- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Heterologous Production of Artemisinin in Physcomitrium patens by Direct in vivo Assembly of Multiple DNA Fragments
Published: Vol 13, Iss 14, Jul 20, 2023 DOI: 10.21769/BioProtoc.4719 Views: 2367
Reviewed by: Alba BlesaNazrin Abd AzizKrishna SaharanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
The sesquiterpene lactone compound artemisinin is a natural medicinal product of commercial importance. This Artemisia annua–derived secondary metabolite is well known for its antimalarial activity and has been studied in several other biological assays. However, the major shortcoming in its production and commercialization is its low accumulation in the native plant. Moreover, the chemical synthesis of artemisinin is difficult and expensive due to its complex structure. Hence, an alternative and sustainable production system of artemisinin in a heterologous host is required. Previously, heterologous production of artemisinin was achieved by Agrobacterium-mediated transformation. However, this requires extensive bioengineering of modified Nicotiana plants. Recently, a technique involving direct in vivo assembly of multiple DNA fragments in the moss, P. patens, has been successfully established. We utilized this technique to engineer artemisinin biosynthetic pathway genes into the moss, and artemisinin was obtained without further modifications with high initial production. Here, we provide protocols for establishing moss culture accumulating artemisinin, including culture preparation, transformation method, and compound detection via HS-SPME, UPLC-MRM-MS, and LC-QTOF-MS. The bioengineering of moss opens up a more sustainable, cost effective, and scalable platform not only in artemisinin production but also other high-value specialized metabolites in the future.
Keywords: Physcomitrium patensBackground
Artemisinin is a secondary metabolite produced by the plant Artemisia annua. It is a sesquiterpene lactone with a unique endoperoxide 1,2,4-trioxane ring. This natural compound has been demonstrated to possess a range of biological activities, of which the most known is as an antimalarial drug. However, its low accumulation in the native plant resulted in a high market price, which becomes a major hindrance in its commercial production. Chemical synthesis is difficult due to its complex chemical structure, hence being not economically feasible. Efforts have been made toward the utilization of microbial production systems for increased artemisinin yield, which resulted in a partial synthesis of the compound requiring additional chemical steps (Ro et al., 2006; Paddon and Keasling, 2014). Plant heterologous hosts such as tobacco have been explored, requiring extensive bioengineering (Ting et al., 2013; Malhotra et al., 2016; Wang et al., 2016), yet producing limited artemisinin (Ikram et al., 2015).
Physcomitrium patens is a plant-based production system with important applications in biotechnology research (Simonsen et al., 2009; Ikram et al., 2015). It is a non-vascular plant that has low metabolic and chemical diversity compared to the higher plants (Bach et al., 2014), due to the low number of cytochromes P450 and UDP glycosyltransferases present in its genome (Yonekura-Sakakibara and Hanada, 2011; Hamberger and Bak, 2013). This attribute confers the advantage of reduced risks in unspecific modification by endogenous enzymes and products via pathways in higher plants for the detoxification of xenobiotics (Bach et al., 2014). P. patens is characterized by a simple terpenoid profile with a genome that possesses only a single diterpene synthase (Chen et al., 2011). Its genome is fully sequenced, has a haploid life cycle, and has an efficient homologous recombination machinery similar to yeast, making it an attractive industrial production system compared to other plant heterologous hosts (Lang et al., 2018; Decker and Reski, 2020). In addition, King et al. (2016) developed an orthodox transformation technology that involved the in vivo arrangement of DNA fragments in P. patens, further giving credence to its application as a photosynthetic chassis for heterologous natural products.
To produce artemisinin in P. patens, five genes—namely amorpha-4,11-diene synthase (ADS), cytochrome P450 (CYP71AV1), alcohol dehydrogenase 1 (ADH1), double-bond reductase 2 (DBR2), and aldehyde dehydrogenase 1 (ALDH1)—were introduced into the moss protoplast, being responsible for the biosynthesis of dihydroartemisinic acid that is subsequently converted into artemisinin by photooxidation (Figure 1). The five genes, consisting of three transformation sets, were transformed into P. patens using in vivo homologous recombination that allows multiple DNA fragments to be transformed at once into the genome (King et al., 2016; Khairul Ikram et al., 2017; Ikram et al., 2019). Here, we describe the techniques employed toward the stable production of artemisinin using P. patens as a heterologous host. P. patens culture preparation and maintenance, transformation technique, and metabolite analysis of artemisinin and intermediate compounds are provided. The graphical representation of the overall procedure is presented in Figure 2.
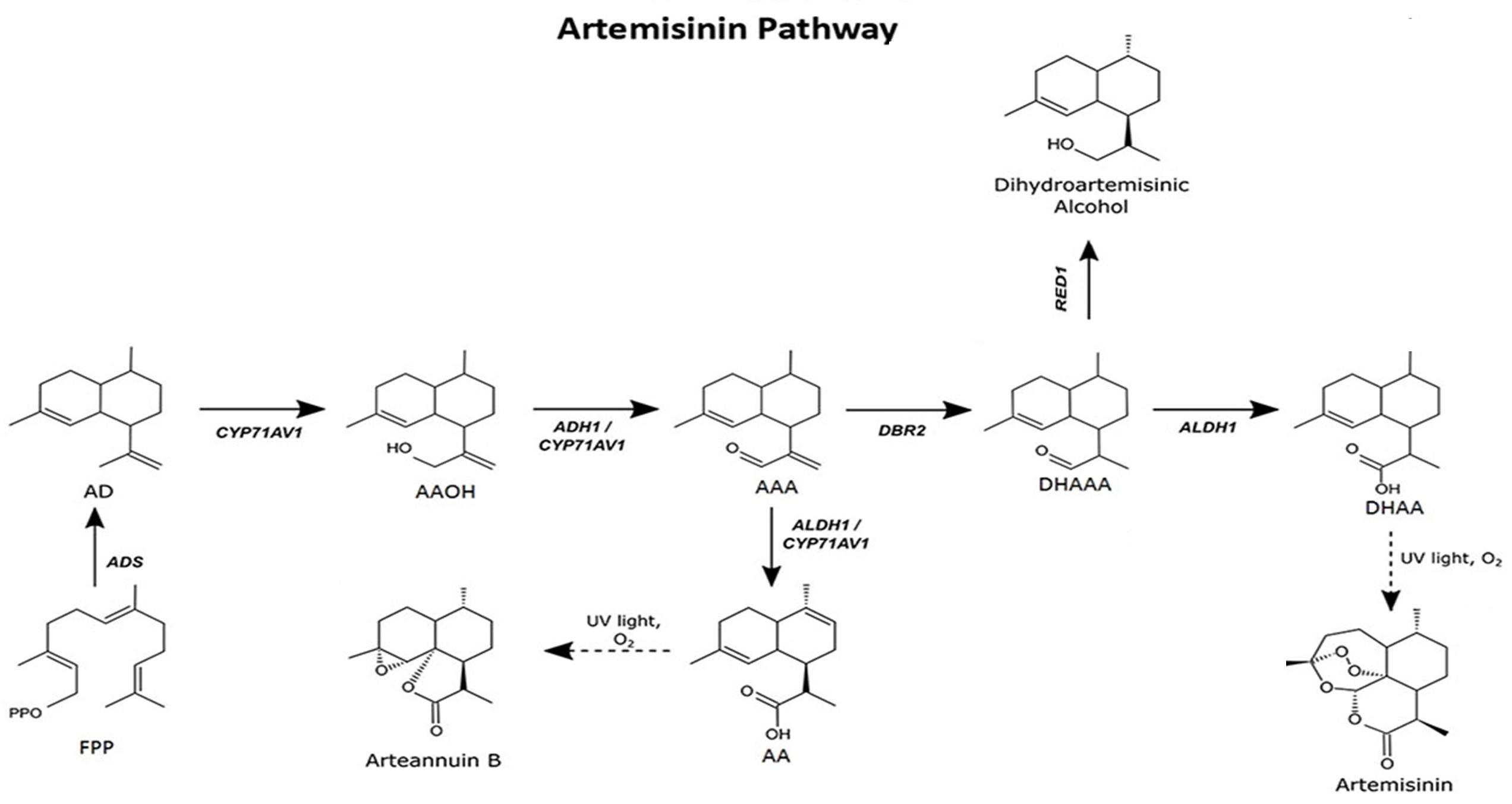
Figure 1. Artemisinin biosynthesis pathway occurs in the glandular trichomes of Artemisia annua. The pathway intermediates are defined as: FPP, farnesyl diphosphate; AD, amorpha-4,11-diene; AAOH, artemisinic alcohol; AAA, artemisinic aldehyde; AA, artemisinic acid; DHAAA, dihydroartemisinic aldehyde; DHAA, dihydroartemisinic acid. Figure adapted from Ikram et al. (2019).
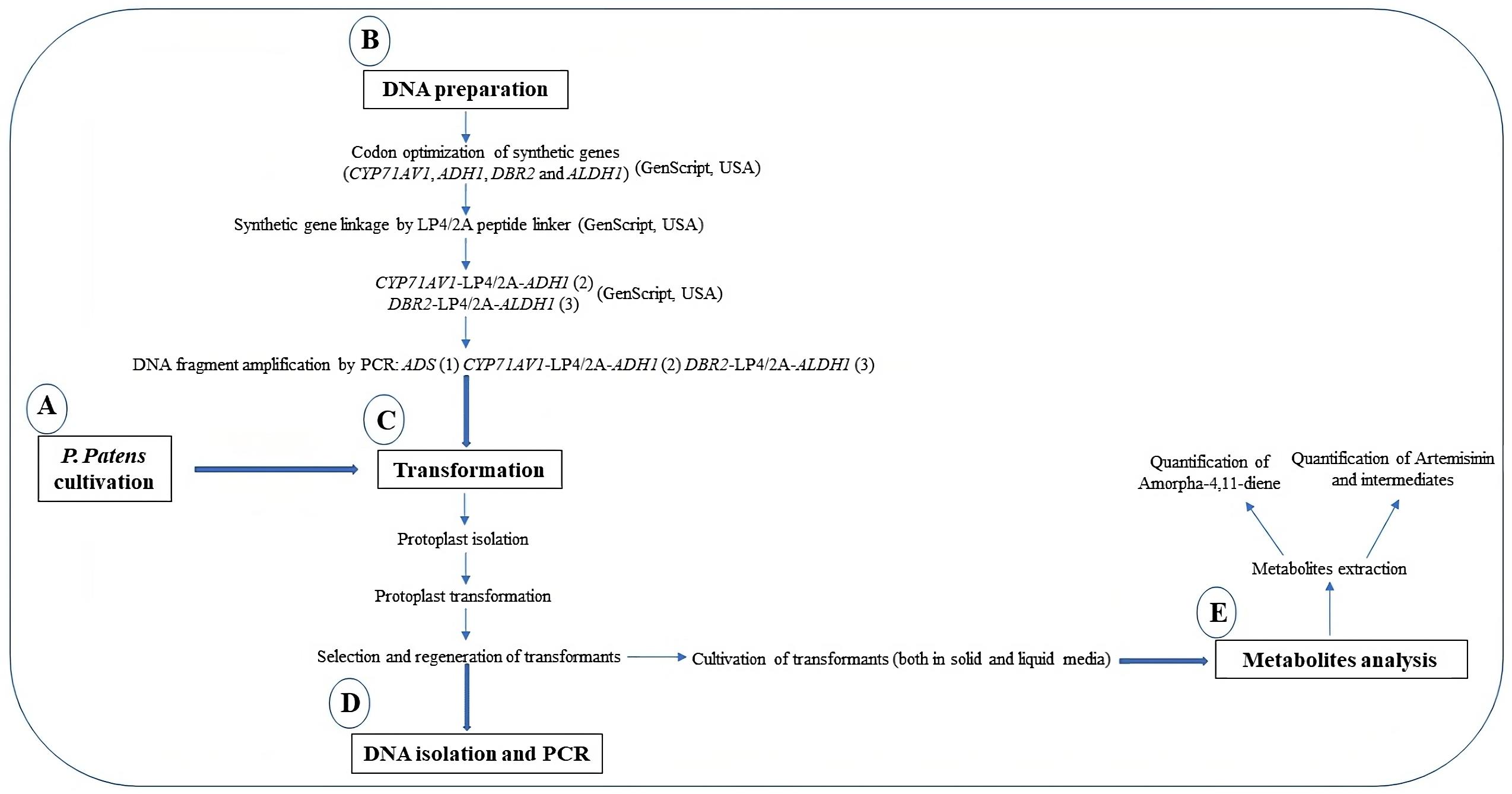
Figure 2. Graphical representation of the protocols
Materials and reagents
Cultivation of Physcomitrium patens
Wildtype Physcomitrium patens (Gransden ecotype #40001) can be obtained from the International Moss Stock Centre at the University of Freiburg, Germany (https://www.moss-stock-center.org/).
Growth room or growth chamber with standard growth condition of a continuous light source with light intensities of 20–50 W/m2 and temperature of 25 °C.
Petri dishes, 90 mm
Serological pipette (Eppendorf)
Sterile pipette (5–50 mL)
Forceps
Note: Wrap forceps separately in foil and autoclave at 121 °C for 20 min.
Cellophane discs
Note: Interleave cellophane discs with filter paper, wrap them in foil paper, and autoclave at 121 °C for 20 min.
Falcon tubes, 50 mL (Thermo Fisher)
Distilled water
Aluminum foil
Erlenmeyer flasks, 50–250 mL
3M surgical tape, 1.25 cm (Micropore, 3M 1533-0)
Tube rack
Parafilm
Calcium chloride (CaCl2) (Sigma-Aldrich, catalog number: C5670-500G)
Ca(NO3)2·4H2O (Sigma-Aldrich, catalog number: C4955-500G)
MgSO4·7H2O (Sigma-Aldrich, catalog number: 63138-1KG)
FeSO4·7H2O (Sigma-Aldrich, catalog number: 215422-250G)
(NH4)2C4H4O6 (Sigma-Aldrich, catalog number: 09985-1KG)
KH2PO4 (Sigma-Aldrich, catalog number: 229806-250G)
KOH (Sigma-Aldrich, catalog number: 60377-1KG)
CuSO4·5H2O (Sigma-Aldrich, catalog number: C3036-250G)
ZnSO4·7H2O (Sigma-Aldrich, catalog number: 221376-500G)
H3BO3 (Sigma-Aldrich, catalog number: B0394-500G)
MnCl2·4H2O (Sigma-Aldrich, catalog number: M3634-500G)
CoCl2·6H2O (Sigma-Aldrich, catalog number: 31277-100G)
KI (Sigma-Aldrich, catalog number: 221945-5G)
Na2MoO4·2H2O (Sigma-Aldrich, catalog number: M1003-100G)
Agar (Duchefa, catalog number: G1101-0500G)
MES (Sigma-Aldrich, catalog number: M3671)
PhyB medium (see Recipes)
1 M CaCl2 (see Recipes)
DNA preparation for Physcomitrium patens transformation
ADS (a gift from Professor Dae Kyun Ro, University of Calgary, Canada)
CYP71AV1 (DQ268763) (GenScript)
ADH1 (JF910157.1) (GenScript)
DBR2 (EU704257.1) (GenScript)
ALDH1 (FJ809784.1) (GenScript)
LP4/2A peptide linker (GenScript)
Ubiquitin promoter from Arabidopsis thaliana (CP002686.1)
Ubiquitin terminator from Arabidopsis thaliana (CP002686.1)
Maize Ubiquitin 1 promoter from the pMP1355 vector
G418 selection cassettes from the pMP1355 vector
Rice actin promoter from the pZAG1 vector
Hygromycin selection cassette from the pZAG1 vector
Microfuge tubes (1.5 mL)
Phusion® High Fidelity DNA Polymerase (New England Biolabs, catalog number: M0530L)
DpnI (New England Biolabs, catalog number: R0176S)
Primers (IDT, USA)
QIAquick PCR Purification kit (QIAGEN GmbH, catalog number: 28104)
Physcomitrium patens transformation
Falcon tubes, 15 mL (Thermo Fisher, catalog number: 14-959-53A)
Serological pipette (Eppendorf, catalog number: 0030127714)
Sterile pipette (5–50 mL)
Cell strainer (100 μm pore size mesh) (Sigma-Aldrich, catalog number: CLS352360)
P. patens culture (five days old)
CaCl2 (Sigma-Aldrich, catalog number: C5670-500G)
MgCl2·6H2O (Sigma-Aldrich, catalog number: M2670-500G)
Driselase® enzyme (Sigma-Aldrich, catalog number: D9515-1G)
D-Mannitol (Sigma-Aldrich, catalog number: M4125-100G)
Tris-Cl (Sigma-Aldrich, catalog number: 10812846001-500G)
Polyethylene glycol (PEG) (MW 6000) solution (Sigma-Aldrich, catalog number: 14504-1G-F)
8.5% D-Mannitol (see Recipes)
Driselase® enzyme solution (see Recipes)
Protoplast wash (PW) solution (see Recipes)
MMM solution (see Recipes)
MCT solution (see Recipes)
Polyethylene glycol (PEG) solution (see Recipes)
Protoplast regeneration medium (bottom layer; PRMB) (see Recipes)
Protoplast regeneration medium (top layer; PRMT) (see Recipes)
DNA isolation and PCR
Microfuge tubes (1.5 mL)
Mortar and pestle
Liquid nitrogen
10% SDS (w/v)
3 M sodium acetate (pH 5.2)
EDTA (Sigma-Aldrich, catalog number: E9884-100G)
Tris (Sigma-Aldrich, catalog number: 10708976001-1KG)
NaCl (Sigma-Aldrich, catalog number: S9888-500G)
Ice
Sea sand (Sigma-Aldrich, catalog number: 1.07711)
Isopropanol (Sigma-Aldrich, catalog number: 563935-1L)
Ethanol (Sigma-Aldrich, catalog number: 459836-2L)
Extraction buffer (see Recipes)
Metabolite analysis
Membrane filter (0.45 μm) (Sartorius, Minisart® RC4)
HP-5MS UI column (20.0 m × 0.18 mm × 0.18 μm) for GC-MS (Agilent)
BEHC18 column (100 mm × 2.1 mm × 1.7 μm) UPLC-MS (Waters)
Artemisinin (Dafra Pharma, Belgium)
Decane (Sigma-Aldrich, catalog number: D901-500ML)
Methanol (Sigma-Aldrich, catalog number: 34860-2.5L)
Acetonitrile (Sigma-Aldrich, catalog number: 34851-2.5L)
Formic acid (Sigma-Aldrich, catalog number: F0507-1ML)
Distilled water
Trans-caryophyllene (Sigma-Aldrich, catalog number: 22075-5ML)
Ethyl acetate (Sigma-Aldrich, catalog number: 270989-2L)
Citrate phosphate buffer (Sigma-Aldrich, catalog number: P4809-50TAB)
Viscozyme® (Sigma-Aldrich, catalog number: V2010-50ML)
Filter paper (Whatman® qualitative filter paper, Grade 1) (Sigma-Aldrich, catalog number: WHA1001325)
Separation funnel
75% (v/v) methanol:water
Acetonitrile:water with formic acid [1:1,000 (v/v)]
Dihydroartemisinin (Dafra Pharma, Belgium)
Artemisinic acid (Chiralix, Nijmegen)*
Artemisinic alcohol (Chiralix, Nijmegen)*
Dihydroartemisinic alcohol (Chiralix, Nijmegen)*
*Synthesized from dihydroartemisinic acid by Chiralix (Nijmegen)
Procedure
Cultivation of Physcomitrium patens (Bach et al., 2014)
Note: Cultivation of P. patens protonema tissue on solid and liquid media should be performed under sterile conditions according to the standard method. All materials should be sterilized by autoclaving prior to use. We recommend a dedicated sterile bench (laminar air flow bench or Biosafety cabinet) for plant tissue culture to reduce the risk of microbial contamination, since no antibiotics are supplemented in the culture media.
Cultivation on solid media
Pour 25–30 mL of PhyB media into a 90 mm Petri dish or until it reaches the half-way point of the Petri dish and leave to solidify for 5–10 min.
Overlay solidified media with sterilized cellophane discs and allow it to settle for 5–10 min.
Note: If the cellophane discs wrinkle when touching the agar inside the plate, use the forceps to straighten them out so the entire disc is in contact with agar. Avoid air bubbles.
Scrap protonema tissue from the previous culture using forceps and place it into a 50 mL Falcon tube containing 10 mL of sterilized distilled water.
Note: Be sure to sterilize the forceps using the glass beads sterilizer at intervals. Allow forceps to cool before applying.
Homogenize the tissue for 30 s using a homogenizer.
Note: Be careful not to over-blend the tissue; excessive blending will lead to poor regeneration of the moss tissues.
Clean the homogenizer tips by blending them into sterile distilled water in a Falcon tube.
Transfer 2 mL of homogenized tissue using a serological pipette into each Petri dish overlaid with cellophane discs.
Note: Ensure that the tissue is well distributed on the plates by swirling adequately.
Allow excess water to evaporate by leaving the Petri dish’s lid slightly open in the laminar air flow bench for 5–15 min.
Note: Do not over dry the plates. The cellophane will curl, and the moss will not survive.
Seal the plates with 3M surgical tape and incubate in a growth chamber under standard conditions. For longer storage, use plastic film for sealing.
Note: Using 3 M surgical tape will give a high growth rate, but the media will dry quickly within 2–4 weeks. For longer storage (up to six months), seal the plates with a plastic kitchen cling wrap.
For routine subculture on solid media, repeat steps A1a–A1h.
Note: We recommend subculturing every two weeks.
Cultivation in liquid media
Add 2–5 mL of blended P. patens tissue into 20–100 mL of PhyB liquid media in a 250 mL Erlenmeyer flask.
Cover the flask with aluminum foil and seal using parafilm.
Place it on a rotary shaker at 90 rpm in a growth chamber under standard growth conditions (25 °C with continuous 20–50 W/m2 light intensity).
After every 1–2 weeks, harvest the tissues, homogenize them for 30 s using homogenizer, and inoculate in fresh PhyB liquid media.
Note: This will enhance growth rates and maintain the tissue in the haploid stage.
DNA preparation for Physcomitrium patens transformation
Note: P. patens has an efficient homologous recombinant machinery comparable to the yeast Saccharomyces cerevisiae. It can assemble multiple DNA fragments in one single transformation event. The five artemisinin genes are transformed into P. patens using the novel in vivo homologous recombination (King et al., 2016).
The synthetic genes CYP71AV1 (DQ268763), ADH1 (JF910157.1), DBR2 (EU704257.1), and ALDH1 (FJ809784.1) are codon-optimized according to the P. patens codon usage by GenScript, USA.
The genes were synthesized together with the LP4/2A peptide linker from Impatiens balsamina and foot-and-mouth disease virus (FMDV) (François et al., 2004).
Note: The synthetic artemisinin genes are linked with LP4/2A linker to facilitate the expression of several proteins under the control of a single promoter: CYP71AV1-LP4/2A-ADH1 and DBR2-LP4/2A-ALDH1. Furthermore, it minimizes the transformation event from five to three (1) ADS, (2) CYP71AV1-LP4/2A-ADH1, and (3) DBR2-LP4/2A-ALDH1).
The DNA fragment is amplified with short overlapping sequences by adding 25–50 base overhangs via PCR primers without pre-assembly of vectors in E. coli (King et al., 2016). The DNA fragments are amplified using Phusion® High Fidelity DNA Polymerase.
For PCR reactions using 1 μL of plasmid DNA as a template, the PCR product is digested with 1 μL of DpnI for 1 h at 37 °C, followed by inactivation at 65 °C for 20 min.
Note: PCR reaction using plasmid as a template is treated with DpnI to digest the methylated DNA template and select the newly synthesized DNA. The primer list is available in Ikram et al. (2017).
The PCR products are purified using QIAquick PCR Purification kit.
Physcomitrium patens transformation
The five artemisinin genes are transformed into P. patens via three independent transformation events (Figure 3). The first transformation cassette is ADS, under the control of maize Ubiquitin1 with a geneticin (G418) selection marker. The second transformation cassette is CYP71AV1-LP4/2A-ADH1, controlled by the rice actin promoter with hygromycin selection marker; the third transformation cassette is DBR2-LP4/2A-ALDH1 construct, controlled by the Arabidopsis Ubiquitin promoter and the G418 selection marker. The genome homologous overhang for the second transformation is targeted to remove the previously integrated G418 cassette, while the third transformation is targeted to remove the second selection cassette, hygromycin.
Note: The targeted integration in the P. patens nuclear genome utilizes 500 bases or longer homologous sequences between the genome and inserted DNA. We recommend using a minimum of 750 base pairs of homologous genomic sequences flanking the targeted insertion site.
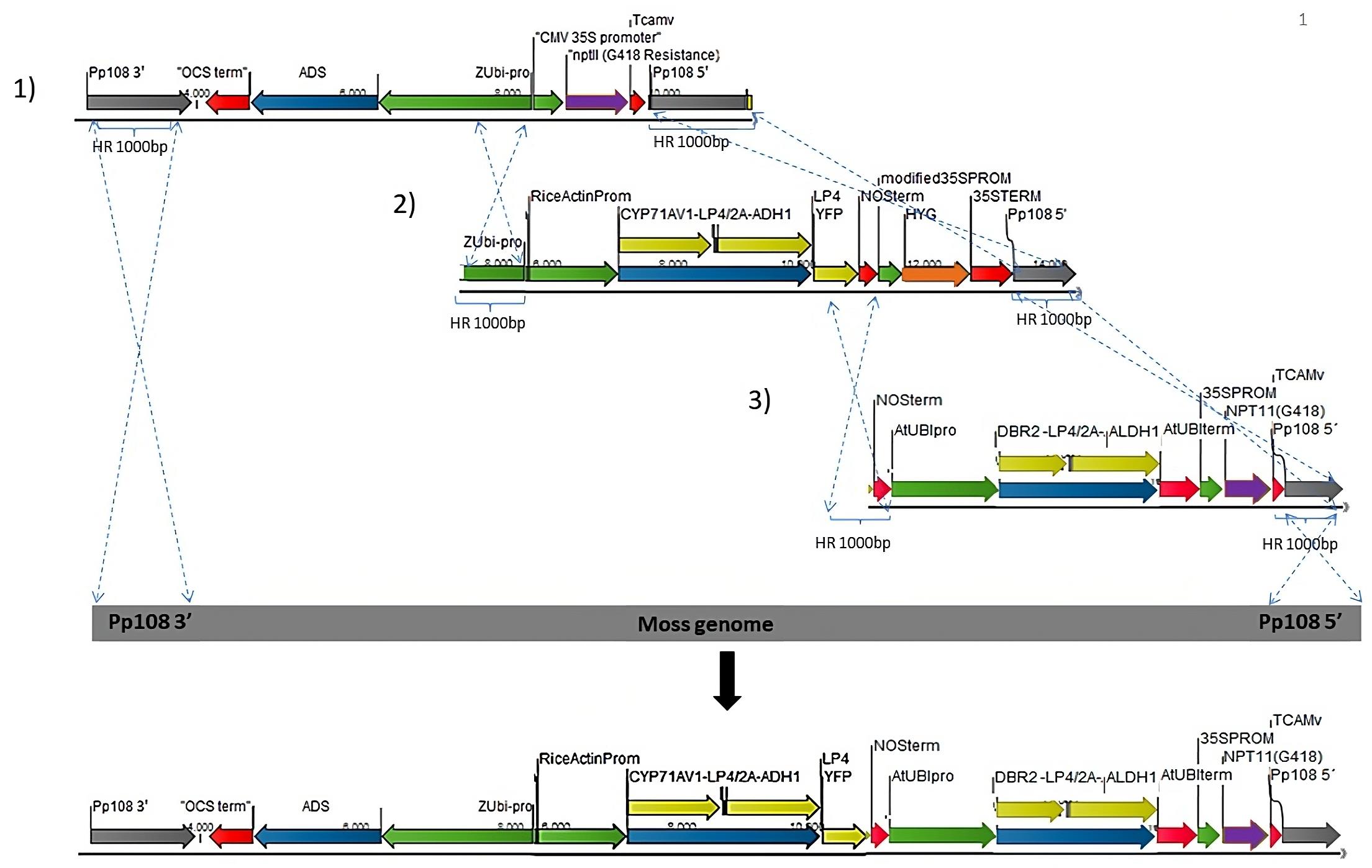
Figure 3. Schematic representation of the different transformation sets into P. patens genome (1–3) using homologous recombination. Grey arrow: P. patens genomic locus (Pp108) homologous recombination flanking regions. Blue: artemisinin genes. Yellow: artemisinin genes linked with 2A/LP4 linker. Green: Promoters. Red: Terminators. Purple: G418/NPTII selection cassette. Orange: hygromycin selection cassette. The dotted lines point to the insertion between each transformation cassette and the P. patens genomic locus.Protoplast isolation (Liu and Vidali, 2011; Bach et al., 2014)
Prepare a fresh PEG solution and let it sit for 2–3 h. Filter sterilize using a 0.22 μm syringe filter just before use.
Note: For transformation, all working solutions (PEG, Mannitol, Driselase, MMM, PW) need to be filter-sterilized using a 0.22 or 0.45 μM syringe filter just before use.
Pour PRMB into the Petri dishes and overlay them with a sterile cellophane disc.
Scrape 5-day-old protonema tissue from the agar plates overlaid with cellophane (approximately 1.5 g fresh weight) and add 1 mL of Driselase® enzyme solution for every 40 mg of P. patens tissue.
Note: We had inconsistent Driselase®enzyme quality from different batches. Hence, it is best to check the efficacy of the Driselase®upfront, or use our new method using Cellulase ONOZUKA R10 and Macerozyme R10 (Batth et al., 2021).
Incubate the tissue at room temperature in normal laboratory light conditions with occasional gentle shaking for 30–60 min.
After incubation, filter the tissue through a sterilized cell strainer (100 μm pore mesh filter) to collect the protoplast and separate it from the undigested tissues. Transfer the protoplast into a 15 mL tube.
Centrifuge the filtrate at 150–200× g for 4 min at 22 °C, with slow acceleration or breaking.
Note: Protoplast is highly sensitive to external force; hence, centrifugation needs to be slow with gentle breaking.
Slowly remove the supernatant with a serological pipette. Be careful not to disturb the protoplast pellet.
Resuspend the protoplast pellet in PW solution using the same volume of Driselase solution that was used in step C1c.
Repeat steps C1f–C1g.
Resuspend the protoplasts in 8.5% D-mannitol (i.e., half of the original volume) and estimate the protoplast density using a hemacytometer (Batth et al., 2021).
Note: The number of protoplasts is used to calculate the final protoplasts concentration in step C2b (Liu and Vidali, 2011; Batth et al., 2021).
Protoplast transformation
Centrifuge at 150–200× g for 4 min at 22 °C with slow breaking and remove the supernatant (see steps C1f and C1g).
Resuspend the protoplast in sterile MMM solution at a concentration of 1.6 × 106 protoplast/mL.
Add 10 μg of total DNA to the bottom of a 15 mL tube, followed by adding 300 μL of protoplast suspension from step C2b and 300 μL of sterilized PEG solution (see step C1a). Mix by gently flicking the tube.
Note: The multi-DNA fragment is added in an equimolar ratio. The total volume of DNA should be less than 30 μL for a successful transformation.
Incubate the mixture in the water bath for 5 min at 45 °C and another 5 min at room temperature.
Dilute protoplast suspension with 300 μL of 8.5% D-mannitol five times with 1 min waiting interval between each dilution.
Add 1 mL of 8.5% D-mannitol another five times with 1 min waiting interval between each dilution.
Pellet the transformed protoplast by centrifugation at 150–200× g for 4 min with slow breaking and remove the supernatant with a pipette.
Resuspend the protoplast in 500 μL of 8.5% D-mannitol, followed by 2.5 mL of protoplast regeneration media (PRMT), and mix by pipetting up and down.
Note: To keep the agar melted, placed the PRMT in a 45 °C water bath, cool enough to ensure protoplast survival.
Evenly dispense 1 mL of the mixture onto the Petri dishes containing protoplast regeneration media (bottom layer) overlaid with cellophane discs (see step C1b). Each transformation will result in three plates.
Seal the plates with 3M surgical tape and incubate them in the growth chamber under standard conditions for 5–7 days.
Selection and regeneration of P. patens transformants
After 5–7 days, transfer the cellophane with regenerating protoplast onto PhyB media containing appropriate selection marker and incubate for two weeks under standard conditions.
After two weeks on PhyB selective media, transfer the cellophane disc with recovered transformants onto a normal PhyB media without antibiotics for another two weeks.
Repeat steps C3a and C3b twice to obtain stable transformants. For characterization, verify transformants using PCR amplification of transgenes and metabolic profiling.
DNA isolation and PCR
Isolation of DNA from transformed P. patens is performed according to the modified method (King et al., 2016; Khairul Ikram et al., 2017).
Place 50–100 mg of fresh P. patens tissue together with a few grains of sea sand into 1.5 mL microfuge tubes and snap-freeze in liquid nitrogen.
Grind the frozen tissue using a microfuge tube pestle and add 400 μL of extraction buffer followed by 80 μL of 10% SDS.
Vortex the mixture and incubate at 65 °C for 30 min.
After incubation, add 180 μL of 3 M sodium acetate (pH 5.2) to the mixture and mix by pipetting up and down.
Centrifuge mixture at 1,500× g at room temperature for 10 min to pellet the debris.
Transfer supernatant to a new tube, add an equal volume of isopropanol, and mix by inversion.
Incubate mixture on ice for 20 min.
Pellet the DNA by centrifugation at 1,500× g for 30 min and discard the supernatant.
Wash the pellet with 80% (v/v) ethanol.
Air dry the sample and re-suspend in 50 μL of sterile water. Store at -20 °C for later use.
For PCR, use 1 μL of DNA as a template.
Metabolite analysis
Extraction of metabolites from P. patens cells
Snap-freeze one-week-old fresh P. patens tissue with liquid nitrogen and pulverize into fine powder using mortar and pestle.
Transfer 3,000 mg of the tissue powder into a 15 mL Falcon tube and add 3 mL of citrate phosphate buffer (pH 5.4).
Vortex and sonicate the mixture for 15 min.
Add 1 mL of Viscozyme® to the mixture and incubate at 37 °C overnight.
Extract the mixture by adding 3 mL of ethyl acetate into the mixture and mix by inverting the tube multiple times.
Centrifuge the mixture at 2,000× g at room temperature for 10 min.
Transfer the extract (supernatant) to a new tube.
Repeat steps E1e–E1f three times and combine all extract into one tube.
Concentrate and dry the extract using nitrogen gas flow in the fume hood. Store at -20 °C until further analysis.
Before analysis, resuspend the dried extract in 300 μL of 75% v/v methanol:water and filter through a 0.45 μm membrane filter.
Extraction of metabolites from P. patens liquid culture
Harvest 500 mL of P. patens liquid culture by passing through a filter paper (Whatman® qualitative filter paper, Grade 1).
Extract the liquid culture with 200 mL of ethyl acetate in a separation funnel.
Concentrate ethyl acetate portion using a rotary evaporator to a volume of 1 mL.
Repeat steps E1f and E1g.
Quantification of Amorpha-4,11-diene in transformed P. patens
Amorpha-4,11-diene is the first product of amorphadiene synthase (ADS), the first gene in the artemisinin biosynthetic pathway. It is a volatile compound, and its analysis can be conducted using a HS-SPME (headspace solid phase microextraction) coupled to a GCMS (Shimadzu, QP2010 Plus) (Drew et al., 2011; Andersen et al., 2015). Quantification of amorpha-4,11-diene is performed according to the published protocol Rodriguez et al. (2014).
Blend one-week-old P. patens in sterile distilled water using a homogenizer.
Normalize the tissue concentration to 0.2 mg/mL (fresh weight).
Add 2 mL of the tissue homogenate into 20 mL of liquid PhyB media in a 100 mL Erlenmeyer flask.
Incubate on a shaker at 90 rpm under standard growth conditions for four days.
Add 2 mL of decane into the culture and allow for a two-week incubation on a shaker (90 rpm) under standard growth conditions.
After two weeks, harvest 100 μL of decane and dilute it twice with ethyl acetate spiked with trans-caryophyllene as internal standard.
Inject 1 μL of the extract in split mode and separate with a HP-5MS UI column (20.0 m × 0.18 mm × 0.18 μm). Hydrogen gas is used as the carrier gas, with injection temperature of 250 °C, oven temperatures of 60 °C (3 min), and 60–320 °C at 40 °C/min.
The concentration of amorpha 4,11-diene can be calculated based on the internal standard run in parallel [see Rodriguez et al. (2014)].
Quantification of artemisinin and intermediates using UPLC-MRM-MS
Artemisinin and artemisinin biosynthesis intermediates can be measured in a targeted approach using tandem quadrupole mass spectrometer that is equipped with an electrospray ionization source and coupled to an Acuity UPLC system (Ting et al., 2013).
We use a BEH C18 column (100 mm × 2.1 mm × 1.7 μm) for chromatographic separation by applying acetonitrile (solvent A) and water with formic acid [1:1,000 (v/v)] (Solvent B) in gradient system.
The gradient starts with solvent A, 5% (v/v) for 1.25 min, followed by a gradient increase to 50% in 2.35 min, and further increase to 90% (v/v) at 3.65 min.
Maintain for 0.75 min before returning to 5% (v/v) solvent A for 0.15 min gradient.
Use the same solvent system as in step E4b to equilibrate the column for 1.85 min at a flow rate of 0.5 mL/min while maintaining the column temperature at 50 °C.
UPLC-MRM-MS condition: set injection volume at 10 μL; desolvation and cone gas flow set to 1,000 and 50 L/h; desolvation and temperatures set to 650 and 150 °C, respectively; capillary voltage set to 3.0 kV; and mass spectrometer operated in positive ionization mode.
Use collision induced dissociation by Argon gas for fragmentation.
Artemisinin and compound intermediates are identified and quantified by multiple reaction monitoring (MRM) with optimized measurement settings for MRM channels (see Table 1).
Relative level of artemisinin and compound intermediates are measured using external calibration curves of reference standards.
Perform metabolite profiling twice and two months apart using the same original cell lines sub-cultivated on PhyB media.
Table 1. Optimized multiple reaction monitoring transition settings for UPLC-MRM-MS measurement of artemisinin, artemisinic acid, dihydroartemisinic acid, artemisinic alcohol, and dihydroartemisinic alcohol (Khairul Ikram et al., 2017)
Parent (m/z) Daughter (m/z) Cone voltage Collision voltage Artemisinin 283.19 265.22 247.19
219.22
12
12
12
14
8
8
Artemisinic acid 235.16 217.21
199.25
189.22
18
18
18
14
16
10
Dihydroartemisinic acid 237.16 163.17
107.12
81.10
16
16
16
28
26
18
Artemisinic alcohol 221.16 203.27
147.09
14
14
20
10
Dihydroartemisinic alcohol 223.22 205.27
109.13
95.07
14
14
14
24
14
12
Data analysis
Validation of transformants
The first transformation event resulted in 11 transformants; all were positive transformants containing the first gene, ADS. The ADS product amorpha-4,11-diene was detected in the 11 lines, with an average content of 200 mg/L. The second transformation resulted in 47 transformants. Eleven out of the 47 were selected for genotyping and all 11 had the ADS and CYP71AV1-ADH1 genes. The final transformation event resulted in three transformants, all having the five genes, ADS, CYP71AV1-ADH1, and DBR2-ALDH1. Figure 4 shows the stable transformants of the three transformation events after two rounds of selection. Genotyping of the three independent transformants showed that the five genes in the biosynthesis of dihydroartemisinic acid were integrated into the genome [Khairul Ikram et al. (2017), refer to Figure S1 in Supplementary Material]. The PCR analysis showed there was no untargeted integration for the selected lines, indicating a uniform integration of the five genes with one copy of each.

Figure 4. Stable P. patens transformants of the three transformation events after two rounds of selection. (A) transformants with ADS gene; (B) transformants with ADS and CYP71AV1-ADH1 genes; (C) transformants with ADS, CYP71AV1-ADH1, and DBR2-ALDH1.
Metabolite profiling
Artemisinin was detected in the transgenic P. patens extracts using ultra-high performance liquid chromatography coupled with a triple quadrupole mass spectrometer operated in MRM mode (UPLC-MRM-MS) (Figure 5). Only artemisinin was detected with no intermediates, and this was confirmed by comparison with an artemisinin standard as shown in Figure 5. The analysis was performed twice, two months apart, in triplicates each time. Quantification of artemisinin yield using external calibration was 0.21 mg/g dry weight (DW), which is higher than what was initially obtained via other plant heterologous expression such as Nicotiana tabacum (0.0068 mg/g DW) (Farhi et al., 2011) and Nicotiana benthamiana (0.003 mg/g DW) (Wang et al., 2016). Analysis by liquid chromatography coupled to quadrupole time-of-flight mass spectrometry (LC-QTOF-MS) shows no glycosylated or glutathione-conjugated products were detected in the P. patens extracts and culture medium. This could be due to the lack of glycosyltransferase 1 (GT1) genes in P. patens compared to higher vascular plants. As no pathway intermediates (conjugated or not) were detected in our extracts, we conclude that the pathway operates efficiently in P. patens.
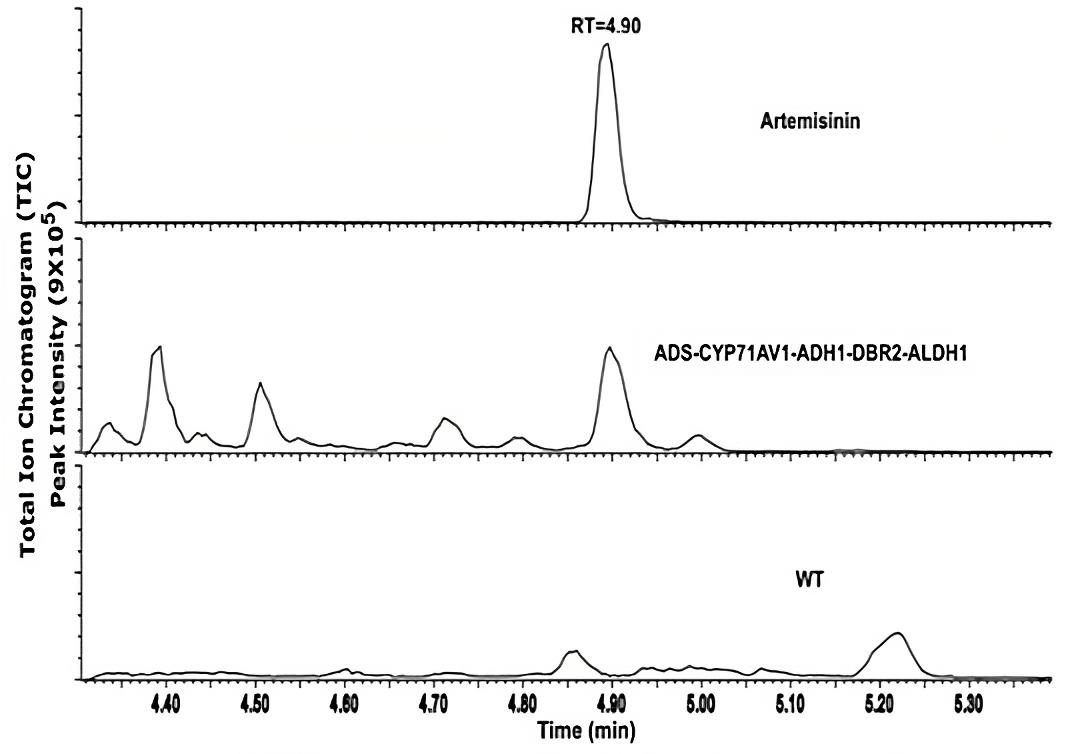
Figure 5. UPLC-MRM-MS analysis of transgenic Physcomitrium patens. Chromatogram of artemisinin standard (top), transgenic P. patens (middle), and wildtype P. patens (bottom). Figure taken from Khairul Ikram et al. (2017).
Recipes
PhyB media
Reagent Final concentration Amount (for 1 L) Ca(NO3)2 4.59 mM 800 mg MgSO4·7H2O 1.01 mM 250 mg FeSO4·7H2O 44.96 μM 12.5 mg (NH4)2C4H4O 2.72 mM 500 mg KH2PO4 1.84 mM 250 mg CuSO4·5H2O 0.11 μM 0.0275 mg ZnSO4·7H2O 0.10 μM 0.0275 mg H3BO3 4.96 μM 0.307 mg MnCl2·4H2O 0.99 μM 0.195 mg CoCl2·6H2O 0.12 μM 0.0275 mg Na2MoO4·2H2O 0.06 μM 0.0125 mg KI 0.08 μM 0.014 mg Distilled H2O n/a Add up to 1 L Total n/a 1 L Adjust the pH of PhyB media to 6.5 using 4 M KOH. Solidify with 0.7% (w/v) agar and sterilize by autoclaving at 121 °C. Add 1 mL of sterile 1 M CaCl2 solution just before use.
Note: Adding CaCl2 freshly before use minimizes precipitation of the media.
The PhyB media (without the addition of CaCl2) can be stored at room temperature under dark conditions for up to one month. The excess media (with CaCl2) can be used or stored for up to two weeks.
1 M CaCl2
Reagent Final concentration Amount (for 100 mL) CaCl2 1 M 11.1 g Distilled H2O Add up to 100 mL Total n/a 100 mL 8.5% D-Mannitol
Reagent Final concentration Amount (for 500 mL) D-mannitol 8.5% 25.75 g Distilled H2O Add up to 500 mL Total n/a 500 mL Driselase enzyme solution
Reagent Final concentration Amount Driselase 0.5% 10 mg 8.5% mannitol 8.5% 20 mL Total n/a 20 mL Note: Filter sterilize with 0.22 μm or 0.45 μm before use.
Protoplast wash (PW) solution
Reagent Final concentration Amount (for 500 mL) D-mannitol 8.5% 42.5 g Calcium chloride (1 M CaCl2) 10 mM 5 mL Distilled H2O n/a Add to 500 mL Total n/a 500 mL Note: Add the calcium chloride immediately before use and filter sterilize.
MMM solution
Reagent Final concentration Amount (for 20 mL) D-mannitol 10.3% 9.1% 17.7 mL Magnesium chloride (1M MgCl2) 15 mM 300 μL MES 10% 2 mL Total n/a 20 mL Note: 2-[N-morpholino]ethanesulfonic acid (MES) (1% w/v, pH 5.6). Dissolve the D-mannitol in the H2O, sterilize by autoclaving, and store at room temperate. On the day of transformation, add the MES and MgCl2 to the D-mannitol solution and filter sterilize.
MCT solution
Reagent Final concentration Amount (for 10 mL) Ca(NO3)2·4H2O 100 mM 236 mg 8.5% mannitol 7.65% 10 mL Tris-Cl (1 M, pH 8.0) 10 mM 100 μL Total n/a 10 mL Note: Prepare fresh on the day of transformation and filter sterilize after adding the PEG.
Polyethylene glycol (PEG) solution
Reagent Final concentration Amount (for 5 mL) PEG (MW 6000) 67 mM 2 g MCT solution n/a 5 mL Total n/a 5 mL Note: Mix well as soon as the MCT is added to avoid recrystallization. Allow the mixture to stand for 2–3 h at room temperature and filter sterilize before use.
Protoplast regeneration media bottom layer solution (PRMB)
Reagent Final concentration Amount (for 1 L) (NH4)2C4H4O6 5 mM 920 mg D-Mannitol 6% 120 g PhyB media solution Add up to 1 L Agar 0.7% 7 g Total n/a 1 L Note: Sterilize by autoclaving at 121 °C. Add 1 mL of sterile 1 M CaCl2 solution just before use.
Protoplast regeneration media top layer solution (PRMT)
Reagent Final concentration Amount (for 250 mL) (NH4)2C4H4O6 5 mM 230 mg D-Mannitol 8% 20 g PhyB media solution Add up to 250 mL Agar 0.4% 1 g Total n/a 250 mL Note: Sterilize by autoclaving at 121 °C. Add 5 mL of sterile 1 M CaCl2 solution just before use.
Extraction buffer
Reagent Final concentration Amount (for 250 mL) Tris (1 M, pH 7.5) 200 mM 50 mL NaCl (5M) 250 mM 12.5 mL EDTA (0.5M, pH 8.0) 25 mM 12.5 mL SDS 0.5% 0.0625 mL Distilled H2O n/a Add up to 250 mL Total n/a 250 mL
Acknowledgments
The authors would like to thank Professor Mark Estelle, Professor Yuji Hiwatashi, Professor Dae Kyun Ro and Dafra Pharma (Belgium) for kindly providing the pMP1355, PZAG1 vector, the ADS template and the artemisinin and dihydroartemisinic acid, respectively. The work was supported by Ministry of Higher education Malaysia. The protocol was referred to Khairul Ikram et al. (2017), Bach et al. (2014), and King et al. (2016).
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Andersen, T. B., Cozzi, F. and Simonsen, H. T. (2015). Optimization of Biochemical Screening Methods for Volatile and Unstable Sesquiterpenoids Using HS-SPME-GC-MS. Chromatography 2(2): 277-292.
- Bach, S. S., King, B. C., Zhan, X., Simonsen, H. T. and Hamberger, B. (2014). Heterologous stable expression of terpenoid biosynthetic genes using the moss Physcomitrella patens. Methods Mol Biol 1153: 257-271.
- Batth, R., Cuciurean, I. S., Kodiripaka, S. K., Rothman, S. S., Greisen, C. and Simonsen, H. T. (2021). Cellulase and Macerozyme-PEG-mediated Transformation of Moss Protoplasts. Bio-protocol 11(2): e3782.
- Chen, F., Tholl, D., Bohlmann, J. and Pichersky, E. (2011). The family of terpene synthases in plants: a mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J 66(1): 212-229.
- Decker, E. L. and Reski, R. (2020). Mosses in biotechnology. Curr Opin Biotechnol 61: 21-27.
- Drew, D. P., Rasmussen, S. K., Avatoc, P. and Simonsen, H. T. (2011). A comparison of headspace solid-phase microextraction and classic hydrodistillation for the identification of volatile constituents from Thapsia spp. provides insights into guaianolide biosynthesis in Apiaceae. Phytochem Anal: 23(1): 44-51.
- Farhi, M., Marhevka, E., Ben-Ari, J., Algamas-Dimantov, A., Liang, Z., Zeevi, V., Edelbaum, O., Spitzer-Rimon, B., Abeliovich, H., Schwartz, B., et al. (2011). Generation of the potent anti-malarial drug artemisinin in tobacco. Nat Biotechnol 29(12): 1072-1074.
- François, I. E. J. A., Van Hemelrijck, W., Aerts, A. M., Wouters, P. F. J., Proost, P., Broekaert, W. F. and Cammue, B. P. A. (2004). Processing in Arabidopsis thaliana of a heterologous polyprotein resulting in differential targeting of the individual plant defensins. Plant Sci 166(1): 113-121.
- Hamberger, B. and Bak, S. (2013). Plant P450s as versatile drivers for evolution of species-specific chemical diversity. Philos Trans R Soc Lond B Biol Sci 368(1612): 20120426.
- Ikram, N. K. B. K. and Simonsen, H. T. (2017). A Review of Biotechnological Artemisinin Production in Plants. Front Plant Sci 8: 1966.
- Ikram, N. K. B. K., Zhan, X., Pan, X. W., King, B. C. and Simonsen, H. T. (2015). Stable heterologous expression of biologically active terpenoids in green plant cells. Front Plant Sci 6: 129.
- Ikram, N. K. K., Kashkooli, A. B., Peramuna, A., Krol, A. R. V., Bouwmeester, H. and Simonsen, H. T. (2019). Insights into Heterologous Biosynthesis of Arteannuin B and Artemisinin in Physcomitrella patens. Molecules 24(21): 3822.
- Khairul Ikram, N. K. B., Beyraghdar Kashkooli, A., Peramuna, A. V., van der Krol, A. R., Bouwmeester, H. and Simonsen, H. T. (2017). Stable Production of the Antimalarial Drug Artemisinin in the Moss Physcomitrella patens. Front Bioeng Biotechnol 5: 47.
- King, B. C., Vavitsas, K., Ikram, N. K., Schroder, J., Scharff, L. B., Bassard, J. E., Hamberger, B., Jensen, P. E. and Simonsen, H. T. (2016). In vivo assembly of DNA-fragments in the moss, Physcomitrella patens. Sci Rep 6: 25030.
- Lang, D., Ullrich, K. K., Murat, F., Fuchs, J., Jenkins, J., Haas, F. B., Piednoel, M., Gundlach, H., Van Bel, M., Meyberg, R., et al. (2018). The Physcomitrella patens chromosome-scale assembly reveals moss genome structure and evolution. Plant J 93(3): 515-533.
- Liu, Y. C. and Vidali, L. (2011). Efficient Polyethylene Glycol (PEG) Mediated Transformation of the Moss Physcomitrella patens. J Vis Exp (50): 2560.
- Malhotra, K., Subramaniyan, M., Rawat, K., Kalamuddin, M., Qureshi, M. I., Malhotra, P., Mohmmed, A., Cornish, K., Daniell, H. and Kumar, S. (2016). Compartmentalized Metabolic Engineering for Artemisinin Biosynthesis and Effective Malaria Treatment by Oral Delivery of Plant Cells. Mol Plant 9(11): 1464-1477.
- Paddon, C. J. and Keasling, J. D. (2014). Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development. Nat Rev Microbiol 12(5): 355-367.
- Ro, D. K., Paradise, E. M., Ouellet, M., Fisher, K. J., Newman, K. L., Ndungu, J. M., Ho, K. A., Eachus, R. A., Ham, T. S., Kirby, J., et al. (2006). Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440(7086): 940-943.
- Rodriguez, S., Kirby, J., Denby, C. M. and Keasling, J. D. (2014). Production and quantification of sesquiterpenes in Saccharomyces cerevisiae, including extraction, detection and quantification of terpene products and key related metabolites. Nat Protoc 9(8): 1980-1996.
- Simonsen, H. T., Drew, D. P. and Lunde, C. (2009). Perspectives on using physcomitrella patens as an alternative production platform for thapsigargin and other terpenoid drug candidates. Perspect Medicin Chem 3: 1-6.
- Ting, H.-M., Wang, B., Ryden, A.-M., Woittiez, L., van Herpen, T., Verstappen, F. W. A., Ruyter-Spira, C., Beekwilder, J., Bouwmeester, H. J. and van der Krol, A. (2013). The metabolite chemotype of Nicotiana benthamiana transiently expressing artemisinin biosynthetic pathway genes is a function of CYP71AV1 type and relative gene dosage. New Phytol 199(2): 352-366.
- Wang, B., Kashkooli, A. B., Sallets, A., Ting, H. M., de Ruijter, N. C. A., Olofsson, L., Brodelius, P., Pottier, M., Boutry, M., Bouwmeester, H., et al. (2016). Transient production of artemisinin in Nicotiana benthamiana is boosted by a specific lipid transfer protein from A. annua. Metab Eng 38: 159-169.
- Yonekura-Sakakibara, K. and Hanada, K. (2011). An evolutionary view of functional diversity in family 1 glycosyltransferases. Plant J 66(1): 182-193.
Article Information
Copyright
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Khairul Ikram, N. K., Zakariya, A. M., Saiman, M. Z., Kashkooli, A. B. and Simonsen, H. T. (2023). Heterologous Production of Artemisinin in Physcomitrium patens by Direct in vivo Assembly of Multiple DNA Fragments. Bio-protocol 13(14): e4719. DOI: 10.21769/BioProtoc.4719.
Category
Biological Engineering > Synthetic biology > Genetic modification
Plant Science > Plant metabolism > Other compound
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.