- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In situ Quantification of Cytosine Modification Levels in Heterochromatic Domains of Cultured Mammalian Cells
(*contributed equally to this work) Published: Vol 13, Iss 14, Jul 20, 2023 DOI: 10.21769/BioProtoc.4716 Views: 1725
Reviewed by: Gal HaimovichVeena PadmanabanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2022
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Epigenetic modifications of DNA, and especially cytosine, play a crucial role in regulating basic cellular processes and thereby the overall cellular metabolism. Their levels change during organismic and cellular development, but especially also in pathogenic aberrations such as cancer. Levels of respective modifications are often addressed in bulk by specialized mass spectrometry techniques or by employing dedicated ChIP-seq protocols, with the latter giving information about the sequence context of the modification. However, to address modification levels on a single cell basis, high- or low-content microscopy techniques remain the preferred methodology. The protocol presented here describes a straightforward method to detect and quantify different DNA modifications in human cell lines, which can also be adapted to other cultured mammalian cell types. To this end, cells are immunostained against two different cytosine modifications in combination with DNA counterstaining. Image acquisition takes place on a confocal microscopy system. A semi-automated analysis pipeline helps to gather data in a fast and reliable fashion. The protocol is comparatively simple, fast, and cost effective. By employing methodologies that are often well established in most molecular biology laboratories, many researchers are able to apply the described protocol straight away in-house.
Keywords: DNA methylationBackground
Methylation of the fifth carbon atom of cytosine (5′-methylcytosine; 5mC) is found in the DNA of most eukaryotes and in all human cell types. It is one of the most abundant and conserved epigenetic modifications and regulates gene expression by silencing promoter regions and facilitating the compaction of heterochromatin (Lande-Diner et al., 2007; Smith and Meissner, 2013). Mammalian DNA methylation occurs predominantly in the context of symmetric CpG dinucleotides. Consequently, of the 28 million CpG sites in the human genome, between 60% and 80% are methylated, which corresponds to approximately 5% of all cytosines (Smith and Meissner, 2013). However, up to 200 base-pair long stretches of these dinucleotides, so-called CpG-islands, are often found in the context of promoters and transcription start sites, where they are sparsely methylated (Bird, 1986; Smith and Meissner, 2013).
Moreover, DNA methylation contributes to the integrity of heterochromatin by serving as a binding platform for various proteins that can recognize and bind 5mC, thereby shaping the heterochromatin landscape (Ludwig et al., 2016). On the other hand, the members of the Tet (Ten-eleven-translocation)-protein family, Tet1, Tet2, and Tet3, were found to oxidize 5mC to 5′-hydroxymethylcytosine (5hmC), 5′-formylcytosine (5fC), and 5′-carboxylcytosine (5caC). All oxidative derivatives of 5mC were found to act as intermediates in the active removal of DNA methylation and are themselves removed via different glycosylases (Ludwig et al., 2016). Tet proteins share the same catalytic activity (Bauer et al., 2015) but different physiological roles (Santiago et al., 2014). An example is Tet1 and its short isoform, which exhibit distinct nuclear localization during DNA replication. This results in aberrant cytosine modification levels in heterochromatic regions of mouse and human cells and LINE 1 activation (Arroyo et al., 2022). While in murine cells heterochromatin rich domains are found on most chromosomes, in human cells long stretches of pericentric heterochromatin are mostly found on chromosomes 1, 9, and 16 (Meneveri et al., 1993; Bizhanova and Kaufman, 2021).
Changes in cytosine modifications and, in particular, hypomethylation, are a hallmark of many cancers (Vidal et al., 2017). A reproducible method to measure changes in cytosine modification levels in euchromatic vs. heterochromatic regions is highly desirable in this context. For this purpose, microscopy techniques are more revealing than mass spectrometry or ChIP-seq methods, since direct images from single cells provide information about the abundance, subnuclear distribution, and cell cycle variation of different epigenetic marks. Therefore, we developed a pipeline to address cytosine modification levels and applied it to MCF7 human breast adenocarcinoma cells, expressing higher levels of TET1 proteins, and MCF10a non-transformed human breast epithelial cells (Figure 1). Our method, performed in Arroyo et al. (2022), includes immunodetection of cytosine modifications with antibodies against 5mC and either 5hmC, 5fC, or 5caC, followed by confocal microscopy imaging and image analysis. The implemented image analysis includes nucleus and heterochromatin and (euchromatic) nucleoplasm segmentation, achieved by creating different masks based on the DNA counterstaining (DAPI) signal. Heterochromatin segmentation using DAPI as a proxy for differences in chromatin compaction has been extensively used in quantitative analyses of the nuclear landscape (Schmid et al., 2017; Pradhan and Cardoso, 2023). The following protocol can be applied to a variety of experimental models and cell lines in order to study and quantify differences in cytosine modification patterns in different cellular backgrounds.
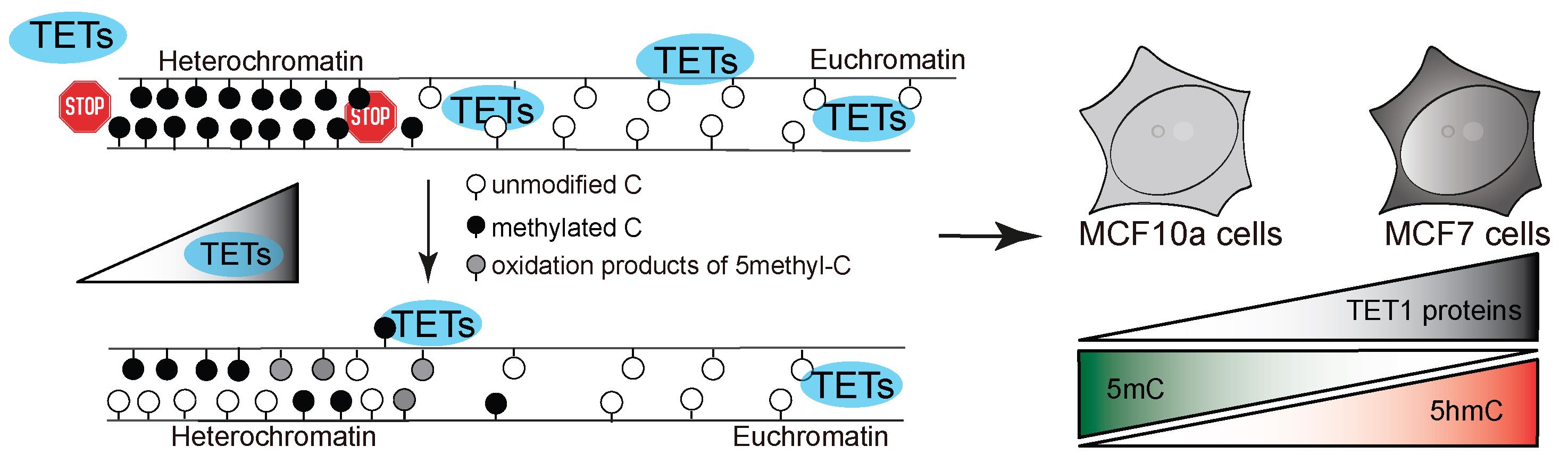
Figure 1. Model of study. TET protein catalytic activity in heterochromatin regions results in aberrant oxidation of 5-methylcytosine in human cells, depending on the level of TET proteins.
Materials and reagents
P100 dishes (Merck KGaA, catalog number: CLS430293)
6-well dishes (Merck KGaA, catalog number: CC302)
Glass coverslips (Carl Roth, catalog number: P232.1)
Microscopy slides (Thermo Fisher Scientific, catalog number: ABAA000080)
Petri dish (Carl Roth, catalog number: EL50.1)
Gel-blotting paper (Carl Roth, catalog number: A126.1)
Aluminum foil (Carl Roth, catalog number: 1770.1)
Parafilm (Merck KGaA, catalog number: P7793)
MCF7 cells (ATCC, catalog number: HTB-22) (Soule et al., 1973)
MCF10a cells (ATCC, catalog number: CRL-10317) (Soule et al., 1990)
DMEM (Merck KGaA, catalog number: D6429), store at 4 °C
DMEM/F12 (Merck KGaA, catalog number: D6421), store at 4 °C
Fetal bovine serum (FBS) (Gibco, catalog number: A5256701), aliquot and store at -20 °C
Horse serum (Merck KGaA, catalog number: H1138), aliquot and store at -20 °C
Pen/Strep (Merck KGaA, catalog number: P4333), store at 4 °C
L-Glutamine (Merck KGaA, catalog number: G7513), store at 4 °C
hEGF (Merck KGaA, catalog number: E9644)
Hydrocortisone (Merck KGaA, catalog number: H0888)
Cholera toxin (Merck KGaA, catalog number: C8052)
Insulin human (Merck KGaA, catalog number: I2643)
PBS (VWR International, catalog number: 0780)
Trypsin (Merck KGaA, catalog number: T4049), store at 4 °C
Gelatine (Sigma-Aldrich, catalog number: G2500)
Formaldehyde (Merck KGaA, catalog number: F8775)
EDTA (Merck KGaA, catalog number: E5134)
MgCl2·6H2O (Merck KGaA, catalog number: 1374248)
CaCl2 (Merck KGaA, catalog number: C1016)
Trizma® hydrochloride (Merck KGaA, catalog number: T2319)
Tween 20 (Merck KGaA, catalog number: P9416)
Triton X-100 (Merck KGaA, catalog number: X100)
Methanol (Merck KGaA, catalog number: 32213)
RNase A (Merck KGaA, catalog number: 10109169001)
BSA (Merck KGaA, catalog number: 10735094001)
DNase I (Merck KGaA, catalog number: D5025)
Monoclonal mouse α-5mC (CF-MAB - Helmholtz Munich, 32E2) (Weichmann et al., 2020)
Polyclonal rabbit α-5-hmC (Active Motif, catalog number: 39769)
Polyclonal rabbit α-5-fC (Active Motif, catalog number: 61223)
Polyclonal rabbit α-5-caC (Active Motif, catalog number: 61225)
Goat α-mouse IgG (H+L) Alexa Fluor 488-conjugated (Thermo Fisher Scientific, catalog number: A32723) (suggested antibody, which worked well in this protocol)
Goat α-rabbit IgG (H+L) Alexa Fluor 594-conjugated (Thermo Fisher Scientific, catalog number: A32740) (suggested antibody, which worked well in this protocol)
DAPI, 4’,6-diamidino-2-phenylindole (Merck KGaA, catalog number: D9542)
Mowiol® 4-88 (Merck KGaA, catalog number: 81381)
Wet chamber for staining (Cardoso and Leonhardt, 1996)
Solutions
0.1% gelatine (see Recipes)
3.7% formaldehyde (see Recipes)
88% methanol (see Recipes)
0.5% Triton X-100 in PBS (see Recipes)
0.01% PBS-T (see Recipes)
0.01% PBS-TE (see Recipes)
4%, 2%, 1% BSA in PBS (see Recipes)
100 µg/mL RNase in 1× PBS (see Recipes)
2× DNase I Buffer (see Recipes)
Recipes
0.1% gelatine
Reagent Final concentration Amount Gelatine 0.1% 0.5 g 1× PBS 1× 500 mL Total n/a 500 mL Sterilize by autoclaving at 121 °C for 30 min. 3.7% formaldehyde
Reagent Final concentration Amount 36.5%–38% formaldehyde solution 3.7 % 2.5 mL 1× PBS 1× 22.5 mL Total n/a 25 mL Prepare fresh and use within one week. 88% methanol
Reagent Final concentration Amount Methanol 88% 88 mL ddH2O 12% 12 mL Total n/a 100 mL Store at -20 °C 0.5% Triton X-100 in PBS
Reagent Final concentration Amount 1× PBS 1× 99.5 mL Triton X-100 0.5% 0.5 mL Total n/a 100 mL Prepare at least two days prior to use and store at room temperature. 0.01% PBS-T
Reagent Final concentration Amount 1× PBS 1× 249.975 mL Tween 20 0.01% 0.025 mL Total n/a 250 mL Store at room temperature. 0.01% PBS-TE
Reagent Final concentration Amount 1× PBS 1× 249.75 mL Tween 20 0.01% 0.025 mL EDTA 100 mM Total n/a 250 mL Store at room temperature 4% BSA in PBS
Reagent Final concentration Amount BSA 4% 0.6 g 1× PBS 1× 15 mL Total n/a 15 mL Dissolve BSA completely, prepare 1.5 mL aliquots, and store at -20 °C. If 1% or 2% BSA are needed, prepare appropriate dilutions with 1× PBS. 100 µg/mL RNase in 1× PBS
Reagent Final concentration Amount RNase A - 0.025 g ddH2O - 250 µL Total 100 mg/mL 250 µL Dissolve RNase A powder completely in ddH2O and store in 25 µL aliquots at -20 °C.
Before use, prepare a 1:1,000 dilution with 1× PBS to yield a concentration of 100 µg/mL.2× DNase I Buffer
Reagent Final concentration Amount Trizma® hydrochloride pH 7.5 20 mM 2 mL MgCl2 5 mM 0.102 g CaCl2 1 mM 0.011 g ddH2O - 98 mL Total n/a 100 mL Prepare 2 mL aliquots and store at -20 °C.
Equipment
Laminar flow cabinet (neoLab Migge GmbH, BDK 1200-SK, catalog number: 83 99 51200)
Cell culture CO2 incubator (Binder, catalog number: C150-230V)
Water bath (GFL Technology, catalog number: 10865892E)
Hybridization oven (Appligene, catalog number: 22012105); alternatively, use 37 °C incubator
Confocal microscope (Perkin Elmer Spinning Disk/Nikon Ti-E, PerkinElmer Life Sciences) equipped with high magnification objective (oil immersion 100× Plan-Apochromat, NA 1.49)
Software
Image acquisition software [in our case, Volocity 6.3 (Perkin Elmer, USA) was used] “Fiji Is Just ImageJ” Fiji (NIH/http://Fiji.sc/Fiji) or ImageJ (NIH/https://imagej.nih.gov/ij/download.html) for image pre-processing and analysis
RStudio (https://rstudio.com/) for plotting and statistical analysis
Procedure
Cell culture (all steps in laminar flow cabinet)
Culture cells by following established protocols for adherent cell lines (for more details see Note 1).
Split cells 2–3 times per week or as needed (for more details see Note 2).
Seeding cells for immunostaining (all steps in laminar flow cabinet)
Fill appropriate cell culture dish (6-well) with ddH2O (2 mL).
Sterilize glass coverslips by dipping multiple times into 70% ethanol using sterilized tweezers.
Transfer glass coverslips to a ddH2O-filled dish. Make sure to submerge coverslips.
Aspirate ddH2O.
Cover glass coverslips with 100 μL of 0.1% gelatine each and incubate for at least 15 min (preferably overnight) at 37 °C.
Aspirate remaining gelatine solution.
Split cells as usual and seed 0.25 × 106 cells in a glass coverslip–containing dish.
Incubate for at least 18 h at 37 °C, 5% CO2.
Immunostaining for DNA base modifications
Remove medium from cells that were seeded the day before at a density of 0.25 × 106 per p35 dish.
Carefully wash cells with 1 mL of 1× PBS and discard PBS.
Fix cells with 1 mL of 3.7% formaldehyde for 10 min at room temperature.
Remove formaldehyde solution.
Wash three times with 2 mL of PBS-T (for more details see Note 3).
Permeabilize cells with 1 mL of 0.5% Triton X-100 in PBS for 20 min.
Remove Triton X-100.
Add ice-cold 88% methanol and incubate for 5 min at room temperature.
Remove methanol.
Wash as before three times with PBS-T.
Prepare a wet chamber with moist gel-blotting paper and Parafilm on top (for more details see Note 4 and Figure 2).
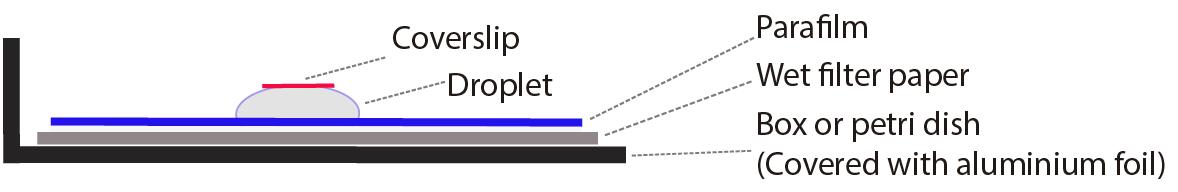
Figure 2. Diagram of a wet chamber for immunostainingPrepare a 100 μg/mL RNase A in 1× PBS solution.
Place coverslips cell side down in 20 μL droplets of RNase A solution on Parafilm and incubate for 1 h at 37 °C.
Wash three times with PBS-T, by carefully adding 100 μL of PBS-T droplets under the coverslips (for more details see Note 5).
Block for 30 min at 37 °C by placing coverslips in 20 μL of 1% BSA in 1× PBS cell side down.
Prepare primary antibody solution (detection solution) per coverslip. For one coverslip use:
10 μL of 2% BSA in PBS.
10 μL of DNase buffer (2×).
0.25 μL of DNase I (2,000 U/mL).
Antibody against 5mC (1:250).
Antibody against 5-hmC, 5-fC, or 5-caC (1:250, 1:100, or 1:100, respectively).
Place coverslips cell side down in detection solution.
Incubate at 37 °C for 70 min.
Wash three times with 100 μL droplets of PBS-TE (0, 5, and 10 min).
Dilute secondary antibodies.
Prepare 20 μL of secondary antibody solution (detection solution) per coverslip. For one coverslip use:
20 μL of 2% BSA in PBS.
Secondary antibody goat α-mouse IgG (H+L) Alexa Fluor 488-conjugated (1:500).
Secondary antibody goat α-rabbit IgG (H+L) Alexa Fluor 594-conjugated (1:500).
Incubate with secondary antibodies at room temperature for 45 min.
Wash three times with 100 μL droplets of PBS-T (0, 5, and 10 min).
Counterstain with 20 μL of 1 μg/mL DAPI for 10 min.
Meanwhile, take out a glass slide and put a 20 μL drop of Mowiol mounting medium on top.
After DAPI staining, briefly wash coverslip by dipping in ddH2O.
Dry excess water by carefully tipping the edge of the coverslip on a paper tissue.
Place on top of the mounting medium with the cells side facing down.
Microscopy
Set up the image acquisition settings for each channel and use the same settings for all images, avoiding oversaturation.
Acquire images from higher to lower wavelength, starting with red, green, and blue at the end. See Note 6 for setting details.
Acquire Z-stack images with a fixed Z-step length, preferably 0.5 μm. Acquire enough Z-stack to image the whole nuclei from “top” to “bottom.” For the majority of the cells, 10–12 Z-stacks are enough.
For further image processing in Fiji, data should be exported as .tiff files as Multicolor Z-stack.
Image analysis (manual procedure using Fiji)
The purpose of this image analysis pipeline is to use DAPI intensities to segment the cell nuclei, creating a nuclear mask, and then segment heterochromatic regions (HCmask). Euchromatic regions are defined by heterochromatin subtraction from the nucleus mask. Once these subnuclear compartments are defined, the final aim of the analysis is to measure cytosine modification levels within these different regions. Minor optimizations to the following procedure can be applied for different cell types or imaging systems.
Pre-processing
Analyze → Set Measurements → select the desired properties to be measured (see Note 7).
Edit → Options → Colors… → Foreground: black; Background: black; Selection: white.
Open a Multicolor Z-stack image.
Image → Stacks → Z Project… → Choose first slice as Start slice and last slice as Stop slice and Max Intensity from Dropdown Menu.
Right-mouse click → Rename to “Master.”
Right-mouse click → Duplicate → Title: “nucleus_mask;” Tick: Duplicate hyperstack; Channels (c): leave as is.
Nucleus segmentation
Select window “nucleus_mask.”
Choose DAPI channel using channel slider.
Image → Type → 8-bit.
Process → Filters → Gaussian Blur… → Sigma (Radius): 1.0 → “OK” → Click “Yes” in dialogue “Process Stack?”.
Process → Enhance Contrast… → Saturated pixels: 0.3% → “OK.”
Right-mouse click → Duplicate → Title: “forHCmask;” Tick: Duplicate hyperstack; Channels (c): leave as is.
Select window “nucleus_mask.”
Process → Binary → Make Binary → Method: Triangle; Background: Dark; Tick: Black background (of binary masks).
Process → Binary → Watershed → “OK” → Click “Yes” in dialogue “Process Stack?”.
Select “Wand (tracing) tool.”
Select window “nucleus_mask.”
Select nucleus of interest.
Edit → Clear Outside → Click “Yes” in dialogue “Process Stack?”.
Analyze → Tools → ROI manager… → Add [t] → Rename… to “nucleus_mask.”
Heterochromatin segmentation
Select window “forHCmask.”
Select window “ROI manager.”
Select ROI “nucleus_mask.”
Edit → Clear Outside → Click “Yes” in dialogue “Process Stack?”.
Image → Adjust → Threshold… → Dropdown 1: Triangle; Dropdown 2: B&W; Tick: Dark background → adjust to DAPI dense areas → Apply.
Click “OK” in dialogue “Convert Stack to Binary.”
Edit → Selection → Create selection.
Select window “ROI manager” → Add [t] → Rename… to “HCmask.”
Close window “Threshold.”
Heterochromatin subtraction
Select window “nucleus_mask.”
Select window “ROI manager” and select ROI “HCmask.”
Edit → Clear.
Edit → Selection → Create selection.
Select window “ROI manager” → Add [t] → Rename… to “Nucleoplasm_mask.”
Close windows “nucleus_mask” and “forHCmask.”
Measure intensities in the respective regions of interest (ROIs)
Select window “Master.”
Select window “ROI manager.”
Select desired ROI.
Press “Ctrl + M” to measure the first channel.
Use the channel slider to move to the next channel and press “Ctrl + M” to measure again.
Repeat until all ROIs are measured in all channels and remember the series.
Save the measurements as .csv files (Ctrl + S).
The pipeline for image analysis is illustrated in Figure 3.
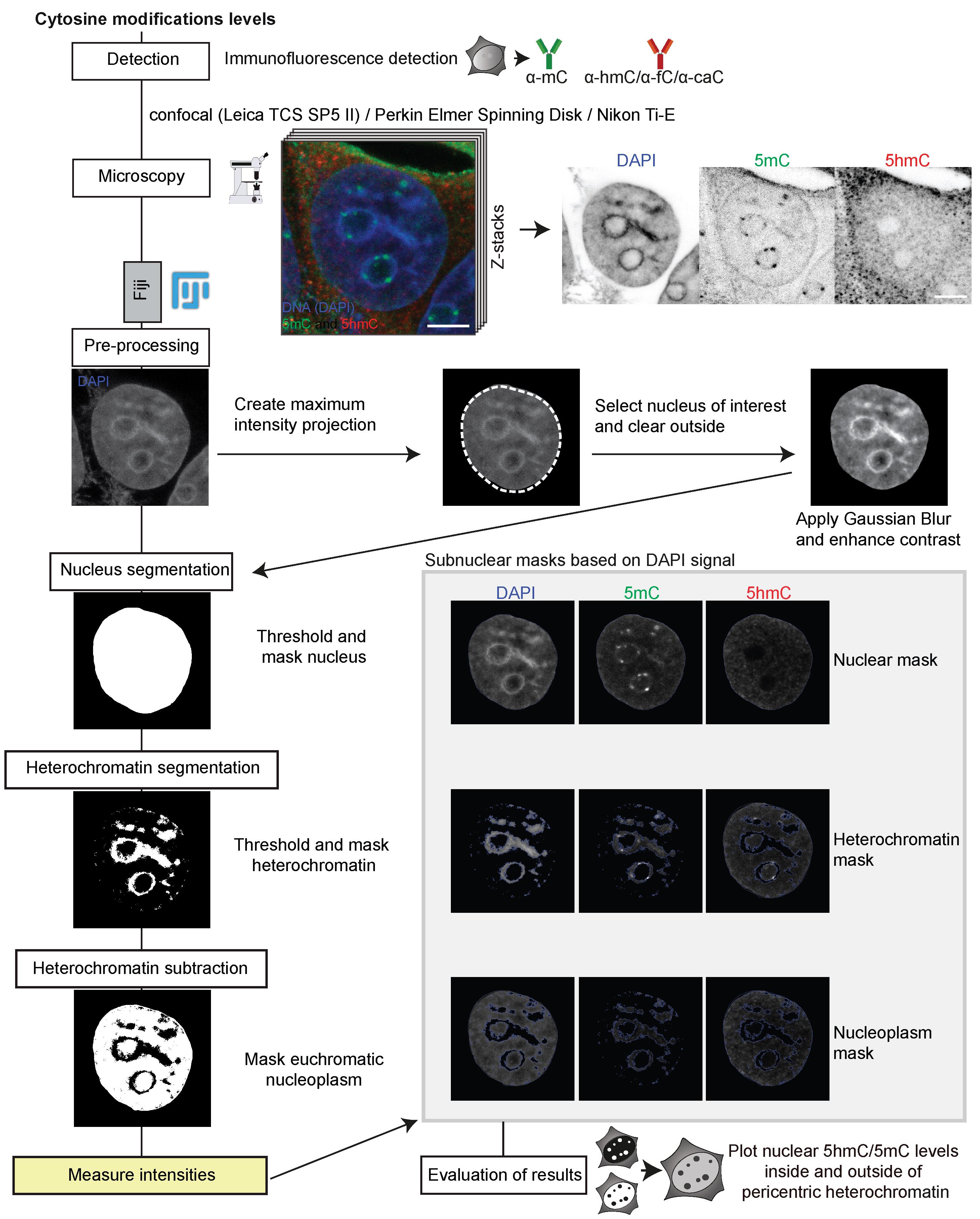
Figure 3. Image processing and in situ quantitative analysis pipeline for cytosine modifications levels. After immunofluorescence detection of DNA base modifications and image acquisition by confocal microscopy, the preprocessing of the multicolor Z-stack is performed in Fiji. Afterwards, the nucleus, heterochromatin, and the euchromatic nucleoplasm are segmented into regions of interest (ROIs) based on their respective DAPI signals, and binary masks are generated. Signals of all channels in the respective ROIs are then measured based on the binary masks and results exported as .csv files.
Image analysis (semi-automated procedure)
In addition to the step-by-step analysis procedure described in section E, in this section we describe a semi-automated analysis pipeline using a macro in Fiji. The semi-automated procedure speeds up the analysis process along with a higher standardization for different samples.
Start the semi-automated image analysis pipeline macro by opening it in Fiji (see File S1).
Open a Multicolor Z-stack image of interest.
Click “Run” in the Macro UI.
You will be prompted to select the DAPI channel. Use the channel slider to move to the DAPI channel and click “OK” in the dialogue box.
You will be prompted to choose a nucleus of interest. After clicking on the nucleus of interest, confirm by clicking “OK” in the dialogue box.
You will be prompted to set a threshold for the heterochromatin regions. Use the sliders in the threshold menu to adjust and confirm by clicking “OK” in the dialogue box.
Based on the DAPI signal, the macro generates ROIs for (I) the nucleus, (II) the heterochromatin, (III) the euchromatic nucleoplasm. Based on these ROIs, the macro quantifies the fluorescence intensities in channel 1 for ROI I, II, and III, in channel 2 for ROI I, II, and III, and in channel 3 for ROI I, II, and III.
You are prompted to save the measurements as a .csv file. Save with “Ctrl + S” and confirm by clicking “OK” in the dialogue box.
All windows will be closed, and you can continue with the next image, starting from step 2.
Data analysis
After the image analysis, we obtain .csv files as output, with a total of nine rows corresponding to measurements in three channels (DAPI, 5mC, and 5hmC) and for the three different masks (nucleus, heterochromatin, nucleoplasm). The output includes values for “Area,” “Mean,” “StdDev,” “IntDen,” etc., which depend on the selected settings of the measurement in Fiji. For our analysis, described in Arroyo et al. (2022), the respective mean cytosine modification levels were normalized against the respective normalized 5mC levels to compensate for the epigenetic heterogeneity in cancer cells (Guo et al., 2019). Normalized mean intensities for at least 20 cells were plotted with RStudio (Version 1.1.447). For the statistics, an independent two-group comparison was made for all groups, using a Wilcoxon–Mann–Whitney test. Boxplots showing normalized mean intensity of cytosine modifications in heterochromatin regions are shown in Figure 4A and representative images in Figure 4B.
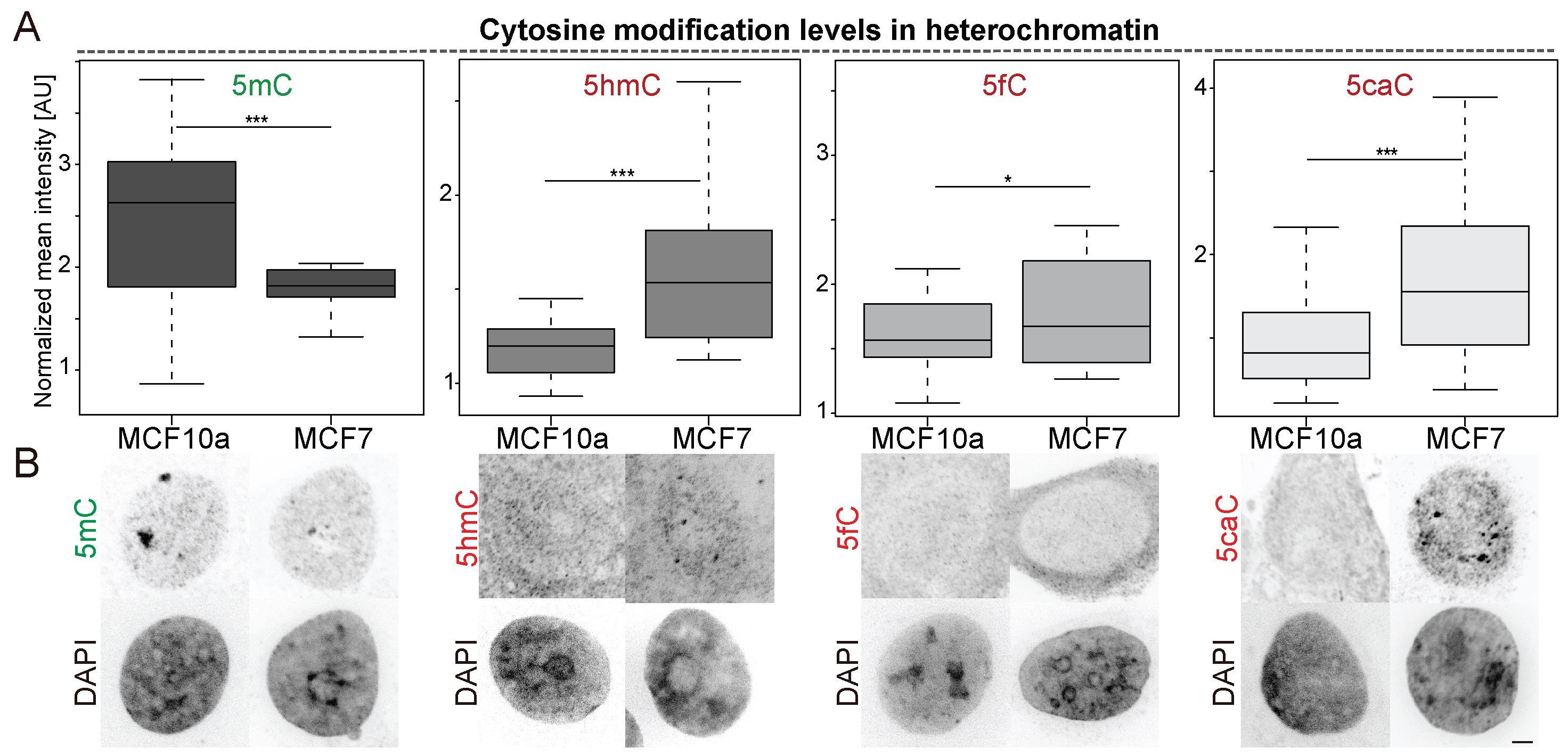
Figure 4. Cytosine modification levels in heterochromatic regions in MCF cell lines. (A) Boxplots showing normalized mean intensities of cytosine modifications in heterochromatin regions. n (5mC) = 28 (MCF10a) - 27 (MCF7), n (5hmC) = 25 (MCF10a) - 24 (MCF7), n (5fC) = 27 (MCF10a) - 19 (MCF7), n (5caC) = 18 (MCF10a) - 19 (MCF7) cells. For all boxplots, the box represents 50% of the data, starting in the first quartile (25%) and ending in the third (75%). The line inside represents the median. The whiskers represent the upper and lower quartile. Statistical significance was tested with a paired two-samples Wilcoxon test [n.s. (not significant) is given for p-values ≥ 0.05; one star (*) for p-values < 0.05 and ≥ 0.005; two stars (**) is given for values < 0.005 and ≥ 0.0005; three stars (***) is given for values < 0.0005]. (B) Representative confocal mid-Z sections of DAPI-stained nuclei of MCF10a and MCF7 cells showing levels of 5mC and 5hmC, or 5fC or 5caC, respectively. Confocal Z-stacks were analyzed with Fiji. Scale bar = 2 μm.
General notes and troubleshooting
Cells are cultured in a humidified atmosphere under 5% CO2 at 37 °C. Media are supplemented with pen/strep (100 IU/mL penicillin and 100 μg/mL streptomycin) and 2.5 mM L-glutamine. MCF10a non-tumorigenic mammary gland cells are cultured in DMEM/F12 supplemented with final concentrations of 5% horse serum, 20 ng/mL EGF, 0.5 mg/mL hydrocortisone, 100 ng/mL cholera toxin, and 10 mg/mL insulin. MCF7 breast cancer cells are cultured in DMEM containing 10% FBS.
Trypsinization of cells: Aspirate the medium and discard; wash cells with PBS/EDTA 0.02% (w/v), aspirate, and discard; add enough warmed 1× trypsin–EDTA solution to cover the cell monolayer and place the dish in a CO2 incubator at 37 °C for 2–5 min; once detached, resuspend the cells in growth medium containing serum; gently pipette the cells up and down to disrupt cell clumps and transfer an appropriate amount of cell suspension to a new dish and top up with normal medium.
No incubation time is necessary. PBS-T is added and removed from the dish.
As a wet chamber, a plastic box of different dimensions can be used. For example, a box of 1.5 cm × 12 cm × 12 cm (depth × width × height) covered with aluminum foil to protect the sample from light exposure. Soak one extra thick gel-blotting paper (10 cm × 10 cm) with ddH2O inside the box and remove excess water. Then, cut a 10 cm piece of parafilm and place it on top of the soaked gel-blotting paper.
For washing steps in droplets, lift the coverslips with tweezers, carefully remove the droplet of PBS-T, and add a new one, using a pipette.
Image acquisition settings for confocal microscope (Perkin Elmer Spinning Disk/Nikon Ti-E):
Channel 1: TexasRed (Set “Binning” to “x1.” Set “Exposure” to “350 ms.” Set “UltraVIEW 561 nm Laser Power” to “15.0%.”
Channel 2: GFP (Set “Binning” to “x1.” Set “Exposure” to “350 ms.” Set “UltraVIEW 488 nm Laser Power” to “15.0%.”
Channel 3: DAPI (Set “Binning” to “x1.” Set “Exposure” to “350 ms.” Set “UltraVIEW 405 nm Laser Power” to “16.5%.”
We recommend measuring the following properties: Area, Standard deviation, Min and max gray values, integrated density, and Mean gray value. However, depending on the image analysis and the user, additional properties can be required, for example Shape descriptors.
Acknowledgments
This protocol was used in Arroyo et al. (2022). The research was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) grants CA 198/10-1 Project number 326470517 and CA 198/16-1 Project number 425470807 to M.C.C.
Competing interests
The authors declare no competing interests.
References
- Arroyo, M., Hastert, F. D., Zhadan, A., Schelter, F., Zimbelmann, S., Rausch, C., Ludwig, A. K., Carell, T. and Cardoso, M. C. (2022). Isoform-specific and ubiquitination dependent recruitment of Tet1 to replicating heterochromatin modulates methylcytosine oxidation. Nat Commun 13(1): 5173.
- Bauer, C., Göbel, K., Nagaraj, N., Colantuoni, C., Wang, M., Müller, U., Kremmer, E., Rottach, A. and Leonhardt, H. (2015). Phosphorylation of TET proteins is regulated via O-GlcNAcylation by the O-linked N-acetylglucosamine transferase (OGT). J Biol Chem 290(8): 4801-4812.
- Bird, A. P. (1986). CpG-rich islands and the function of DNA methylation. Nature 321(6067): 209-213.
- Bizhanova, A. and Kaufman, P. D. (2021). Close to the edge: Heterochromatin at the nucleolar and nuclear peripheries. Biochim Biophys Acta Gene Regul Mech 1864(1): 194666.
- Cardoso, M. C. and Leonhardt, H. (1996). Immunofluorescence Techniques in Cell Cycle Studies. In Cell Cycle — Materials and Methods (pp. 15-28). Springer Berlin Heidelberg.
- Guo, M., Peng, Y., Gao, A., Du, C. and Herman, J. G. (2019). Epigenetic heterogeneity in cancer. Biomark Res 7: 23.
- Lande-Diner, L., Zhang, J., Ben-Porath, I., Amariglio, N., Keshet, I., Hecht, M., Azuara, V., Fisher, A. G., Rechavi, G. and Cedar, H. (2007). Role of DNA methylation in stable gene repression. J Biol Chem 282(16): 12194-12200.
- Ludwig, A. K., Zhang, P. and Cardoso, M. C. (2016). Modifiers and readers of DNA modifications and their impact on genome structure, expression, and stability in disease. Front Genet 7: 115.
- Meneveri, R., Agresti, A., Marozzi, A., Saccone, S., Rocchi, M., Archidiacono, N., Corneo, G., Della Valle, G. and Ginelli, E. (1993). Molecular organization and chromosomal location of human GC-rich heterochromatic blocks. Gene 123(2): 227-234.
- Pradhan, S. K. and Cardoso, M. C. (2023). Analysis of cell cycle and DNA compaction dependent subnuclear distribution of histone marks. Methods Mol Biol 2589: 225-239.
- Santiago, M., Antunes, C., Guedes, M., Sousa, N. and Marques, C. J. (2014). TET enzymes and DNA hydroxymethylation in neural development and function - how critical are they? Genomics 104(5): 334-340.
- Schmid, V. J., Cremer, M. and Cremer, T. (2017). Quantitative analyses of the 3D nuclear landscape recorded with super-resolved fluorescence microscopy. Methods 123: 33-46.
- Smith, Z. D. and Meissner, A. (2013). DNA methylation: roles in mammalian development. Nat Rev Genet 14(3): 204-220.
- Soule, H. D., Maloney, T. M., Wolman, S. R., Peterson, W. D., Brenz, R., McGrath, C. M., Russo, J., Pauley, R. J., Jones, R. F. and Brooks, S. C. (1990). Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res 50(18): 6075-6086.
- Soule, H. D., Vazguez, J., Long, A., Albert, S. and Brennan, M. (1973). A human cell line from a pleural effusion derived from a breast carcinoma. J Natl Cancer Inst 51(5): 1409-1416.
- Vidal, E., Sayols, S., Moran, S., Guillaumet-Adkins, A., Schroeder, M. P., Royo, R., Orozco, M., Gut, M., Gut, I., Lopez-Bigas, N., et al. (2017). A DNA methylation map of human cancer at single base-pair resolution. Oncogene 36(40): 5648-5657.
- Weichmann, F., Hett, R., Schepers, A., Ito-Kureha, T., Flatley, A., Slama, K., Hastert, F. D., Angstman, N. B., Cardoso, M. C., König, J., et al. (2020). Validation strategies for antibodies targeting modified ribonucleotides. RNA 26(10): 1489-1506.
Supplementary information
The following supporting information can be downloaded at here:
- File S1.
Article Information
Copyright
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Arroyo, M., Cardoso, M. C. and Hastert, F. D. (2023). In situ Quantification of Cytosine Modification Levels in Heterochromatic Domains of Cultured Mammalian Cells. Bio-protocol 13(14): e4716. DOI: 10.21769/BioProtoc.4716.
Category
Molecular Biology > DNA > DNA modification
Cell Biology > Cell imaging > Confocal microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.