months at -20 °C
1 M HEPES (VWR, catalog number: 97064-360); store at room temperature
DMEM (Thermo Fisher, catalog number: 11995); store at 4 °C
Bovine serum albumin (BSA) (fraction V) (Roche, catalog number: 10775835001); store at 4 °C
CaCl2·2H2O (Sigma-Aldrich, catalog number: C5080); store at room temperature
MgSO4·7H2O (Sigma-Aldrich, catalog number: M5921); store at room temperature
KCl (Sigma-Aldrich, catalog number: P8041); store at room temperature
NaHCO3 (Fisher Scientific, catalog number: S233-500); store at room temperature
NaCl (Fisher Scientific, catalog number: BP358); store at room temperature
NaH2PO4·H2O (Sigma, catalog number: 9763); store at room temperature
100 mM sodium pyruvate (Thermo Fisher, catalog number: 11360070); store at 4 °C
D-Glucose (Sigma-Aldrich, catalog number: G7528); store at room temperature
MEM amino acids (Thermo Fisher, catalog number: 11130051); store at 4 °C
L-serine (Sigma, catalog number: S4311); store at room temperature
Glycine (Sigma, catalog number: G4392); store at room temperature
L-glutamine (Thermo Fisher, catalog number: 25030081); aliquot and store at -20 °C
Penicillin/streptomycin (Thermo Fisher, catalog number: 15140122); aliquot and store at -20 °C
Digestion buffer (see Recipes)
Imaging medium (optional) (see Recipes)
Equipment
Electroporator (BTX, model: ECM 830), Tweezertrodes platinum plated 7 mm (BTX, catalog number: 45-0488), or needle electrodes (2-needle array electrode 5 mm; BTX, catalog number: 45-0206)
Isoflurane flow regulator (Pro 5 oxygen concentrator 625), induction chamber, and nose cone
Cell culture incubator or bead bath at 37 °C (e.g., Fisher Scientific, model: Isotemp 210)
Procedure
Inject plasmids and electroporate FDB
Prepare a solution of 0.5 mg/mL hyaluronidase in sterile deionized H2O (< 18 MOhm). Aliquot and store at ≤ -20 °C.
Note: It is best to prepare ~100 μL per mouse, to allow 20 μL injection into each footpad and dead volume in the syringe needle.
Anesthetize the mouse in an induction chamber with 3% isoflurane and 0.8 L/min of O2. Once the mouse has stopped moving and breathing has slowed, depth of anesthesia should be confirmed through toe-pinch reflex.
Note: We have performed this procedure on mice ranging from three to six months of age, but generally 16–20 weeks; even so, the procedure is expected to be appropriate for mice outside this range.
During the induction period, load a syringe (1 mL or less) with hyaluronidase solution from step A1. Inject 20 μL into each footpad.
Transfer the mouse from the induction chamber to a nose cone maintaining flow of 1.5% isoflurane. The mouse should lay belly down with legs extended away from its head. Clean the skin of the foot with isopropanol.
The FDB is a long, thin muscle spanning the length of the mouse foot. It is the most superficial of the foot muscles in the mouse. Hyaluronidase should be injected beneath the skin but above this muscle. There is an excess of skin at the heel of the mouse foot that can guide injection into the footbed. The syringe needle can be pressed upward against the excess skin as the needle is inserted down the length of the foot, to ensure that this is achieved. Insert the needle to a point just above the branching of the foot into toes. Eject 20 μL of hyaluronidase solution while slowly pulling the needle out. This helps to deliver the solution evenly throughout the footbed.
Remove the mouse from isoflurane and observe its recovery. Mice should be allowed to recover for several minutes on a heated pad. Once the mouse is awake and alert, return it to its home cage. Allow 2 h for hyaluronic acid to digest extracellular matrix before injecting plasmids.
Prepare plasmids for injection. Injection of 20 μg per foot is standard, although more or less may be desirable (this must be determined empirically). Injection volumes should range between 10 and 20 μL. Therefore, dilute plasmid to a concentration between 1 and 2 μg/mL (if necessary) in sterile water.
Note: Overexpression of some proteins can be toxic, which manifests experimentally as relatively few transfected fibers.
Repeat steps A1–A4 to anesthetize the mouse and prepare for plasmid injection to the FDB. Additionally, while anesthesia is induced, prepare the electroporator, connect electrodes, adjust settings, and sterilize electrode tips.
Inject plasmid into one foot, as in step A5. Immediately position the needle tips of the electrode at opposite ends of the foot (one near the heel and one near the toes). Electroporate the muscle with 20 pulses of 150 V and 20 ms duration separated by 980 ms. The foot should twitch with each pulse. If the voltage is too high, the mouse may convulse throughout its body; if the voltage is too low, the foot will not twitch, and transfection will be poor.
Note: These settings are guidelines that have worked well in our hands but may require adjusting. Transduction efficiency will depend on accuracy of injections and electrode placement as well as the strength of electroporation used.
Allow the mouse to recover as in step A6. Robust expression of plasmids is typically seen within 7–10 days after the procedure.
Dissect FDB and isolate individual muscle fibers
Prepare stocks of 1% collagenase in sterile DMEM (high glucose, with L-glutamine and sodium pyruvate).
Prepare digestion buffer (Recipe 1) and warm to 37 °C until FDB muscles are dissected. Prepare 1 mL of digestion buffer per mouse. Both FDB muscles can be digested in a single microfuge tube with 1 mL of digestion buffer, or individual muscles can be digested in 500 μL each.
Euthanize the mouse with an inhalation overdose of isoflurane and perform a secondary means of euthanasia (e.g., cervical dislocation). Pin the foot to a dissecting board just above the branching of the digits using a dissection pin.
Find the excess skin at the heel of the foot and grasp with forceps. Using dissecting scissors, clip the excess skin. This creates a small hole in the skin at the heel of the foot. Using forceps, gently pull up on the skin at this incision. The skin should be a thin layer, distinct from the muscle underneath. Holding the skin away from the muscle with forceps will help to avoid cutting into and damaging the muscle in the next step.
Using dissecting scissors, cut through the skin on either side of the foot from heel to toe. Leave the skin attached at the toes. The skin should now form a triangle, point detached at the heel, and base attached at the toes. From the tip, peel the skin back to expose the underlying foot muscles.
The FDB tendon is found at the base of the foot near the heel. Grasp the tendon with forceps and cut such that the forceps are still holding the tendon after it is released from the bone. The FDB is the long, superficial muscle directly beneath the skin. It can be released from underlying muscles by pulling up on the tendon with forceps and clipping at the sides along the length of the muscle. It terminates where the toes branch. Cut the tendon to fully release the muscle from the foot. OPTIONAL STOPPING POINT: If individual muscle fibers are not required for downstream analysis, the following steps are not necessary. Transfection efficiency is generally low, so detection of the target protein may require a biochemical tag with strong detection reagents. If the target protein is fluorescently tagged, the following steps are recommended.
Note: See Video 1 for a demonstration of the dissection.
 Video 1. Dissection of mouse flexor digitorum brevis muscle
Video 1. Dissection of mouse flexor digitorum brevis muscleTransfer the whole muscle to a Petri dish containing ~20 mL of sterile HBSS. With forceps, briefly rinse the muscle by moving it through the solution to remove any blood, hair, or other contaminants. Move rinsed FDB muscles to digestion buffer. Incubate at 37 °C for 3 h.
Prepare 10% BSA in DMEM (digestion stop solution, 1 mL per FDB). Warm to 37 °C.
Note: Fetal bovine serum (FBS) can replace BSA, depending on the application.
Transfer FDB muscle from digestion buffer to 1 mL of digestion stop solution. Using a wide-bore P200 pipette tip (cut the narrow tip of a normal P200 tip to make a tip diameter of approximately 2 mm), dissociate the fibers with smooth pipetting, 10 times. Repeat this process twice more, using consecutively narrower pipette tip bores. Finally, using a standard P200 pipette tip, pipette the solution 40 times to fully release individual muscle fibers.
Allow the contents of the tube to settle for 2 min and then remove the supernatant containing individual muscle fibers.
Allow the supernatant to settle for an additional 15 min. The muscle fibers will settle, while small debris, dead cells, and single cells from the tissue will remain floating. Remove and discard the supernatant. Resuspend the individual muscle fibers in a buffer suitable for the desired analysis. For any experiment involving visualization of a fluorescent protein, we recommend modified DMEM imaging buffer (Recipe 2) or commercially available phenol-free DMEM, because phenol red and several vitamins contained in standard cell culture media fluoresce when excited with ~488 nm light.
Data analysis
This method can be used to set up a variety of downstream analyses, including molecular biology and biochemistry, which should be adapted to the preferred protocols for individual labs. We find that the procedure described above has particular utility in live-cell imaging applications. Single muscle fibers from step B11 can be resuspended in a small volume (< 100 μL) of imaging buffer and seeded directly onto a Matrigel-coated glass-bottom culture dish. The low plating volume facilitates adherence within approximately 30 min, at which point additional imaging buffer should be applied to the dish. We provide some examples of experiments that can be done on individual muscle fibers, but these are only a small sampling of what is possible. Detailed instructions for analysis can be found in the methods and Figure S5 of Foltz et al. (2021).
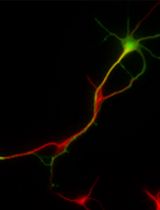
Muscle fibers can be electroporated with plasmids to modulate gene expression (e.g., short-hairpin or micro-RNAs) and a reporter gene (in this case, green fluorescent protein), and then analyzed for phenotype seven days later. In this example, a small molecule sensor for phosphatidylserine (fusion protein of lactadherin C2-domain and mCherry fluorescent protein) was included in imaging medium, the muscle fiber membrane was damaged by 405 nm light, and accumulation of the sensor was observed at the damage site with timelapse confocal microscopy. Images are shown from approximately 7 min post-laser injury. In this example, the shRNA did not influence the localization of the phosphatidylserine probe after injury (Figure 1). Note that the ability of a given plasmid to modulate gene expression should be validated in vitro before in vivo use.

Figure 1. Exposure of intracellular membrane lipids after membrane injury
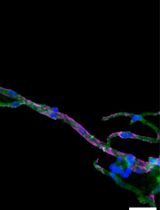
Amphyphysin-2 (BIN1) is a membrane-remodeling protein with a putative role in transverse-tubule biogenesis. Expression of BIN1-mCherry shows expression apparently in t-tubules (Figure 2A), which is in agreement with subcellular localization reported (Prokic et al., 2020). Seven minutes after damage to the sarcolemma, BIN1 becomes enriched at the plasma membrane near the site of injury (white arrowheads, Figure 2B).
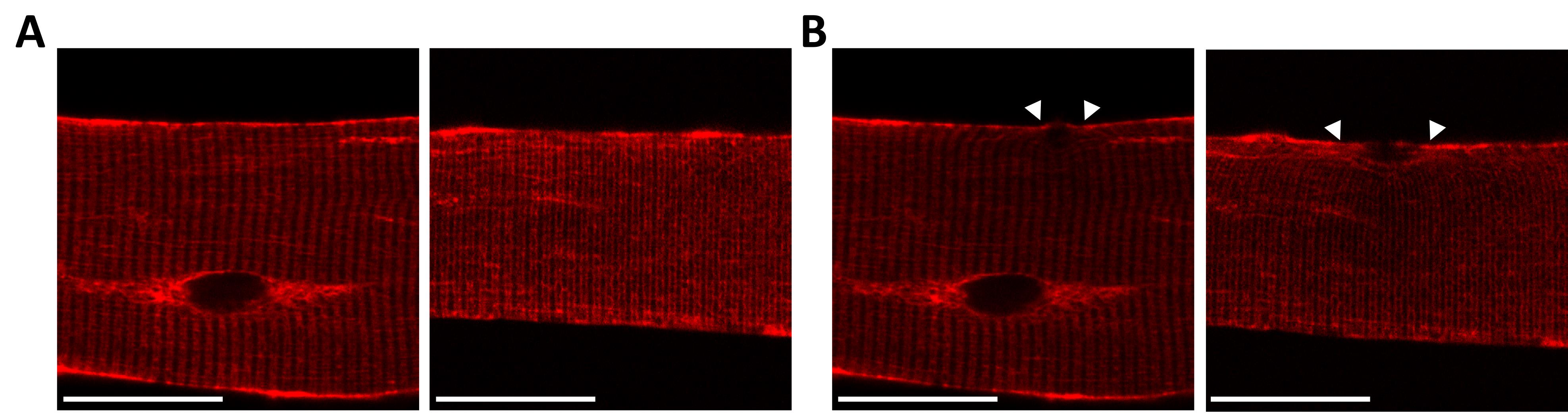
Figure 2. Protein localization in vivo
Notes
We have injected up to two plasmids per FDB in mice, for total expression of four plasmids per animal. When two plasmids were injected into a single FDB, the plasmids were pre-mixed and injected simultaneously. We have observed variability in terms of the efficiency of expression. The likeliest causes are imprecise injections of the plasmid or placement of the electroporation electrodes. However, there is a strong possibility that it is also dependent on the gene expressed. Overexpression of some genes may be toxic. In rare cases, few to no fibers may be expressing the plasmid. Users should keep note of transfection efficiency with all plasmids to track emergent patterns. Changes in the expression vector, for example the promoter, can reduce the level of gene expression and mitigate potential toxicity.
Successful electroporation requires the use of a strong electrical field. It poses a high risk of pain to the mouse and should only be performed on animals maintained under deep anesthesia. Every care should be taken to minimize distress to the animals.
Recipes
Digestion buffer
Reagent Final concentration Amount Collagenase A 0.2% 200 μL HEPES (1 M) 25 mM 25 μL DMEM n/a 775 μL Total n/a 1 mL Imaging medium (optional)
Reagent Final concentration Amount CaCl2·2H2O 1.8 mM 26.4 mg MgSO4·7H2O 0.8 mM 197.2 mg KCl 5.3 mM 39.5 mg NaHCO3 44 mM 369.6 mg NaCl 110 mM 642.8 mg NaH2PO4·H2O 0.9 mM 127.7 mg Sodium pyruvate 1 mM 1 mL D-Glucose 5.3 mM 100 mg MEM amino acids 2× 4 mL L-serine 0.4 mM 42 mg Glycine 0.4 mM 30 mg L-glutamine 4 mM 2 mL Penicillin/streptomycin 100 U/L 1 mL H2O 92 mL Total 100 mL
Acknowledgments
This protocol is derived from the original research paper (Foltz et al., 2021; DOI: 10.1083/jcb.202007059). This work was supported by grants from the National Institutes of Health: the National Institute of Arthritis and Musculoskeletal and Skin Diseases awards R01AR067786 (to H. Criss Hartzell), R01AR071397 (to H.J. Choo), and F32AR074249 (to S.J. Foltz); National Eye Institute award R01EY114852 (to H. Criss Hartzell); and National Institute of General Medical Sciences grant R01GM132598 (to H. Criss Hartzell). Figure prepared in BioRender.com.
Competing interests
The authors declare no competing interests.
Ethics
All procedures involving animals were approved by the Emory Institutional Care and Use Committee under protocols 201800130 (Hartzell) and 201700233 (Choo).
References
- Angelini, C., Giaretta, L. and Marozzo, R. (2018). An update on diagnostic options and considerations in limb-girdle dystrophies. Expert Rev Neurother 18(9): 693-703.
- Biancalana, V. and Laporte, J. (2015). Diagnostic use of Massively Parallel Sequencing in Neuromuscular Diseases: Towards an Integrated Diagnosis. J Neuromuscul Dis 2(3): 193-203.
- Demonbreun, A. R. and McNally, E. M.(2015). DNAElectroporation, Isolation and Imaging of Myofibers. J Vis Exp (106): e53551.
- Denes, L. T., Riley, L. A., Mijares,J. R., Arboleda, J. D., McKee, K., Esser, K. A. and Wang, E. T. (2019). Culturing C2C12 myotubes on micromolded gelatin hydrogelsaccelerates myotube maturation. Skelet Muscle 9(1): 17.
- Deshmukh, A. S., Murgia, M., Nagaraj,N., Treebak, J. T., Cox, J. and Mann, M. (2015). Deep proteomics of mouse skeletal muscle enables quantitation of protein isoforms,metabolic pathways, and transcription factors. Mol Cell Proteomics 14(4): 841-853.
- Dessauge, F., Schleder, C., Perruchot,M. H. and Rouger, K. (2021). 3Din vitro models of skeletal muscle: myopshere, myobundle and bioprinted muscle construct. Vet Res 52(1): 72.
- Fichna, J. P., Macias, A., Piechota, M., Korostynski, M., Potulska-Chromik, A., Redowicz, M. J. and Zekanowski, C. (2018). Whole-exome sequencing identifies novel pathogenic mutations and putative phenotype-influencing variants in Polish limb-girdle muscular dystrophy patients. Hum Genomics 12(1): 34.
- Foltz, S. J., Cui, Y. Y., Choo, H. J. and Hartzell, H. C. (2021). ANO5 ensures trafficking of annexins in wounded myofibers. J Cell Biol 220(3): e202007059.
- Gregorevic, P., Blankinship, M. J., Allen, J. M., Crawford, R. W., Meuse, L., Miller, D. G., Russell, D. W. andChamberlain, J. S. (2004). Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med 10(8): 828-834.
- Janssen, I., Heymsfield, S. B., Wang, Z. M. and Ross, R. (2000). Skeletal muscle mass and distribution in 468 men and women aged 18-88 yr. J Appl Physiol (1985) 89(1): 81-88.
- Lek, M. and MacArthur, D. (2014). The Challenge of Next Generation Sequencing in the Context of Neuromuscular Diseases. J Neuromuscul Dis 1(2): 135-149.
- Li, S., Kimura, E., Fall, B. M., Reyes, M., Angello, J. C., Welikson, R., Hauschka, S. D. and Chamberlain, J. S.
(2005). Stable transduction of myogenic cells with lentiviral vectors expressing a minidystrophin. Gene Ther 12(14): 1099-1108. - Ousterout, D. G., Kabadi, A. M., Thakore, P. I., Majoros, W. H., Reddy, T. E. and Gersbach, C. A. (2015). Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat Commun 6: 6244.
- Prokic, I., Cowling, B. S., Kutchukian, C., Kretz, C., Tasfaout, H., Gache, V., Hergueux, J., Wendling, O., Ferry, A., Toussaint, A., et al. (2020). Differential physiological roles for BIN1 isoforms in skeletal muscle development, function and regeneration. Dis Model Mech 13(11): dmm044354.
- Qiao, C., Koo, T., Li, J., Xiao, X. and Dickson, J. G. (2011). Gene therapy in skeletal muscle mediated by adeno-associated virus vectors. Methods Mol Biol 807: 119-140.
Article Information
Copyright
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Foltz, S. J., Hartzell, H. C. and Choo, H. J. (2023). In vivo Electroporation of Skeletal Muscle Fibers in Mice. Bio-protocol 13(13): e4759. DOI: 10.21769/BioProtoc.4759.
- Foltz, S. J., Cui, Y. Y., Choo, H. J. and Hartzell, H. C. (2021). ANO5 ensures trafficking of annexins in wounded myofibers. J Cell Biol 220(3): e202007059.
Category
Molecular Biology > DNA > Transfection
Developmental Biology > Cell growth and fate > Myofiber
Cell Biology > Tissue analysis > Tissue isolation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.