- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Rapid Identification of Pathogens in Severe Pneumonia by Species-specific Bacterial Detector (SSBD)
(*contributed equally to this work) Published: Vol 13, Iss 10, May 20, 2023 DOI: 10.21769/BioProtoc.4681 Views: 1908
Reviewed by: Alka MehraLionel Schiavolin
Original research article
The authors used this protocol in:

Oct 2022
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Fast and accurate detection of pathogenic bacterial infection in patients with severe pneumonia is significant to its treatment. The traditional culture method currently used by most medical institutions relies on a time-consuming culture process (over two days) that is unable to meet clinical needs. Rapid, accurate, and convenient species-specific bacterial detector (SSBD) has been developed to provide timely information on pathogenic bacteria. The SSBD was designed based on the fact that Cas12a indiscriminately cleaves any DNA following the binding of the crRNA-Cas12a complex to the target DNA molecule. SSBD involves two processes, starting with PCR of the target DNA using primers specific for the pathogen, followed by detection of the existence of pathogen target DNA in the PCR product using the corresponding crRNA and Cas12a protein. Compared to the culture test, the SSBD can obtain accurate pathogenic information in only a few hours, dramatically shortening the detection time and allowing more patients to benefit from timely clinical treatment.
Keywords: Severe pneumoniaBackground
Severe pneumonia is a progressive inflammation of the lungs caused by pathogenic bacterial infection, leading to systemic infection if not treated promptly. The types of pathogens that cause severe pneumonia are complex and diverse, and antibacterial drugs used for different pathogen infections have great differences (De Pascale et al., 2012; Hansen et al., 2016). Therefore, the detection of early pathogenesis is not only the basis for precise diagnosis, but also necessary for accurate treatment and rational drug selection. However, the traditional culture method used by most medical institutions often relies on the process of pathogen culture, which takes several days (Lazcka et al., 2007). Inability to provide timely information about pathogen infection affects the clinical need to treat critically infected patients. Therefore, it is critical to accurately and rapidly identify the pathogens that cause severe pneumonia to enhance the diagnosis and treatment of such severe diseases to the greatest degree feasible.
The CRISPR/Cas system, widely used in the gene editing field, performs endonuclease functions on target nucleic acid molecules by Cas proteins and the guide RNA complementary to the target sequence. Recently, some Cas proteins (e.g., Cas12a and Cas13a) have been found to exhibit nuclease activity that indiscriminately cleaves arbitrary nucleic acids following specific binding to target molecules (Gootenberg et al., 2017; Chen et al., 2018; Li et al., 2018). Thus, by introducing single-stranded reporter nucleic acids that can generate fluorescent signals after cleavage, the detection of target nucleic acid molecules can be enabled by detecting fluorescent signals. Based on this principle, we have developed an accurate and rapid species-specific bacterial detector (SSBD) system (Wang et al., 2022). In previous studies, we have proven through clinical trials that SSBD can accurately detect ten of the most common clinical pathogens, namely Acinetobacter baumannii, Escherichia coli, Klebsiella pneumoniae, Staphylococcus aureus, Pseudomonas aeruginosa, Staphylococcus epidermidis, Enterococcus faecalis, Enterococcus faecium, Stenotrophomonas maltophilia, and Staphylococcus capitis.
The detection target for SSBD needs to be selected for each species. In summary, we identify the detection target through two processes. The first process utilizes bioinformatics to align the 1,791 high-quality genomes of 232 microorganism species (each species includes multiple isolated strains) and screen species-specific DNA fragments of each species. In the second process, the species-specific DNA fragments are manually selected and experimentally validated to identify the optimal DNA targets. For the first bioinformatics analysis process (Figure 1), we first performed genome sequence alignment of multiple strains belonging to the same species to filter the common DNA fragments of the species. Then, these common DNA fragments were aligned with other species' genomes to remove similar DNA fragments present in other species, with the remaining DNA fragments being the species-specific DNA fragments. The screened species-specific DNA fragments of each pathogen had the following features: (1) intra-species conservation (these DNA fragments are present in multiple isolated strains belonging to the same species), and (2) inter-species specificity (these DNA fragments are not present in other species). For each species, dozens to thousands of specific DNA fragments were screened, but not all of these fragments were ideal targets for Cas12a detection. Therefore, the bioinformatics data were then artificially filtered based on the following three principles: DNA length preferably longer than 200 bp to facilitate primer design, absence of repetitive sequences, and the existence of PAM sequences (5′-TTTN-3′) of Cas12a. Then, for 10 common clinical pathogens, several species-specific DNA fragments were randomly selected as the candidate target for detection. Primers and crRNAs were designed according to the selected candidate target DNA fragments sequence of each pathogen. Finally, the specificity and reliability of these primers and crRNAs were verified by crossover validation experiments, and the best-performing primer and crRNA for each species were selected based on the experimental results.
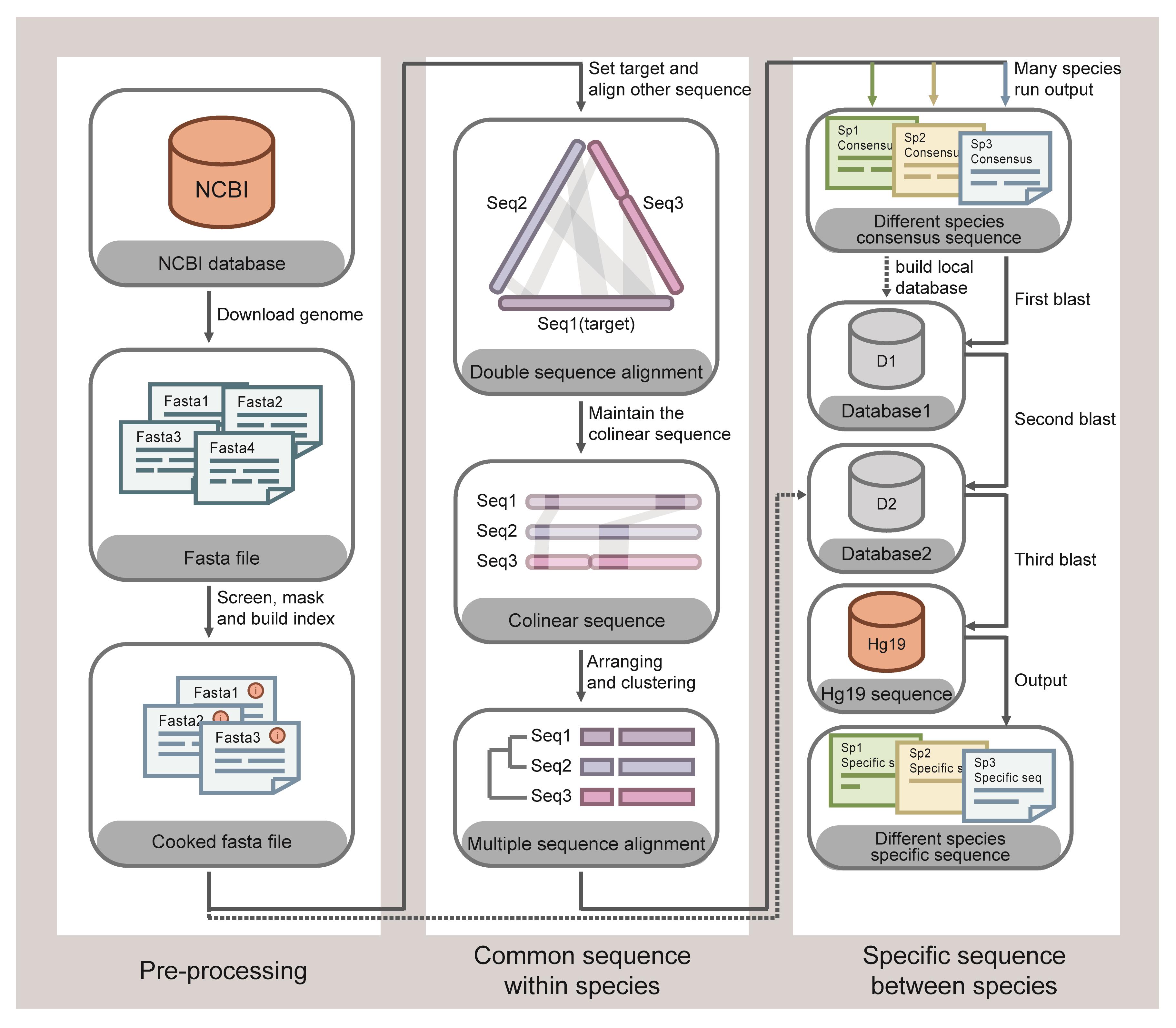
Figure 1. Schematic diagram of screening species-specific DNA fragments by bioinformatics. Reference from our published articles (Wang et al., 2022).
The advantage of the SSBD system is mainly reflected on detection accuracy and speed. The accuracy of the SSBD is reflected by the fact that the target of the detection is the species-specific DNA fragments (intra-species conserved and inter-species specificity) of each pathogen (DNA targets of the 10 pathogens in this protocol are shown in Table 5), as screened by the bioinformatics analysis in our previous study (Wang et al., 2022). The corresponding PCR amplification primers and crRNAs guiding Cas12a for targeted cleavage were designed according to the species-specific DNA fragments. SSBD can be completed in just a few hours, as it does not include a culture process and only involves three steps of sample DNA extraction, PCR amplification, and Cas12a testing. Meanwhile, the method is easy to be replicated, as DNA extraction and PCR amplification are the fundamental experimental assays in many molecular testing departments. Additionally, Cas12a protein and crRNA can be ordered from biotech companies.
In conclusion, SSBD has great advantages as a new rapid detection method for pathogens, showing potential to even replace traditional culture methods, making it well worth to be extended and applied in the clinical diagnosis of patients with severe pneumonia. Meanwhile, the SSBD is based on species-specific DNA targets of pathogenic microbes, which means it can be expanded to any species other than bacteria.
Materials and Reagents
384-well microplate (Corning, catalog number: 3701)
1.5 mL nuclease-free tube (Eppendorf, catalog number: 0030125150)
0.2 mL PCR tube (Eppendorf, catalog number: 0030124332)
10 μL tips (SANFICO, catalog number: TFR810)
10 μL extended tips (SANFICO, catalog number: TFR810-L)
200 μL tips (SANFICO, catalog number: TFR8200)
1,000 μL tips (SANFICO, catalog number: TFR81000)
Quick-DNA/RNA Pathogen Miniprep kit (ZYMO RESEARCH, catalog number: R1042)
TIANSeq HiFi Amplification Mix (TIANGEN, catalog number: NG219)
NEBufferTM 3.1 (New England Biolabs, catalog number: B7203)
Nuclease-free water (Thermo Fisher, catalog number: 10977015)
Cas12a protein
Note: In our experiments, the Cas12a protein expression and purification was performed as described (Li et al., 2018). We also recommend purchasing Cas12a from a biological company (e.g., New England Biolabs).
Primers for PCR amplification (primer sequences are shown in Table 1)
Note: The sequence of primers was published in our previous article in Appendix 1, Table 1 (Wang et al., 2022). The primers can be ordered as PAGE-purified DNA oligonucleotide from a DNA synthesis company (e.g., GenScript Biotech or Sangon Biotech).
Table 1. Primers sequences
Species Primers sequences (5′ > 3′) A. baumannii forward primer CACAGCGTTTAACCCATGCC reverse primer TATCGCCACCTGCACAGAAG E. coli forward primer GTTCCTGACTATCTGGCGGG reverse primer GCTTCCTGACTCCAGACACC K. pneumoniae forward primer CATGGGCATATCGACGGTCA reverse primer CCTGCAACATAGGCCAGTGA S. aureus forward primer AGGTGCAGTAGACGCATAGC reverse primer CATTCGCTGCGCCAATACAA P. aeruginosa forward primer TCTCTCTATCACGCCGGTCA reverse primer TCGCATCGAGGTATTCCAGC S. epidermidis forward primer CACGCATGGCACTAGGTACA reverse primer CGAAAAAGAGTTGTCCTTGTTGA S. capitis forward primer GGTTCAGTCATCCCCACGTT reverse primer CAGCTGCGACAACTGCTTAC E. faecalis forward primer CGGCAAGTTTGGAAGCAGAC reverse primer CAGCGCCTAGTCCTTGTGAT E. faecium forward primer ATCGGAAATCGGTGTGGCTT reverse primer TCAAATGCATCCCTGTGCCT S. maltophilia forward primer CGCCTCCCGTTTACAGATTA reverse primer TCGGCTCCACCACATACAC crRNA used in Cas12a reaction (crRNA sequences are shown in Table 2)
Note: The RNA can be ordered as a desalted RNA oligonucleotide or as PAGE-purified RNA oligonucleotide from an RNA synthesis company (e.g., GenScript Biotech or Sangon Biotech).
Table 2. crRNA sequences
Species crRNA sequences (5′ > 3′) A. baumannii UAAUUUCUACUAAGUGUAGAUGUAAAGAAGAAUUACUUGAA E. coli UAAUUUCUACUAAGUGUAGAUACGGUCGCUAUGAUGGCAAG K. pneumoniae UAAUUUCUACUAAGUGUAGAUACUGCGGUAAUCGCCAUCUU S. aureus UAAUUUCUACUAAGUGUAGAUACAACGUGAACUUGCUGAGG P. aeruginosa UAAUUUCUACUAAGUGUAGAUUUGUAAACACACAUGAGGAG S. epidermidis UAAUUUCUACUAAGUGUAGAUUAAUAUUCAUCAAUUAGAGA S. capitis UAAUUUCUACUAAGUGUAGAUAAUCACCUUAUUAAUCAAUA E. faecalis UAAUUUCUACUAAGUGUAGAUGCACCAAAUAACACAGCUGA E. faecium UAAUUUCUACUAAGUGUAGAUACUACCUGAACUUAUAUACA S. maltophilia UAAUUUCUACUAAGUGUAGAUGCAUGACACCUGCGACCAGG Note: The backbone sequence of crRNA is 5′-UAAUUUCUACUAAGUGUAGAU-3′, and the following sequence is used to recognize the target DNA.
ssDNA-reporter (synthesized by Sangon Biotech)
Note: Sequence of ssDNA-reporter: 5′-FAM-TTATT-BHQ1-3′. The feature of the ssDNA-reporter is that a fluorescent molecule that can generate a fluorescent signal (e.g., FAM) is coupled at one DNA end, and a quencher molecule (e.g., BHQ1) that can block the fluorescent molecule producing a fluorescent signal is coupled at the opposite DNA end.
Ethanol (Sangon Biotech, catalog number: A500737-0500)
Equipment
Centrifuge (Eppendorf, Centrifuge 5418 R)
Centrifuge for 0.2 mL PCR tube (Scilogex, catalog number: 914041419999)
PCR thermocycler (LongGene, T20D)
Fluorescence plate reader (Austria Tecan, Infinite M200 Pro)
NanoDrop One (Thermo Scientific, catalog number: ND-ONE-W)
Biological safety cabinet (Thermo Scientific, catalog number: 51028226)
Vortexer (Scilogex, catalog number: 821200049999)
1–10 μL pipettes (Eppendorf, catalog number: 3123000020)
2–20 μL pipettes (Eppendorf, catalog number: 3123000098)
20–200 μL pipettes (Eppendorf, catalog number: 3123000055)
100–1,000 μL pipettes (Eppendorf, catalog number: 3123000063)
Software
Primer design software, NCBI Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/)
CrRNA design software, CHOPCHOP (https://chopchop.cbu.uib.no/)
Software to control the fluorescence plate reader, Tecan i-control (Austria Tecan)
Software for data analysis, Microsoft Excel
Procedure
Detection targets design
Note: The primers and crRNAs used in this protocol for 10 pathogens (Table 1) have been experimentally and clinically validated with excellent effectiveness.
Select detection targets. Manual screening of species-specific DNA data from bioinformatics; the screening principles are:
To facilitate primer design, DNA length is preferably longer than 200 bp.
DNA fragment without repetitive sequences.
DNA fragment contains PAM sequences (5′-TTTN-3′) of Cas12a.
Design crRNA.
Find PAM sequences (5′-TTTN-3′) within the DNA target region.
Select the sequence 20 bp downstream of the PAM sequence as the target of crRNA. Replace the T-base in the 20 bp target sequence with a U-base. Place the target sequence downstream of the crRNA backbone sequence (5′-UAAUUUCUACUAAGUGUAGAU-3′).
Note: The base distribution of the target sequence should be balanced, with a GC content of 40%–80%. We also recommend using crRNA design software, e.g., CHOPCHOP (https://chopchop.cbu.uib.no/).
Use NCBI Primer-BLAST primer design software to select primers.
The input sequence is the upstream and downstream sequence of the crRNA target site.
Set the Primer parameters as default parameters.
Set Primer Pair Specificity Checking Parameters Database as Refseq representative genomes, and Organism as Homo sapiens (taxid: 9606).
Pick a primer pair predicted without non-specific amplification.
Experimental validation
For example, for the 10 sets of primers and crRNAs used in this protocol, each set of primers and crRNAs needs to ensure that no non-specific signals are detected for the remaining pathogens and human genomic DNA.
DNA extraction from bronchoalveolar lavage fluid (BALF) sample
Biosafety notes: DNA extraction procedure should be performed in a biological safety cabinet to ensure user safety. Wear biological protecting clothing, gloves, and masks during operation.
DNA extraction was performed referring to the Quick-DNA/RNA Pathogen Miniprep kit instructions. The procedure is as follows:
Add 800 μL of DNA/RNA shield to each 200 μL of BALF sample and mix well by using vortexer.
Centrifuge at 16,000× g for 1 min. Transfer up to 200 μL of the supernatant into a 1.5 mL nuclease-free tube.
Add 2 μL of proteinase K and mix well.
Add 400 μL of pathogen DNA/RNA buffer. Mix well and incubate at room temperature for 5 min.
Transfer the mixture into a Zymo-Spin IIC column in a collection tube and centrifuge at 16,000× g for 30 s. Discard the flowthrough.
Add 500 μL of pathogen DNA/RNA wash buffer to the Zymo-Spin IIC column and centrifuge at 16,000× g for 30 s. Discard the flowthrough.
Repeat the previous step.
Add 500 μL of ethanol (95%–100%) to the Zymo-SpinTM IIC column and centrifuge at 16,000× g for 1 min to ensure that the ethanol is removed. Discard the collection tube and transfer the Zymo-SpinTM IIC column into a new nuclease-free 1.5 mL tube.
Add 25–50 μL of nuclease-free water at 65 °C directly to the matrix of the column and centrifuge at 16,000× g for 1 min to elute the DNA.
Measure DNA concentration using a NanoDrop and adjust DNA concentration to 100 ng/μL.
Note: The DNA yield extracted from 200 μL of BALF is 8–43 μg. Depending on the patient's situation and operating procedure, typically 200 μL of BALF is sufficient for subsequent analysis. If the DNA yield is low, centrifuge the BALF sample at 12,000× g for 5 min, discard the supernatant, and retain 200 μL of bottom liquid.
PCR amplification enrichment of target DNA sequences to be tested
For one BALF sample DNA, prepare 10 PCR reactions using amplification primers, one for each of the 10 pathogenic bacteria.
Setting up the positive and negative controls: the templates for the positive and negative controls are the genomic DNA of the corresponding pathogen and nuclease-free water, respectively.
Prepare the PCR reactions in a 0.2 mL PCR tube.
Component 50 μL reaction 2× HiFi amplification mix 25 μL Forward and reverse primers (10 μM) 2 μL of each Template DNA (100 ng/μL) 1 μL Nuclease-free water 20 μL Mix well and pulse-spin in a microcentrifuge.
Place a 0.2 mL PCR tube in a PCR thermocycler.
PCR procedure:
98 °C 2 min 98 °C 10 s 38 cycles 52 °C 10 s 72 °C 15 s 72 °C 5 min 4 °C Forever After the PCR reaction, without the PCR purification step, directly use the PCR reaction solution as the detection substrate for the Cas12a reaction.
Detection of PCR products using Cas12a
Prepare the reactions in a 384-well microplate on ice:
Component 20 μL reaction ssDNA-reporter (10 μM) 1 μL PCR reaction solution 2 μL 10× NEBuffer 3.1 2 μL Cas12a (10 μM) 1 μL crRNA (10 μM) 1 μL Nuclease-free water 13 μL Note: The PCR reaction solution for each pathogen requires the use of the corresponding crRNA. Preparation of premixes is recommended to improve experimental efficiency.
With a pipette set at 15 μL and keeping the plate on ice, mix the reaction by pipetting the entire solution up and down 10 times.
Note: Pipetting 10 times can ensure sufficient mixing of the reaction solution. Avoid excessive bubbles in the reaction solution; any trace bubbles will clear after 30 min of incubation at 37 °C in the next step.
Place the plate in a fluorescence plate reader. Incubate at 37 °C for 30 min, then measure the fluorescence values of each reaction well (excitation wavelength: 485 nm; emission wavelength: 535 nm).
Data analysis
Note: The data analysis of this protocol is applied to the detection of pathogenic bacteria in patients with severe pneumonia in the ICU. For other studies, the analysis standards require adjustments accordingly.
For each sample, analyze the fluorescence values for each pathogen separately.
For each pathogen detected, the positive control (PC) is the genomic DNA of corresponding pathogen, and the negative control (NC) is nuclease-free water.
The fluorescence value of BALF sample (shown as F) is two-fold larger than the negative control, indicating that the SSBD method detected the corresponding pathogen.
In our previous article, the application of SSBD was for pathogen detection in ICU patients, so the degree of pathogenic infection is required to guide drug usage. The pathogen strength is determined to some extent by the fluorescence signal (Table 3). Set I (interval) as the point of distinction. I = (PC - 2NC)/3. When 2NC < F < 2NC+I, the pathogen strength level is defined as level I. When 2NC+I < F < 2NC+2I, the pathogen strength level is defined as level II. When F > 2NC+2I, the pathogen strength level is defined as level III.
Table 3. Determining the presence of bacteria by fluorescence signal
Fluorescence value Test results Bacterial DNA molarity F < 2NC Undetected N.A. 2NC < F < 2NC+I Detected level I 10-15 M–10-14 M 2NC+I < F < 2NC+2I Detected level II 10-14 M–10-13 M F > 2NC+2I Detected level III over 10-13 M NC: Valuenegative control; F: ValueSample; I = (Valuepositive control - 2 Valuenegative control)/3
Example results
Using the data of two samples from our previously published article, we calculated the values of 2NC, 2NC+I, and 2NC+2I according to the fluorescence values of PC and NC, respectively. The fluorescence values of the samples are shown in Table 4.
Table 4. Fluorescence values of two samples
Species Fluorescence value Detection result PC NC Sample 1 Sample 2 2NC 2NC+I 2NC+2I Sample 1 Sample 2 A. baumannii 9788 903 9300 9398 1806 4467 7127 Detected Level III Detected Level III E. coli 8435 821 657 630 1642 3906 6171 Undetected Undetected K. pneumoniae 8285 1791 1718 1730 3582 5150 6717 Undetected Undetected S. aureus 8316 1008 1018 1003 2016 4116 6216 Undetected Undetected P. aeruginosa 9323 1170 1164 1069 2340 4668 6995 Undetected Undetected S. epidermidis 9309 1237 1210 1183 2474 4752 7031 Undetected Undetected S. capitis 8832 1633 1949 1772 3266 5121 6977 Undetected Undetected E. faecalis 8241 614 512 505 1228 3566 5903 Undetected Undetected E. faecium 8432 1936 1692 1700 3872 5392 6912 Undetected Undetected S. maltophilia 8194 1620 1549 1579 3240 4891 6543 Undetected Undetected The fluorescence value of A. baumannii in these two samples was higher than 2NC+2I. Therefore, the presence of A. baumannii in these two samples was detected, with a level III bacterial strength. For the rest of the pathogens, the fluorescence values of the samples were lower than 2NC, which indicates that the rest of the pathogens were not detected.
Notes
Optimizing PCR reactions can significantly avoid the generation of non-specific signals. When designing primers, BLAST the primers and select primers with excellent specificity. The suitable PCR reaction program is confirmed by experiment. Moreover, it is basically impossible for crRNA recognition sequences to exist in the non-specific amplification products generated during PCR. Therefore, combining the crRNA-Cas12a reaction with the PCR reaction greatly ensures the specificity of the detection. It is noteworthy that if crRNA is synthesized by in vitro transcription with RNA polymerase, the DNA used as in vitro transcription template needs to be removed to avoid contaminating crRNA (since the DNA templates can bind to crRNA to activate Cas12a).
Features considered important while selecting target:
Target sequences are present in multiple strains of this species' genomes.
Target sequences are not found in other species' genomes.
Target without repetitive sequences.
Target including PAM sequences.
Table 5. Information of target DNA region used in the protocol
Species GenBank of genome Target sequences (5′ > 3′) Corresponding gene A. baumannii CP115629.1 CACAGCGTTTAACCCATGCCATTGGTAAAGCTAAAGCAATGGAAATGTGCCTTACCGCTCGTCAAATGGGTGCAGTAGAGGCAGAACAAAGTGGTTTGGTTGCACGTGTGTTTAGTAAAGAAGAATTACTTGAACAAACCTTGCAAGCCGCTGAAAAAATCGCAGCACGGTCTTTAACTGCAAACATGATGCTCAAAGAAACCATTAATCGAGCTTTTGAGGTGAATCTCACCGAAGGTTTACGTTTTGAACGCCGCATGTTCCACTCCATTTTTGCAACTGCGGACCAAAAAGAAGGCATGCAGGCATTTGTTGAAAAACGGCAGGCAAATTTTAAAAATCAGTAAGAGATCAATAAAATGAATACAGAAAATCACTTAATGATTGAGCGACAAGGTAAGCTTGGCGTGATCACCTTAGATCGTGTGACTCATCTCAATGCATTGTCGCTAGATATGATTGAAGGAATTGGCGCCCAATTGGAGTTGTGGCGAAATGATGCTGCTGTTCAGGCGATCTTAATCAAATCAAATAGCCCAAAAGCCTTCTGTGCAGGTGGCGATA enoyl-CoA hydratase/isomerase family protein E. coli CP115173.1 GTTCCTGACTATCTGGCGGGGAATGGTGTGGTTTATCAAACCAGTGATGTGAAGTATGTGATTGCCAACAACAACTTGTGGGCCAGCCCGTTGGATCAACAGTTGCGCAACACCCTGGTTGCCAACCTGAGTACGCAACTGCCCGGCTGGGTGGTTGCCTCCCAGCCTCTGGGAAGCGCCCAGGACACGCTCAATGTTACCGTAACGGAGTTTAACGGTCGCTATGATGGCAAGGTCATTGTCAGTGGTGAGTGGCTGTTGAACCACCAGGGACAACTGATCAAACGTCCGTTCCGTCTGGAAGGAGTGCAAACTCAGGATGGTTACGATGAGATGGTTAAAGTGCTGGCCGGTGTCTGGAGTCAGGAAGC membrane integrity-associated transporter subunit PqiC K. pneumoniae CP115714.1 CATGGGCATATCGACGGTCACCGATTCGCCCGGACGCAGGCGAACATCTTTTTCCGCCACCCGGTCCGCCTGCAGCGCCTGGCCATCGCCGGCAAACAGCGACGGGTAGTCAGTATTGTCGAACGCTTGCCGGTCCTTCAGCTGATAAATACGCACCACCGTCGCCAGGGAGGCGCCTTTGGCGTTGTTATTCACGCCTTCCCGGGCGCGAAGATCCAGATGCAGGGTTTTCACCTGGGGGTAAAAAATGGACTGGGTCACGGAAACGGCGCCGTCTTTCACGGTCTGCGTCAGGCCGCAGCCGGTTAAGACGGTAACCATGAGGAATGCCAGCAACCTGGCGGAGGGTTTAACTGCGGTAATCGCCATCTTCATCGCTCTCCCTGCGGTGGATATTTTCCCGGACGCGCTGATAGCGCCCCAGATAAATCGTGACTCTGTCGTCAGCCTGTTTCTGCGCATCCAGAGGACGCAGCACAGCGGTACGGCCGAGCTGGACGGCATGCTCCTGCTGGCAGCAAAGTTGCGCATCCGGCAGTAAATGGCGGGCGACGCAAAGCTGCAGCCGCACGTCAAGATGAGATCCGAGCCAGACGTGCAGAAGCGCCATCAGGTCGCTGAACAGTTCGCCGCCGGGCAGCCAGCCGCGAACCTCATCAGGCTTTTCCGTGGCCAGCTGCAGCAATACCTGACCGTTCACATCCGTGGCGTGGGTTCCCATCACTGGCCTATGTTGCAGG type VI secretion system baseplate subunit TssG; type VI secretion system lipoprotein TssJ S. aureus CP063802.1 AGGTGCAGTAGACGCATAGCATCATCATATTGAATAGTAAAAACAAATAAAACATAGTAACGTGATTCAGTCGATGTAACAGTCGATAATGAGTCACGTTTTTTTATAGAAAAATATAAGACATAAAAATGTCATAATTTATAGTCGACAAATATCATACTGTATAAACATTTATCATTTTCTCAAGTGCCTTTTACACGATGGAATGAACTTACTTTTTACGAAATTATGCGTATTTTATAAACAAATATCATTGATATAACGGTAAATGTAAGCGTTTACAACAGAAATAACTGCATGCTACGATATTTTTGTAAATTCACTGATTCAAGTATTTTAAGTCAATATGAGGAGGGATGTTATGAGCGATTCTGAGAAAGAAATTTTAAAAAGAATTAAAGATAATCCGTTTATTTCACAACGTGAACTTGCTGAGGCAATTGGATTATCTAGACCCAGCGTAGCAAACATTATTTCAGGATTAATACAAAAGGAATATGTTATGGGAAAGGCATATGTTTTAAATGAAGATTATCCTATTGTTTGTATTGGCGCAGCGAATG winged helix-turn-helix transcriptional regulator P. aeruginosa CP080405.1 TCAGTGACATGAAGCCCGTCCCCGCGGCCAGGCTGCGCAGTTGCGCCTACGAGATGGGTTTTTCTCTCTATCACGCCGGTCAATGCTTGCGCGTGGTTCTCTAGGTTCGACCGAATCAAGAGTGTGTTTTCAGCAAAATAATGATAGTTTGTTGTAAACACACATGAGGAGGTCGTCATGAGCGCTCTCATCAAGGAACGTCCCAGCGCCGATGCCGTCCTGGCCAAGGCCGTCCTGGCCGCGCGCGAGCAATTGGGGCTGACGCAGCTCGAACTGGCCGGCATCGTCGGCGTCGATCGCAGCGCCATCAGCCGCTGGAAGACCCAGGGCCTGCGGGTGGACAGCAAGACCGGCGAGCTGGCTCTGCTGCTGGTGCGAGTCTATCGCGCACTGTATGCCCTGTTCGGCGGGCAGCAGGAGGACATGCGCCACTTCCTGCGCACTCCCAACCATCACCTGGCGGGCGAGCCGCTGGCACTGATGGGACAGGTGCAGGGCCTGGTTCATGTGCTGGAATACCTCGATGCGA DUF2384 domain-containing protein; FAD-dependent oxidoreductase S. epidermidis CP106834.1 CACGCATGGCACTAGGTACAAATCCAGTATAAACAGCGTAGATTAATAATAGTAAAATAGTTACACCTTTAATGAGAACTAGAGGTATATTTAAACGTACTAACAGTTGATAAACGAGGACAATAATTGTACCAACATGTGTACCACTTATAGCTAGTAAATGGTAAATACCTATATCTTTGATGTTAGATTTGTAATATTCATCAATTAGAGAGGTATCACCTGTTATCAACGCTAATATTCTTTCAGGATGCGTGACCCCAGATTTATGAATTATTTTGGTGATATAATTTTGATGATGATATATAGGAGTGAAAATATTGTTTTCTTGGCAAGACTTAAATTGAACTGTTGAAATATTCAACAAGGACAACTCTTTTTCG DNA internalization-related competence protein ComEC/Rec2 S. capitis AP014956.1 GGTTCAGTCATCCCCACGTTTTGATCTTCAATATTCATGTTATAATTTTCTCCTTTTTAAAAGGTTTCATTTACCATTGTTTACTACAGTAAAGGTTGTTTTAAACATATTCGTTTTAATATTTTTAGAGTGAAGATTCTAAAACGTGTGATAAAGGTGGAATATACACATAAAAATTTCCGATATAGAAAAATAAAAAGCTTTCAATCACCTTATTAATCAATAAAAAGTGATTAAAAGCTTTAAATTAAAAGAGGATAAACATCAATTCATATCTCAAACGCTTCTAACATTTTTGAGTTAGATGACTGAATTTGAACTGTGTTTATATCAAGCTTTTGTCTATTCTAGCCATCTTAGAAAACTATATAGAATGAATTCTTATTTAAGAAATCAATGCAGTATTTCTTTAAACTTCTGTTACTTCGACTTCGATGTGTTCGATGCGATGATATGCACCATGATCAACAGGTCCTACAAATTCATTCATTTTCCAACCGTTTTTGATTGCAGATGCTACGAAAGCTTTAGCAGCGATGACTGCTTCTCTTGGAGATTTGCCGTTAGCTAGGTAAGCAGTTGTCGCAGCTG putative pyridoxine kinase; FAD binding superfamily protein E. faecalis CP046113.1 CGGCAAGTTTGGAAGCAGACAAAGGTATTTTTGCAGTATTGAATGTCATCATTGGGATTAATGGTTTTGTTTGGAATGGTACATTGGCTATCGCTGGTTTGATTTTCGCTTTTTCTTGGGGCTATAACTTAGCGAAAGCATACAATGTGAATGAGCTAGCTGGTGGGATTGTCAGTTTAGCTACCTTAATTTCAGGTGTTGCTTTTGCACCAAATAACACAGCTGAATTAGCTGTAAAAGTGCCAGAAAAGATTGCCAACGCAATTAATGGGGCAGAAATTGGTGCGTCAATTGCTAATAATCAATTGACTGTCAATCCATGGGGTTGGTTAAATCTCAACCATCTAAATGGTAACGCTTTCTTTACGGTGATGATTATGGGCGCTTTATCAACCATTATTTTTTGTAAATTAATGCAAGCGGATTTAACAATAAAAATGCCTGATTCTGTACCACCAGCAGTTTCAAAAGCGTTTGCGGCAATTTTACCAGCAACAATCGCATTGTATGTCGTAGCAATTATTAACTTTACGGTGTCAAAACTTTCAGGCGGTCAATTATTAATTGACTTAATTCAAAAATATATCGCGGAACCGTTCCTTGGGTTATCACAAGGACTAGGCGCT PTS sugar transporter subunit IIC E. faecium CP112862.1 ATCGGAAATCGGTGTGGCTTATGCGAAAGGCATCCCAGTGATCGGCTTATATACTGATACTCGACAGCAAGGCGGAACCCACCCGAAAAAAATCGCTGCACTACAAGAAACAGCTGAAAATCAATTTCACTACCTGAACTTATATACAATAGGTTTGATCAAATTGAACGGAAAAGTGGTTTCTTCAGAAATTGAGTTGCTTTCAGAAGTAAAAAGATTTTTAGATGGAGGGACTTTCAGTGATTAAAGAAATCAAAAAGGTGCAAATAGCTTTACTTGCTTTTGGTGTATTCGTTGTCTGCTATAATTTTTATGAATTTATCACACAAAAATATTCGACTTCTCAAGGAATCACATTTATCGTGGAATCTTTATTAGGGATTGCATTGATTTTCATGCCACAAGTCATTTTGAAAGTTTTCAAACTTAAAATACCAGCAGCAATCGTTTTGTTTTACTGGTTTTTCTTGTTCATCTCTGTCTTTTTAGGCACAGGGATGCATTTGA nucleoside 2-deoxyribosyltransferase; hypothetical protein S. maltophilia CP040440.1 CCCGCGAGAAGCTGGATGCGCTGGAAACCGCCGTGCGCGAGCTGGAAGGGCGCGGCGCCGCAGAATGAAAAAAGGAGGCGCACCGCGCCTCCCGTTTACAGATTATTTCCCGGATGGGGCCTTGCAGCAAGGCCCACCGGGCGCCCTAGACTCGCCGGCCTGTCGATCCGGCAGCACAGAGAGGGGCAGTGCCGATGGCGCAGGACACCGAATGGACACCCGTTTCGCATGACACCTGCGACCAGGTCGCCGGGCCGTTCTATCACGGCACCCGCGCTGACCTGGCGGTGGGCGAGCTGCTGAGCGCGGGCTTCCGCTCCAACTATCGCGACAGCGTGGTGATGAACCACATCTACTTCACCACGATTGCCAAGGGCGCCGGGTTGGCTGCGGAGATGGCCCGGGGTGAAGGGCGACCGCGCGTGTATGTGGTGGAGCCGA accessory factor UbiK family protein; NAD(+)--rifampin ADP-ribosyltransferase
Acknowledgments
This protocol was adapted from our previously published work (Wang et al., 2022). This work was supported by National Natural Science Foundation of China (81927808).
Competing interests
The authors declare no competing interests.
Ethics
This study was registered in English at https://clinicaltrials.gov/ (NCT04178382) in November 2019.
References
- Chen, J. S., Ma, E., Harrington, L. B., Da Costa, M., Tian, X., Palefsky, J. M. and Doudna, J. A. (2018). CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360(6387): 436-439.
- De Pascale, G., Bello, G., Tumbarello, M. and Antonelli, M. (2012). Severe pneumonia in intensive care: cause, diagnosis, treatment and management: a review of the literature. Curr Opin Pulm Med 18(3): 213-221.
- Gootenberg, J. S., Abudayyeh, O. O., Lee, J. W., Essletzbichler, P., Dy, A. J., Joung, J., Verdine, V., Donghia, N., Daringer, N. M., Freije, C. A., et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356(6336): 438-442.
- Hansen, V., Oren, E., Dennis, L. K. and Brown, H. E. (2016). Infectious Disease Mortality Trends in the United States, 1980-2014. JAMA 316(20): 2149-2151.
- Lazcka, O., Del Campo, F. J. and Munoz, F. X. (2007). Pathogen detection: a perspective of traditional methods and biosensors. Biosens Bioelectron 22(7): 1205-1217.
- Li, S.-Y., Cheng, Q.-X., Liu, J.-K., Nie, X.-Q., Zhao, G.-P. and Wang, J. (2018). CRISPR-Cas12a has both cis-and trans-cleavage activities on single-stranded DNA. Cell Res 28(4): 491-493.
- Wang, Y., Liang, X., Jiang, Y., Dong, D., Zhang, C., Song, T., Chen, M., You, Y., Liu, H., Ge, M., et al. (2022). Novel fast pathogen diagnosis method for severe pneumonia patients in the intensive care unit: randomized clinical trial. eLife 11: e79014.
Article Information
Copyright
© 2023 The Author(s); This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Zhang, C., Liang, X., Qin, Y., Yu, W. and Chen, Q. (2023). Rapid Identification of Pathogens in Severe Pneumonia by Species-specific Bacterial Detector (SSBD). Bio-protocol 13(10): e4681. DOI: 10.21769/BioProtoc.4681.
- Wang, Y., Liang, X., Jiang, Y., Dong, D., Zhang, C., Song, T., Chen, M., You, Y., Liu, H., Ge, M., et al. (2022). Novel fast pathogen diagnosis method for severe pneumonia patients in the intensive care unit: randomized clinical trial. eLife 11: e79014.
Category
Microbiology > Pathogen detection > PCR
Molecular Biology > RNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.