- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CRISPR/Cas9-based Engineering of Immunoglobulin Loci in Hybridoma Cells
(*contributed equally to this work) Published: Vol 13, Iss 4, Feb 20, 2023 DOI: 10.21769/BioProtoc.4613 Views: 3414
Reviewed by: Luis Alberto Sánchez VargasDay-Yu ChaoAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Development of the hybridoma technology by Köhler and Milstein (1975) has revolutionized the immunological field by enabling routine use of monoclonal antibodies (mAbs) in research and development efforts, resulting in their successful application in the clinic today. While recombinant good manufacturing practices production technologies are required to produce clinical grade mAbs, academic laboratories and biotechnology companies still rely on the original hybridoma lines to stably and effortlessly produce high antibody yields at a modest price. In our own work, we were confronted with a major issue when using hybridoma-derived mAbs: there was no control over the antibody format that was produced, a flexibility that recombinant production does allow. We set out to remove this hurdle by genetically engineering antibodies directly in the immunoglobulin (Ig) locus of hybridoma cells. We used clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) and homology-directed repair (HDR) to modify antibody’s format [mAb or antigen-binding fragment (Fab’)] and isotype. This protocol describes a straightforward approach, with little hands-on time, leading to stable cell lines secreting high levels of engineered antibodies. Parental hybridoma cells are maintained in culture, transfected with a guide RNA (gRNA) targeting the site of interest in the Ig locus and an HDR template to knock in the desired insert and an antibiotic resistance gene. By applying antibiotic pressure, resistant clones are expanded and characterized at the genetic and protein level for their ability to produce modified mAbs instead of the parental protein. Finally, the modified antibody is characterized in functional assays. To demonstrate the versatility of our strategy, we illustrate this protocol with examples where we have (i) exchanged the constant heavy region of the antibody, creating chimeric mAb of a novel isotype, (ii) truncated the antibody to create an antigenic peptide-fused Fab’ fragment to produce a dendritic cell–targeted vaccine, and (iii) modified both the constant heavy (CH)1 domain of the heavy chain (HC) and the constant kappa (Cκ) light chain (LC) to introduce site-selective modification tags for further derivatization of the purified protein. Only standard laboratory equipment is required, which facilitates its application across various labs. We hope that this protocol will further disseminate our technology and help other researchers.
Graphical abstract

Background
Production of antibodies using transient recombinant systems is laborious and confronted with reproducibility issues due to variable transfection efficacy and requires knowledge of the sequence of the variable domain of the antibodies. While this is routine for specialized companies and laboratories equipped with dedicated biofactories, typical academic laboratories are usually not equipped with these infrastructures. For most laboratories, hybridoma represent the cheapest and least labor-intensive way of producing monoclonal antibodies (mAbs).
As immunized animals, from which the original cell line was generated, underwent multiple rounds of immunization and class-switch recombination, there is minor diversity among the isotypes, which are almost exclusively immunoglobulin (Ig) G subtypes. While the isotype might not always be relevant, it is becoming increasingly evident that it can be crucial for the efficacy of therapies (Waldor et al., 1987; Beers et al., 2016; Evers et al., 2021); isotype engineering could strongly increase the efficacy of mAbs used in cancer treatment, for instance (Sharma et al., 2019). Antigen-binding fragments (Fab’) are valuable tools in research due to their small size, but require post-isolation modification of the mAb using papain (Zhao et al., 2009). Lastly, chemical modification of mAbs is usually achieved by random labeling of lysine residues (Nanda and Lorsch, 2014), which is not site-specific and leads to undesirable heterogeneous products, or by engineering of the sequence to introduce unpaired cysteine residues (Adhikari et al., 2020), although novel methods are being developed (Yamada et al., 2019).
To collectively address these issues, we developed a technique that reproducibly allows the modification of the Ig constant locus in hybridoma using clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9). This nowadays routine method of gene editing enables precise targeting of the constant regions of the Ig loci regardless of the variable region and enables the insertion of virtually any sequence to produce an antibody with any switched or mutant isotype, format, or tag. The low diversity in isotypes among hybridoma is, in this case, an advantage, as homology-directed repair (HDR) templates can be reused easily on many cell lines. While we focused on editing the constant heavy chain (CH)1, hinge (H), and light chain (LC) regions, feasibility of CH3 targeting was demonstrated by others (Khoshnejad et al., 2018). Collectively, this illustrates the versatility of the strategy.
We expect that this protocol can be applied to benefit any field, as mAbs are a backbone of biological research, and hope to see peers successfully applying this method for applications we would not be able to envision.
Materials and Reagents
875 cm2 5-layer culture flask (Corning, Falcon®, catalog number: 353144)
75 cm2 culture flask (Greiner Bio-one, Cellstar, catalog number: 658170)
25 cm2 culture flask (Greiner-Bio one, Cellstar, catalog number: 690175)
10 cm Petri dish, culture-treated (Greiner Bio-one, Cellstar, catalog number: 664161)
6-well plates (Corning, Costar, catalog number: 3516)
24-well plates (Corning, Costar, catalog number: 3524)
96-well plates (Corning, Costar, catalog number: 3799)
50 mL Falcon (Thermo Fisher, BrandTM 114821, catalog number: 10420602)
15 mL Falcon (Thermo Fisher, BrandTM 114818, catalog number: 10345521)
1.5 mL tube (Eppendorf, catalog number: 0030120086)
PCR tubes (Eppendorf, catalog number: 0030124820)
Disposable chromatography column (Bio-Rad, catalog number: 7321010)
pX330-U6-Chimeric_BB-CBh-hSpCas9 (Addgene, catalog number: 42230)
pSMART-HCKan, CloneSmart Blunt Cloning kit (Lucigen, catalog number: 40704-2)
pHybr_r2a>mA-srt-his (Addgene, catalog number: 124806)
pHybr_r2a>m2a(silent)-srt-his (Addgene, catalog number: 124807)
PX458-gR2A_ISO (Addgene, catalog number: 124808)
BbsI-HF® (New England Biolabs, catalog number: R3539)
rSAP (New England Biolabs, catalog number: M0371)
T4 DNA ligase (New England Biolabs, catalog number: M0202)
T4 PNK (New England Biolabs, catalog number: M0201)
Q5 polymerase (New England Biolabs, catalog number: M0491)
dNTP (Thermo Fisher, catalog number: R0192)
ddH2O (Thermo Fisher, Invitrogen, catalog number: AM9935)
Agarose (Sigma-Aldrich, catalog number: A4718)
Nancy-520 (Sigma-Aldrich, catalog number: 01494)
NucleoBond Xtra Midi Plus EF kit (Macherey-Nagel, catalog number: 740422.50)
NucleoSpin Gel and PCR cleanup (Macherey-Nagel, catalog number: 740609.50)
ISOLATE II Genomic DNA isolation kit (Bioline, catalog number: BIO-52067)
Hybridoma: rat IgG2a,λ anti mouse PD-L1 [MIH5] (in house)
Hybridoma: rat IgG2a,κ anti mouse DEC-205 [NLDC-145] (ATCC, catalog number: HB-290TM)
Hybridoma: mouse IgG1,κ anti human CD20 [NKI.B20/1] (in house)
SF Cell Line 4D-NucleofectorTM X kit L (Lonza Biosciences, catalog number: V4XC-2024)
PBS (Fresnius Kabi, catalog number: M087312/01)
RPMI-1640 (Thermo Fisher, GibcoTM, catalog number: 42401018)
CD hybridoma medium (Thermo Fisher, GibcoTM, catalog number: 11279023)
Heat-inactivated fetal bovine serum (FBS) (HyClone, catalog number: SV35959)
Ultraglutamine-1 (Lonza Biosciences, catalog number: BE17-605E/U1)
Antibiotic-antimycotic (Thermo Fisher, GibcoTM, catalog number: 15240062)
β-mercaptoethanol (β-ME) (Sigma-Aldrich, catalog number: 444203)
Trypan blue (Thermo Fisher, GibcoTM, catalog number: 15250061)
CELLine CL 1000 bioreactors (INTEGRA Biosciences AG, catalog number: 90005)
Blasticidin (Invivogen, catalog number: ant-bl-05)
Puromycin (Invivogen, catalog number: ant-pr-5)
BSA (Sigma-Aldrich, catalog number: A9418)
NaN3 (Sigma-Aldrich, catalog number: 26628-22-8)
Recombinant Protein G (Thermo Fisher, PierceTM, catalog number: 21193)
Ni-NTA agarose (Quiagen, catalog number: 30230)
Anti-6×His-tag (clone J095G46) (PE, Biolegend, catalog number: 362603)
Anti-rat IgG2a HC (clone MRG2a-83) (PE, Biolegend, catalog number: 407507)
Anti-mouse IgG2a (clone m2a-15F8) (PE, eBioscience, catalog number: 12-4817-82)
Rabbit anti-6×His-tag (clone RM146, unconjugated, Abcam, catalog number: AB14923)
Goat anti-rabbit IgG (H+L) (polyclonal, IRD800, LI-COR, catalog number: 926-32211)
Goat anti-rat IgG (H+L) (polyclonal, AF680, Thermo Fisher, catalog number: A-21096)
Wash buffer (see Recipes)
PBA (see Recipes)
Culture medium (see Recipes)
1× selection medium (see Recipes)
Equipment
Electroporation device, e.g., AMAXA 4D Nucleofector (Lonza BioSciences, AAF-1002X/AAF-1002B)
Humidified incubator, 5% CO2
Thermocycler (e.g., Bio-Rad, catalog number: 1861096EDU)
Flow cytometer, e.g., FACSLyric (BD Biosciences, catalog number: 663518)
Fluorescent scanner, e.g., Amersham TyphoonTM 5 (Cytiva, catalog number: 29187191)
Sanger Sequencing Service
Software
Snapgene (from Insightful Science; available at snapgene.com)
FlowJoTM Software (Ashland, OR; Becton, Dickinson and Company; 2021)
Procedure
General considerations
Before starting hybridoma modification, knowledge on the host species and antibody isotype is required. We have used cut sites to knock in inserts upstream or downstream of CH1 heavy chain (HC), upstream of the hinge, and downstream of Cκ light chain (LC), but virtually any other cut site can be used to tailor the antibody to the intended use.
It takes 4–6 weeks from the transfection of the target cell lines to the isolation of high levels of modified antibodies (Figure 1).
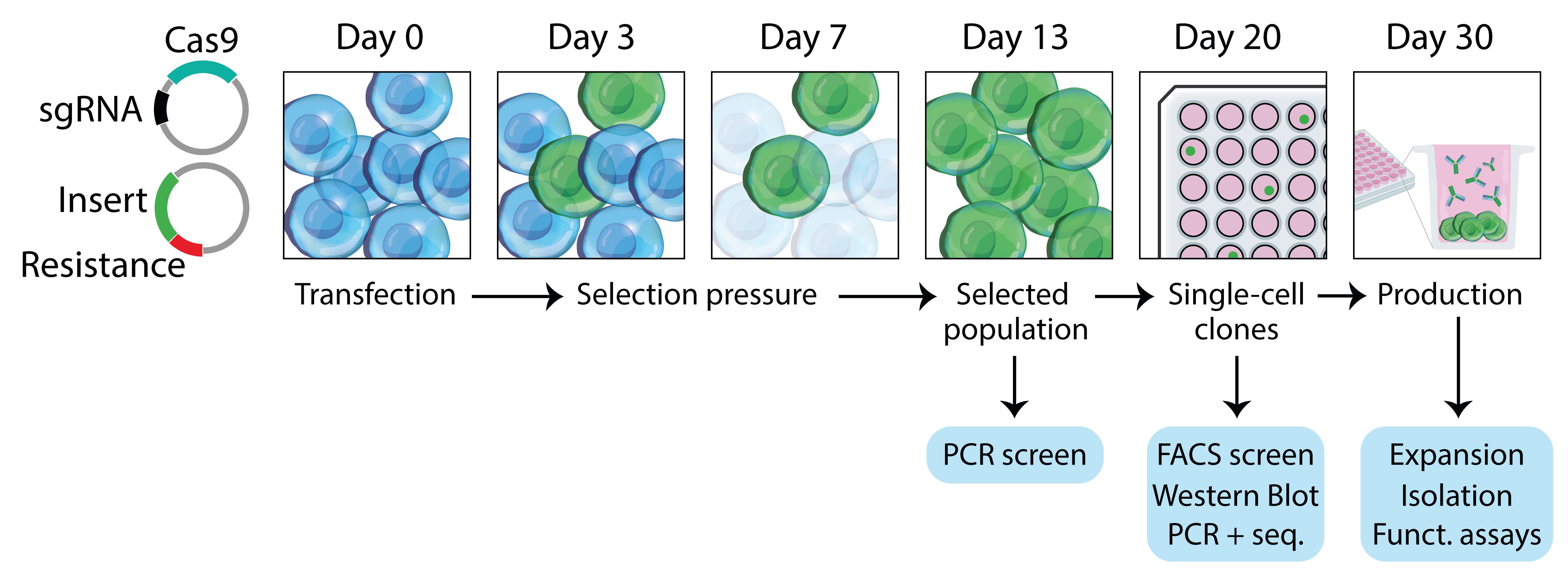
Figure 1. Workflow overview. Hybridoma are transfected on day 0 with a plasmid encoding the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) and guide ribonucleic acid (gRNA), and a plasmid encoding a homology-directed repair (HDR) template containing an antibiotic resistance gene. At day 3, selection pressure is applied, and cells start to grow out. Resistant clones are expanded until reaching confluency, and single-cell clones (SCC) are prepared in 96-well plates. By day 30, positive clones are screened for production of the modified antibody and expanded for production.For the purpose of this protocol and to illustrate the diversity of potential applications, we describe the full characterization of three successfully edited hybridoma: isotype switching of the constant heavy chain (HC) of rat IgG2a (r2a) Ig to a mouse IgG2a (m2a) constant HC and produce a chimeric r2a(V):m2a(C) mAb, introduction of an antigen in the hinge region of r2a HC to produce a Fab’ fragment–antigen fusion, and modification of the mouse IgG1 (m1) HC and mouse kappa (mκ) LC to introduce two site-specific labeling tags.
Design of CRISPR/HDR template and gRNA
For designing an HDR template and gRNA from scratch, the Addgene CRISPR guide or the CRISPR 101 book are good starting points.
In Figures 2–6, the process for designing a CRISPR/HDR template and gRNAs is illustrated step by step. We take the example of targeting the C-terminus of m1 CH1 to engineer a mAb-secreting cell line to produce Fab’ fragments bearing a site-specific labeling site at the C-terminus of the CH1.
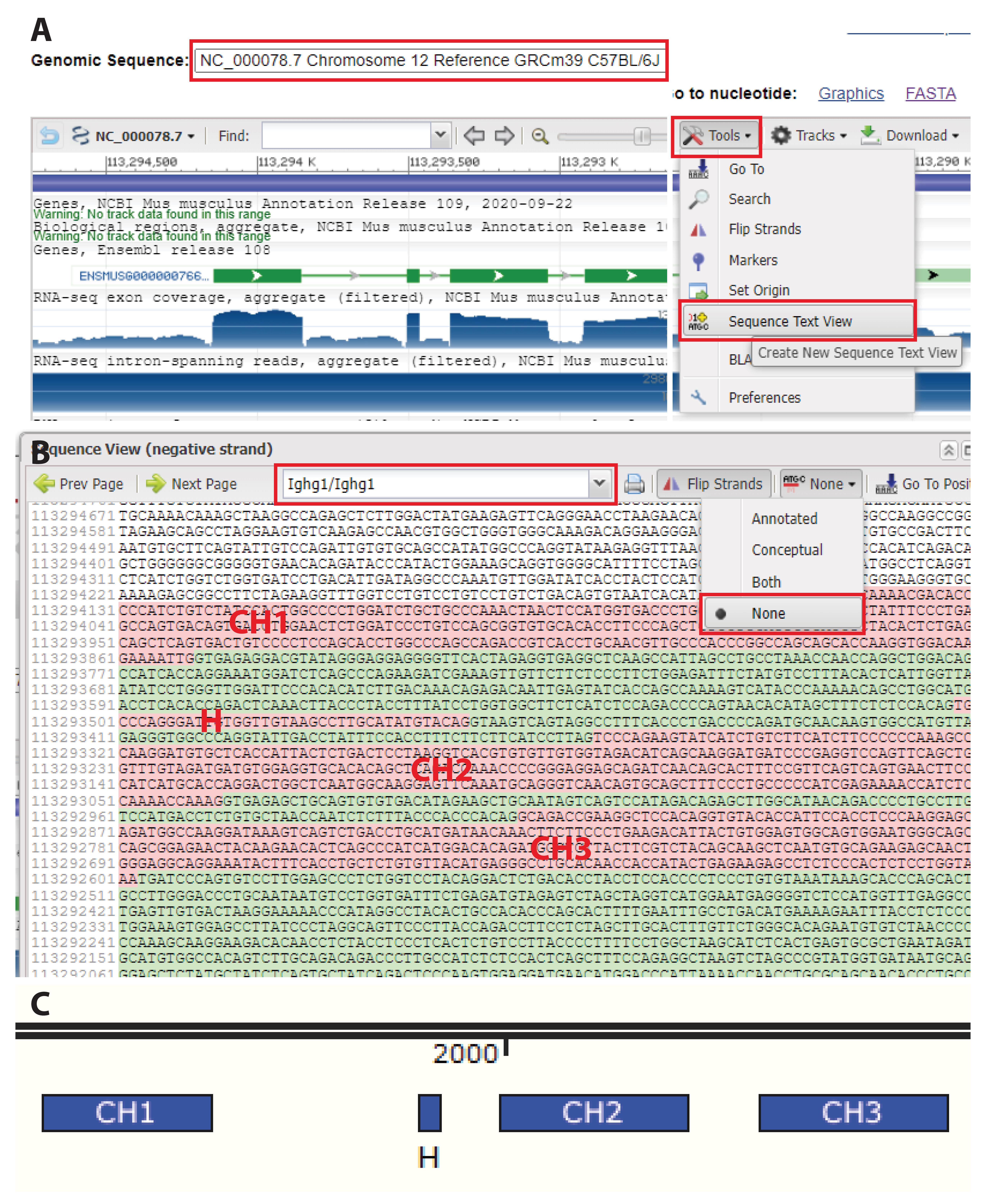
Figure 2. Annotating the target sequence. A. Access the genomic sequence of the locus of interest (e.g., on NCBI Gene). Using “sequence text” view, visualize exons and introns in the target locus. B. Remove annotations to visualize only the nucleotide sequence. For a given locus, exons are marked in red and introns in green. Here are highlighted constant heavy chain (CH)1, hinge, CH2, and CH3 in the Ighg1 gene (mIgG1 constant region). C. Copy the sequence in Snapgene and annotate exons to identify the target region. This typically depends on the intended modification. Here, we target the C-terminus of CH1 of mIgG1.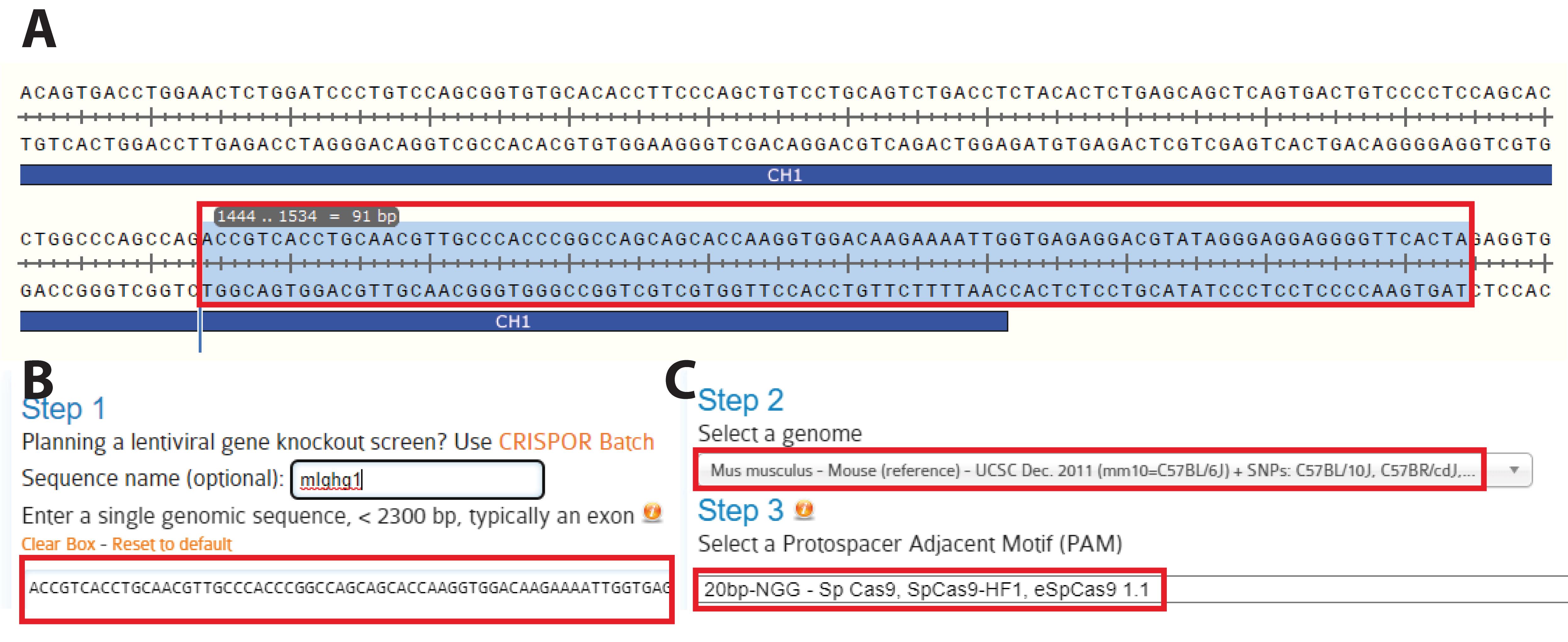
Figure 3. Identification of the target locus. A. Identify the region to target for the knock in. Here, we selected the C-terminus of the CH1 domain to insert part of the hinge region, a short linker, a site-specific labeling site, and a polyhistidine tag. This effectively transforms a mAb into a Fab’ fragment bearing a site-specific labelling site at the C-terminus of its heavy chain (HC). B. Copy the selected sequence to CRISPOR. C. Choose the appropriate host (here, Mus musculus) and protospacer adjacent motif (PAM) (typically, -NGG) and submit.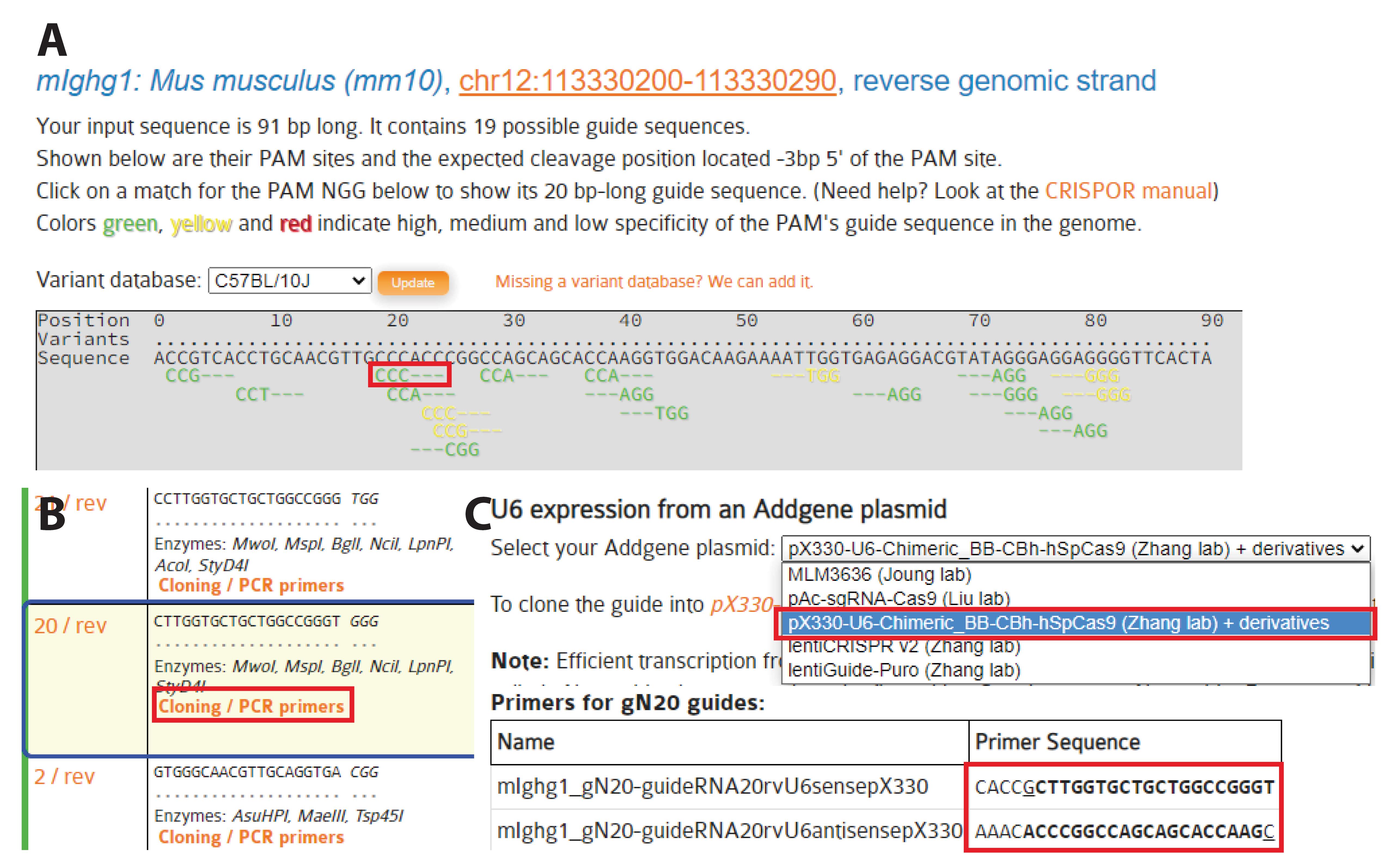
Figure 4. Selection of gRNAs. A. Protospacer adjacent motif (PAM) sequences and corresponding gRNA candidates are identified by CRISPOR. All possible PAM sequences (-NGG here) are identified below the input sequence. B. Sequences and characteristics of potential gRNAs are displayed as a table, ordered by predicted efficacy. The insertion site should be less than 10 bp away from the double strand break if possible. When targeting a new region, select at least two or three gRNAs based on their proximity to the intended site of insertion and precited score. Here, we selected gRNA_m1_HC (5′-CTTGGTGCTGCTGGCCGGGT-3′). By clicking on “cloning,” a new window opens with sequences for cloning into a variety of Cas9-containing vectors. C. We used a U6 expression system (U6 promoter, pX330 vectors, and derivatives) (Cong et al., 2013). Here, we used a 20 nt gRNA, but it was recently proposed to use a 19 nt gRNA for improved efficiency (Kim et al., 2020). Select the appropriate primers and follow the instructions to clone the gRNAs into the pX330 vector at the BbsI site.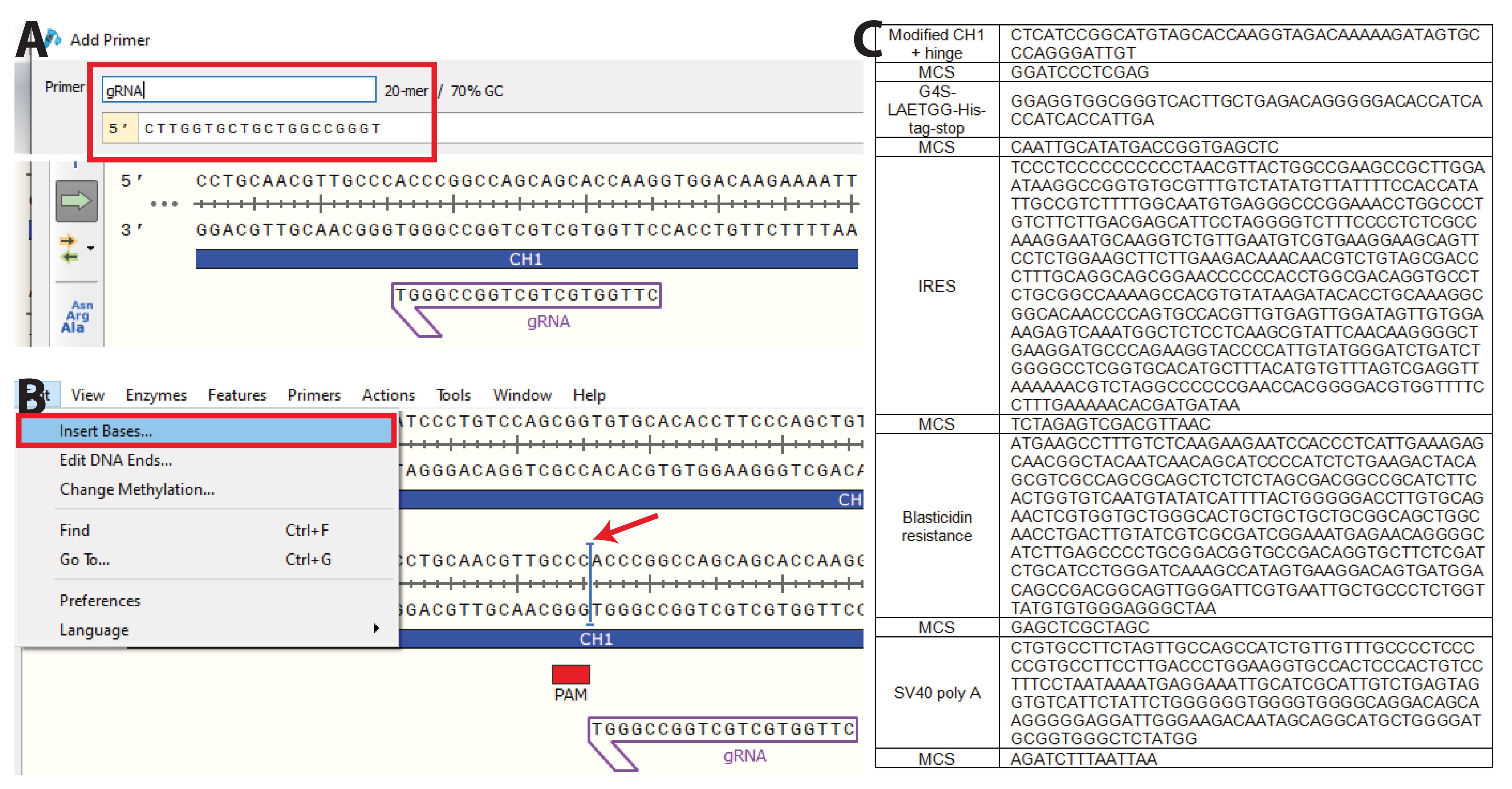
Figure 5. nserting the desired sequence at the knock-in site. A. Copy the gRNA sequence(s) into Snapgene and B. visualize the protospacer adjacent motif (PAM) sequence(s). Insert the sequence to knock in as close as possible to the PAM sequence. C. Insert the predetermined nucleotides containing the desired insert. Here, we inserted part of the hinge region (VPRDC) allowing association with the light chain (LC) downstream of CH1, followed by a multiple cloning site (MCS), a flexible linker (GGGGS), a sortag motif (LAETGG), and a polyhistidine tag (HHHHHH), followed by a stop codon. The insert is followed by an internal ribosomal entry site (IRES) to allow for transcription of blasticidin resistance gene (BSD) and a simian vacuolating virus 40 (SV40) poly A tail to dissociate ribosomes. Each domain is surrounded by a MCS to allow easy exchange of any part of the plasmid. Here, we additionally optimized part of the CH1 to improve association with the LC (PACSTKVDKKI, S>C mutation).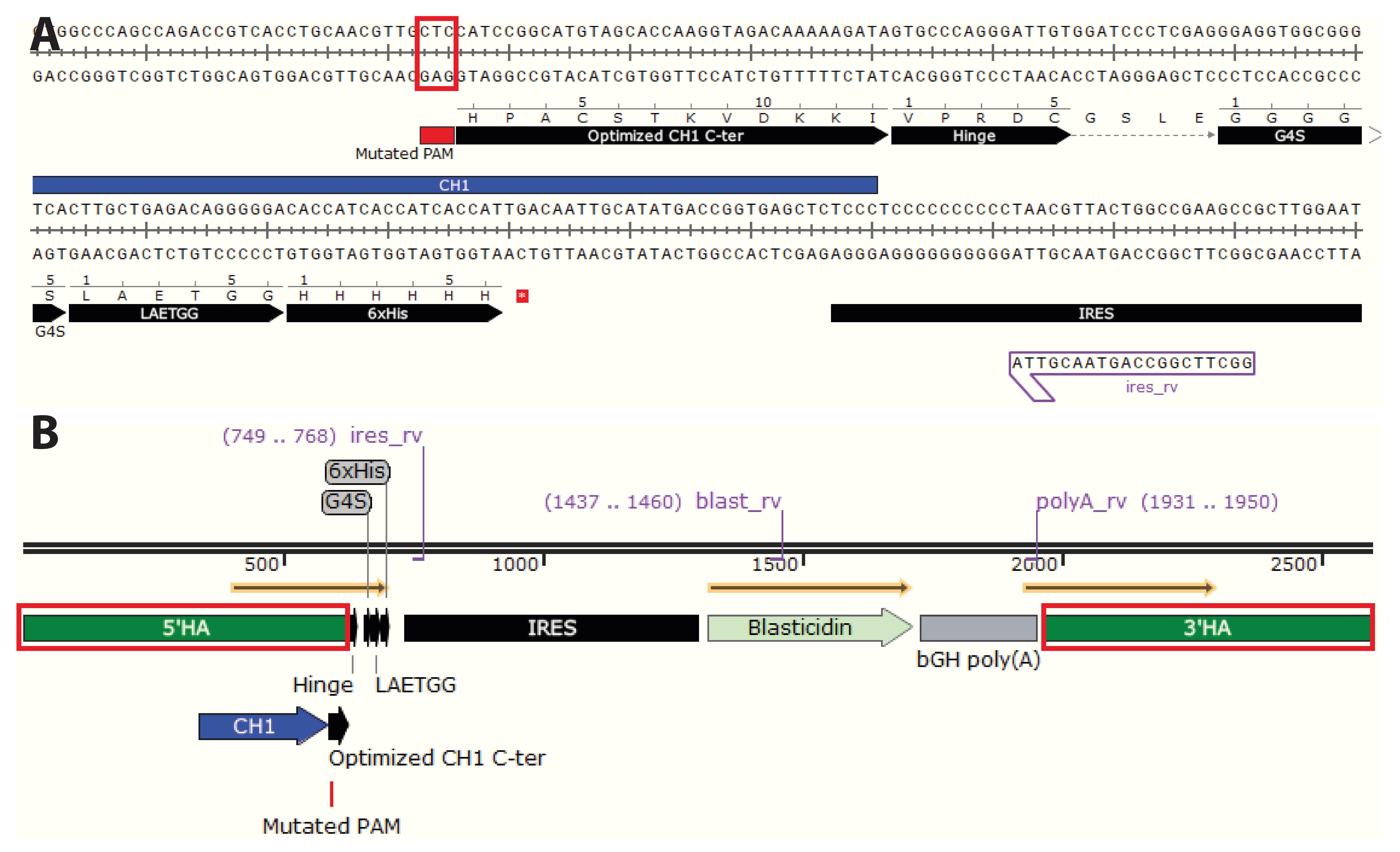
Figure 6. Homology arms (HA) and final HDR template design. The HAs span ca. 0.6 kb on each side of the insert. Their large size allows a few mismatches between the HAs and the target locus without affecting HA annealing to the genomic locus during HDR. Therefore, small adjustments can be done in the HA sequence compared to the germline sequence initially obtained from the genome browser without affecting HDR efficacy. A. It is necessary to mutate the “NGG” protospacer adjacent motif (PAM) sequence(s). To inactivate the PAM sequences, mutate “NGG” to “NGC,” “NGA,” or “NGT.” If the PAM sequence is located within the coding region, ensure that it is a silent mutation. Alternatively, remove the PAM sequences all together. Cells having undergone successful HDR will not contain PAM sequence(s) and the Cas9 will be inactivated. B. To define 5’HA and 3’HA, select ~0.6 kb on each side of the insert.Sequences of the constant immunoglobulin loci can be accessed on NCBI Gene; protospacer adjacent motifs (PAM) sequences and gRNA can be determined using CRISPOR or alternative tools.
The HDR template should contain 5′ and 3′ homology arms (HAs) flanking the desired insert, a stop codon, an internal ribosomal entry site (IRES), an antibiotic resistance gene (blasticidin or puromycin), and a poly A tail (Figure 8A–C). Inserts are typically 1–2 kb in length. While longer inserts are possible, HDR efficacy greatly decreases beyond 2–3 kb. The length of HAs depends on the length of the insert (~0.5 kb HAs per 1 kb insert), and typically ranges between 0.3 kb and 1 kb, although shorter HAs are possible. Here, we used 0.6 kb HAs.
Recommended: Sequence the target region to ensure that no derivation from the germline is present and ensure that PAM sequences and splice acceptor sequences in downstream exons are removed from HAs in the HDR template. General sequencing primers are provided in Table 2 and a general PCR protocol is provided in Supplementary file. Determine gRNA efficacy for the region of interest through a T7 assay.
The HDR template is ordered as double-stranded DNA gBlock and cloned in a pSmart vector (e.g., pSMART-HCKan, CloneSmart Blunt Cloning kit). Candidate gRNAs are ordered as single-stranded DNA oligos that are annealed and cloned in vectors from the pX330 family (Cong et al., 2013). A basic cloning protocol is available in Supplementary file.
After design and cloning, perform midipreps of the plasmid DNA encoding the HDR and gRNA templates using the NucleoBond Xtra Midi Plus EF kit and sequence using standard primers. The A260:A280 ratio should be at least 1.8.
We have developed plasmids enabling modification of the HC of rat IgG2a hybridoma to replace the complete constant region by a new isotype, cleave in the hinge and insert any tag of interest (Fab’ fragment) (van der Schoot et al., 2019), and to modify mIgG1 CH1 (depicted in Figures 2–6 and recorded in the video) and Cκ kappa LC of murine hybridoma (Le Gall et al., 2021). Our published plasmids are available on Addgene or upon request. Inserts between the HAs can easily be swapped using standard molecular cloning techniques and dedicated restriction sites. gRNAs are reported in Table 1 and HAs in Table 5 (notes).
Table 1. gRNA used or mentioned in this protocol. HA sequences for the corresponding HDR templates are reported in Table 5.
gRNA Target Sequence gRNA_iso_r2a Upstream of rat IgG2a CH1 exon TGTAGACAGCCACAGACTTG gRNA_r2a_H Upstream of rat IgG2a hinge exon GACTTACCTGTACATCCACA gRNA_m1_HC C-terminus of mouse IgG1 CTTGGTGCTGCTGGCCGGGT gRNA_mκ_LC C-terminus of mouse κ LC GGAATGAGTGTTAGAGACAA
Culture of hybridoma cells and titration of antibiotic ahead of transfection
Selection of cells that have successfully undergone HDR recombination is performed under antibiotic pressure. Ensuring quality of the parental cell line ahead of the procedure is critical (see Notes). The target hybridoma should be tested for antibiotic (blasticidin/puromycin) sensitivity prior to starting the experiment.
Culture target hybridoma cells, either generated in house or obtained from the American Type Culture Collection, in complete culture media. Make sure to maintain cells at a high viability (> 90%).
When confluent, seed 1 × 106 cells per well in a 6-well plate in 3 mL of culture medium and supplement with 0, 0.5, 1, 2, 4, 8, 10, or 20 μg/mL of the appropriate antibiotic.
Incubate cells at 37 °C for 3–7 days depending on the selected antibiotic (three days for puromycin, seven days for blasticidin).
Determine the lowest concentration at which no viable cells remain in the culture by using Trypan blue and use the determined concentration to perform the selection procedure (Figure 7A–B).
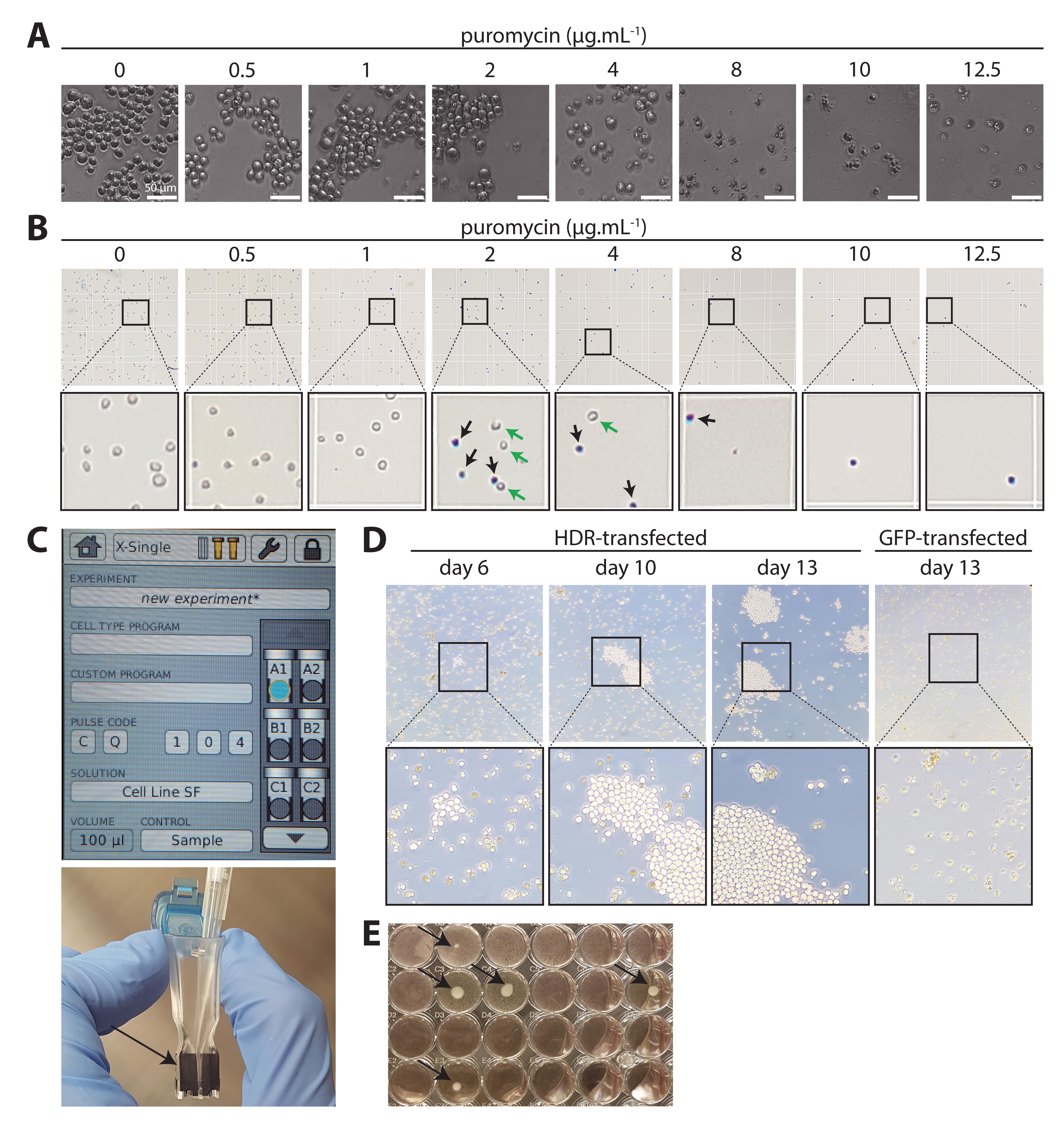
Figure 7. Antibiotic titration, transfection, and expansion of resistant cells. A. Titration of puromycin on wild-type (WT) anti-human CD20 [NKI.B20/1]. Serial dilution of 0–12.5 μg/mL puromycin. After 48 h, cells start dying in the treated conditions. Healthy hybridoma cells are round and regular and grow in clusters. Dead or dying cells lose their round shape and become irregular. Dead cells can be spotted at 0.5 μg/mL, and some live cells remain up to 4 μg/mL. From 8 μg/mL on, cells are clearly dead. A trained eye can easily recognize how viable the cells are. B. To quantify the viability, Trypan blue staining allows to discern live (white) from dead (blue) cells. Here, we selected 8 μg/mL puromycin to conduct the procedure. C. AMAXA cuvette and program (Cell line SF, CQ-104). Carefully pipette the suspension containing cells and DNA between the electrodes (arrow head) without introducing air bubbles. D. After selection, small clusters start to grow out in HDR-transfected plates among a majority of dying or dead cells (day 6). These resistant cells have successfully undergone HDR. Passage the plates until larger clusters start to grow out (day 10) and grow towards confluency (day 13). In contrast, green fluorescent protein (GFP)-transfected hybridoma remain dead and no cluster can be seen (day 13, right). When the plate is full, perform a limiting dilution. E. After limiting dilution, monoclonal cell lines can be easily identified at the bottom of their respective well (arrow heads). Expand and characterize these cell lines.
Transfection of hybridoma cells
This section describes the electroporation of 1 × 106 wild-type hybridoma cells using gRNA/Cas9 + HDR template or green fluorescent protein (GFP) as a mock transfection control. Having cells resuspended for an extended amount of time in SF transfection medium from the SF Cell Line 4D-NucleofectorTM X kit Lis detrimental: work fast and organized and perform transfections sequentially.
On day 0, use confluent wild-type hybridoma cells at a high viability (>90%) to perform the electroporation.
Add 3 mL of culture medium to a 6-well plate and incubate at 37 °C. Pre-warm culture medium in a 50 mL Falcon.
Spin down hybridoma cells from your desired cell line (1,500 rpm, 5 min, 4 °C).
Resuspend cell pellet in an appropriate amount of wash buffer (aim for 1 × 106–2 × 106 cells/mL)
Count the cells using Trypan blue and transfer 1 × 106 cells to a 1.5 mL Eppendorf tube.
Spin down cell suspensions in a tabletop centrifuge [1,500 × g, 5 min, room temperature (RT)].
During this time, prepare two or any desired numbers of DNA mixes in 0.2 mL Eppendorf tubes, by pipetting 1 μg of Cas9/gRNA + 1 μg of HDR template per condition, and 2 μg of pMAX-GFP as mock transfection control (DNA mix tube).
Prepare the 4D-Nucleofector with the parameters: Program CQ-104, Cell line SF.
Carefully aspirate supernatant of the cell pellet.
Add 100 μL of SF transfection medium to the cell pellet and resuspend cells before transferring into DNA mix tube containing HDR template + gRNA or pMAX-GFP.
Resuspend and carefully transfer the suspension to the bottom of a 100 μL electroporation cuvette provided with the Lonza nucleofection kit (Figure 7C). Gently tap the cuvette to ensure that the sample covers the electrodes and the bottom of the cuvette and check that there are no air bubbles.
Close the lid and insert the cuvette in AMAXA nucleofector. Press Start on the display to perform electroporation with the parameters: Program CQ-104, Cell line SF (Figure 7C).
Carefully add 1 mL of pre-warmed medium to the cuvette and transfer cells immediately to the 6-well plate with pre-warmed medium (total volume in well is now ca. 4 mL). Avoid repeated aspiration of the cells.
Wash cuvette with medium to make sure that all cells are transferred.
Proceed to the next transfection (repeat from step 8).
Incubate the 6-well plate at 37 °C in a humidified CO2 incubator.
Selection of engineered hybridoma cells and generation of monoclonal cell lines
This section describes the selection of successfully engineered hybridoma cells to identify HDR-edited clones. This is done by antibiotic selection pressure of HDR- and GFP-transfected hybridoma from day 3 post transfection. Resistant cells are grown to confluency and monoclonal cell lines can be generated.
Transfected cells are cultured for two days in selection-free medium to allow electroporated cells to grow. On day 1, check transfected cells for viability (by eye) and the transfection control cells for GFP expression using a fluorescent microscope. Handle cells carefully to preserve viability.
On day 3, either scrape the cells loose or resuspend until the cells detach and transfer all 4 mL of cell suspension to a 10 cm Petri dish containing 5 mL of pre-warmed culture medium supplemented with the appropriate selection drug at twice the predetermined concentration (2× selection medium). Wash the well with 1 mL of culture medium to collect all the cells and transfer to the Petri dish. After transferring the cells, the final volume in the Petri dish should be adjusted with culture medium to 10 mL to ensure that cells are in 1× selection medium.
Starting on day 5, and two times per week, passage transfected cells. Briefly, scrape cells loose and transfer to 15 mL Falcon tubes. Place 5 mL of new pre-warmed 1× selection medium in the 10 cm Petri dish. Spin down (1,500 rpm, 5 min, RT) and discard supernatant. Resuspend cell pellets in 2 mL of pre-warmed 1× selection medium and transfer suspension back to the Petri dish. Wash the 15 mL Falcon with 3 mL of 1× selection medium and transfer back to the Petri dish.
By day 5–10 depending on the antibiotic, it should be obvious that small clusters are forming among a majority of dead cells in the HDR/gRNA-transfected plate, while GFP-transfected cells are all dead (Figure 7D). If no cell death is observed, antibiotic concentration should be adjusted (see Notes). If no live cells grow out following antibiotic pressure, refer to the listed troubleshooting solutions (see Notes).
Passage cells until these clusters grow out to reach higher confluency and viability (Figure 7D, >90%, typically around day 15–20). When the HDR-transfected cells are full, cells should still be all dead in the GFP plate. At this point, discard the GFP-transfected plate, and proceed to limiting dilution of HDR-transfected cells. After this step, antibiotic pressure is not required anymore. In case few clusters grow out very slowly or some cells are still alive in the GFP-transfected plate, it is best to keep the selected antibiotic in the medium until after identification of monoclonal cell lines.
Pre-plate 100 μL per well of culture medium in five round bottom 96-well plates and incubate at 37 °C.
Scrape confluent HDR-transfected cells loose and spin down the cell suspension (1,500 rpm, 5 min, RT). Keep a 1 mL sample of the supernatant and discard the rest.
Resuspend cells in 5 mL of culture medium and count using Trypan blue. Dilute the cells in culture medium in two steps to obtain a 50 mL suspension at the concentration of 3 cells/mL.
[Example: Initial suspension is at 1.5 × 106 cells/mL → dilution factor: 500,000 (1.5 × 106/3). First dilute 500×, 100 μL of cell suspension in 50 mL of culture medium (3,000 cells/mL). Then take 50 μL of cell suspension (3,000 cells/mL) and dilute 1,000× in 50 mL culture medium (concentration 3 cells/mL).]
Plate 100 μL/well of the suspension in the round bottom 96-well plates. The final concentration is 0.3 cells/well in 200 μL or 33 cells/plate.
From the original cell suspension keep ~500,000 cells for genomic DNA (gDNA) isolation (ISOLATE II Genomic DNA kit isolation kit). Spin down and follow manufacturer's instructions.
Cryo-preserve leftover cells of the bulk population by following a standard cryo-preservation protocol.
While the single-cell clones (SCC) are growing, the bulk population and its supernatant should be characterized immediately for integration of the HDR template. For this, follow section 1–3 in the Data analysis section.
Expansion of modified hybridoma cells
Incubate plates for 10–15 days at 37 °C without changing the medium.
When SCC are visible at the bottom of the wells (Figure 7E), transfer them to a new flat bottom 96-well plate and add 100 μL of new culture medium.
When cells are confluent, collect and freeze supernatant and expand the clones to a 24-well plate and then a T75 flask.
Collect ~500,000 cells for gDNA isolation and cryo-preserve the expanded clones.
Data analysis
Characterization of selected clones (from bulk and/or SCC populations)
Validation that the cell line is producing the right antibody (fragment) is crucial. Screening approaches should be conducted on the bulk population first to ensure success of the procedure and afterwards on the SCC. We typically combine three or more approaches to ensure insert integration and correct protein production: at the genetic, protein, and functional levels. Several examples are provided below to illustrate different methods. This straightforward protocol does not require statistical analysis up until functional characterization of the proteins. If resistant cells do not produce the modified antibodies, refer to the listed troubleshooting solutions (see Notes).
To illustrate this section, we present the full characterization of three distinct cell lines modified using this technique.
MIH5 is a rat IgG2a, λ hybridoma producing monoclonal antibodies binding to murine PD-L1, a protein expressed by cancer cells and myeloid cells. PD-L1 is a therapeutic target in the mAb-based treatment of cancers (checkpoint inhibitors). We engineered MIH5 cell lines that produce chimeric anti-PD-L1 antibodies with the mouse IgG2a (m2a) and mouse IgG2a L234A/L235A/N297A (m2a(silent)) isotypes (Figure 8A) (van der Schoot et al., 2019).
NLDC-145 is a rat IgG2a, κ hybridoma producing monoclonal antibodies binding to murine DEC-205, a C-type lectin expressed by a subset of cross-presenting dendritic cells. Due to its restricted expression, DEC-205 is a promising target for developing targeted vaccines that efficiently deliver antigens to a restricted cell population able to best elicit a T cell response. We engineered NLDC-145 to produce truncated antibodies (Fab’ fragments) bearing CD8 T or CD4 T cell epitopes from the model antigen ovalbumin (OVA) (respectively Fab’OTI and Fab’OTII) (Figure 8B) (unpublished data by Fennemann et al., 2021).
NKI.B20/1 is a mouse IgG1,κ hybridoma producing monoclonal antibodies binding to human CD20. CD20 is the target of Rituximab, a B cell–depleting mAb used to treat B-cell lymphoma, rheumatoid arthritis, or lupus. We generated an NKI.B20/1 cell line that produces a Fab’ fragment bearing two orthogonal site-selective modification sites on the HC and LC (Figure 8C): LAETGG-His-tag on the HC and a LPESGG-Myc-tag motif on the LC. This construct can be used to generate homogenous antibody–drug conjugates bearing two distinct payloads (Le Gall et al., 2021).
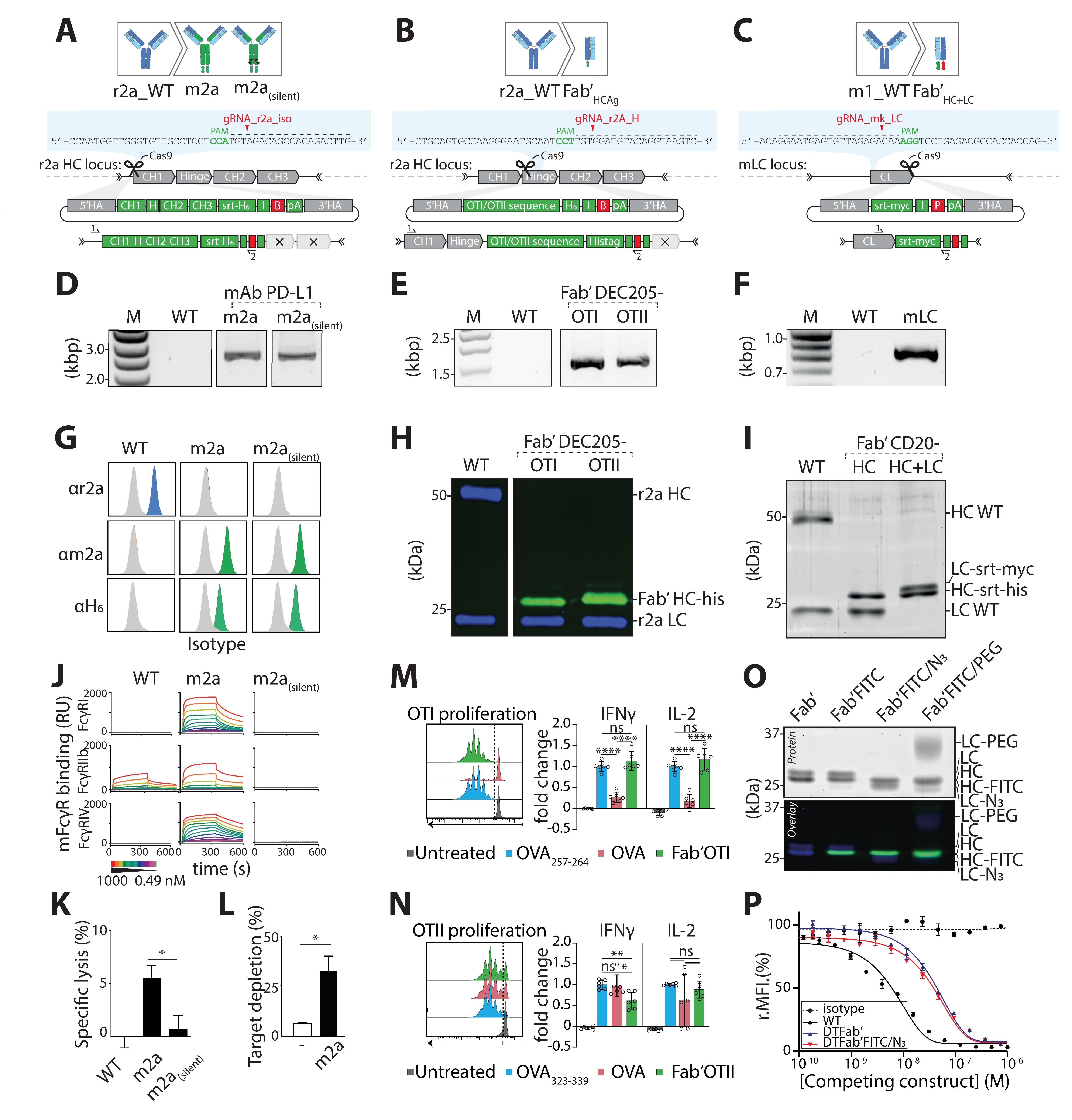
Figure 8. Validation of three engineered cell lines at the genetic, protein, and functional level. A. HDR template and gRNA for isotype switching of rat IgG2a hybridoma to mouse IgG2a. B. HDR template structure and gRNA for rat IgG2a hybridoma to Fab’ antigen (Fab’Ag). Ag presented here, OTI and OTII peptide. C. HDR template and gRNA for editing of the LC of mouse IgG1 hybridoma. In this strategy, HC and LC are both edited to form a Fab’ fragment bearing two site-selective modification sites (LC editing shown here). D. PCR amplification of gDNA isolated from a m2a and m2a(silent) resistant clone using a location specific (1) and HDR specific (2) primer set. E. PCR amplification of gDNA isolated from Fab’OTI and Fab’OTII resistant clones using a location-specific (1) and HDR-specific (2) primer set. F. PCR amplification of gDNA isolated from a mκ resistant clone using a location-specific (1) and HDR-specific (2) primer set. G. After selecting monoclonal hybridoma for each isotype, supernatant was incubated with PD-L1–expressing target cells (CT26) and cells were stained with anti r2a, anti m2a, or anti-His-tag. Displayed plots demonstrate that supernatants exclusively contain the engineered isotype variant with a C-terminal His-tag, while the original rIgG2a mAbs is absent. H. Western blot of Fab’-OTI and -OTII, stained for His-tag (green) and rat IgG2a (blue). Protein G–isolated NLDC-145 antibodies were used as negative control (WT). I. SDS-PAGE in-gel fluorescence (Sypro staining) visualization of WT mAb CD20, Fab’HC, and Fab’HC+LC proteins shows a change in molecular weight after editing of the HC and LC. J. Representative sensograms display interactions of MIH5 WT and MIH5-engineered mAbs (WT r2a, m2a, m2asilent) for immobilized murine FcγR (FcγRI, FcγRIIb, and FcγRIV) at increasing concentrations (0.49–1000 nM). Binding to FcγR is expressed in resonance units (RU). K. In vitro antibody-dependent cellular cytotoxicity (ADCC) assay to compare effector function of MIH5 isotype variants. MC38 cells were 51Cr-labeled, opsonized with m2a or m2a(silent) MIH5 isotype variants, and exposed to whole blood from C57BL/6 mice for 4 h. Specific lysis was quantified by measuring 51Cr release (n = 3, mean ± SEM, * p < 0.05) and shows that m2a induces specific lysis of PD-L1-expressing cells, with no activity of m2a(silent) in vitro. L. In vivo depletion assay. Splenic B cells labeled with Violet and Red tracer dye were used as target cells for in vivo depletion and opsonized with either m2a(silent) variant or m2a. Subsequently, B cells were mixed 1:1 (m2a:m2a(silent) or m2a(silent):m2a(silent)) and injected intravenously into C57BL/6 mice. Twenty-four hours later, spleens were isolated, and ratios between fluorescently labeled cell populations were determined via flow cytometry to quantify the isotype-specific depletion of target cells, showing that m2a and m2a(silent) are both functional in vivo (n = 3, mean ± SEM, * p < 0.05). M. DEC-205-expressing murine dendritic cells were generated from bone marrow using Flt3L and OP9-DL-1 feeder layer (Kirkling et al., 2018). At day 8, CD11c+ cells were isolated using CD11c microbeads and incubated with 1 μM Fab’OTI, 5 nM OVA257-263 (SIINFEKL, positive control) or 10 μM OVA. After 2 h, cells were washed and exposed to freshly isolated fluorescently labeled OTI CD8 T cells in media supplemented with 0.3 μg/mL lipopolysaccharide (LPS). After 72 h, cells were analyzed by flow cytometry for proliferation (cell dye dilution) and activation markers (not shown), and supernatant was analyzed by ELISA for production of pro-inflammatory IFNγ and IL-2 cytokines. Fab’OTI-treated dendritic cells induced CD8 T cell proliferation and activation, showing that it was taken up by DEC-205-expressing dendritic cells and processed, and that the CD8 T cell epitope was presented on class I major histocompatibility complex (MHC) (n = 3, mean ± SD, ns: not significant, **** p < 0.0001, one-way ANOVA with Tukey’s correction for multiple testing). N. Day 8 DEC-205-expressing murine dendritic cells were incubated with 1 μM Fab’OTII, 1 μM OVA323-339 (ISQAVHAAHAEINEAGR, positive control), or 10 μM OVA. After 2 h, cells were washed and exposed to freshly isolated fluorescently labeled OTII CD4 T cells in media supplemented with 0.3 μg/mL LPS. After 72 h, cells were analyzed by flow cytometry for proliferation and activation marker expression (not shown) and supernatant was analyzed by ELISA for production of pro-inflammatory IFNγ and IL-2 cytokines. Fab’OTII-treated dendritic cells induced CD4 T cell proliferation and activation, showing that it was taken up by DEC-205-expressing dendritic cells and processed, and that the CD4 T cell epitope was presented on class II MHC (n = 3, mean ± SD, ns: not significant, * p < 0.05, ** p < 0.01, one-way ANOVA with Tukey’s correction for multiple testing). O. SDS-PAGE analysis of Fab′CD20 sequentially site-specifically labeled with H-GGG-C-K(FITC)-NH2 on the HC and H-GGG-K(N3)-NH2 on the LC confirms introduction of two distinct cargos site-specifically onto the engineered Fab′ and functionality of the introduced motifs. P. Antigen binding competition assay of each engineered protein against mAbNKI.B20/1-AF647 reveals that proteins do not lose binding affinity to their target following CRISPR/Cas9 editing and sequential dual site-specific labeling. D, G, J, K, L. Adapted from van der Schoot et al. (2019) E, H, M, N. Adapted from Fennemann et al. (2021) F, I, O, P. Adapted from Le Gall et al. (2021).B: blasticidin resistance gene, FITC: Fluorescein isothiocyanate, H: hinge, H6: hexahistidine tag (HHHHHH), HA: homology arm, HC: heavy chain, HDR: homology directed repair, I: internal ribosomal entry site (IRES), IFNγ: interferon gamma, IL-2: interleukin-2, LC: light chain, myc: myc-tag (EQKLISEEDL), OVA: ovalbumin, P: puromycin resistance gene, pA: poly A tail, PAM: protospacer adjacent motif, PD-L1: programmed death ligand 1, PEG: polyethylene glycol, srt: sortag motif, WT: wild type.
PCR amplification of target locus to determine HDR integration
PCR screens should be performed on both bulk and SCC populations. For this, use the isolated gDNA from ~500,000 cells and perform amplification of the targeted locus using a primer that only anneals to the inserted sequence in combination with a location-specific primer that anneals around the insertion site outside the HA region. Use ~100 ng of gDNA to amplify the target locus using a standard PCR program (provided in Supplementary file). Table 2 provides sequences of primers that can be used for amplification of murine IgG1 HC, murine kappa LC, and rat IgG2a CH1 HC upon insertion of our HDR templates.
Table 2. Locus-specific and HDR-specific for PCR amplification of target region
Primer Target 5′-3′ Sequence r2a_iso_fw rat IgG2a upstream of CH1 (locus-specific) GGCGACCTGTAACAACTTGG r2a_CH1_fw rat IgG2a CH1 exon (locus-specific) TGTAGGAGCTTGGGTCCAGA m1_CH1_fw mouse IgG1 CH1 (locus-specific) GTGCCGACTTCAATGTGCTT mκ_LC_fw mouse kappa light chain (locus-specific) GTGCTTGTGTTCAGACTCCC blast_rv blasticidin resistance gene (HDR-specific) ATACATTGACACCAGTGAAGATGC ires_rv IRES (HDR-specific) GGCTTCGGCCAGTAACGTTA polyA_rv poly A tail (HDR-specific) CATAGAGCCCACCGCATCCC Run PCR products on a 1% (w/v) agarose gel and visualize DNA using Nancy-520 (1:15,000).
Integration of the insert in the target region is confirmed by the presence of a single band at the expected height for the engineered MIH5 [r2a>m2a and r2a>m2a(silent) (r2a_iso_fw + blast_rv, Figure 8D)], NLDC-145 (r2a>Fab’OTI and r2a>Fab’OTII (r2a_HC_fw + blast_rv, Figure 8E)), and NKI.B20/1 [mκ> mκ-srt-myc (mκ_LC_fw + ires_rv, Figure 8F)] cell lines.
The band at the correct height should be excised and gel-purified to perform Sanger sequencing and confirm correct integration.
Flow cytometry analysis of hybridoma supernatant
A flow cytometry screening can be performed to investigate the expression of e.g., a His-tag/Myc-tag, HC, LC, or a certain isotype.
For a first screen, incubate 50 μL of supernatant of the bulk population or the SCC with 50,000 target-expressing cells for 20 min at 4 °C. After two washes with PBA, proceed with secondary staining using the antibodies indicated in Table 3 to stain the desired part of the produced antibodies (isotype, tag, etc.).
Table 3. Secondary antibodies used to analyze hybridoma production by flow cytometry
Target Clone Fluorophore Company Cat. number Dilution 6×His-tag J095G46 PE Biolegend 362603 25 rat IgG2a HC MRG2a-83 PE Biolegend 407507 400 mouse IgG2a m2a-15F8 PE eBioscience 12-4817-82 500 For a second screen aimed at selecting high-producing clones, use a similar workflow but include serial dilutions of the supernatants of positive clones. It is advised to take along a dilution of known concentrations of the parental antibody to calculate the amount of antibody in the samples.
We modified r2a-producing MIH5 cell lines to produce m2a and m2a(silent) isotypes and incubated undiluted supernatant of SCC with PD-L1-expressing cells (Figure 8G). Flow cytometry analysis shows production of the original isotype by the initial cell line but not selected clones, and production of a m2a isotype with a His-tag, attesting for proper integration.
Western blot analysis of hybridoma supernatant
A flow cytometry screening can be performed to investigate the expression of e.g., His-tag/Myc-tag, heavy chain, light chain, or a certain isotype without requiring high numbers of target-expressing cells. Binding to the target needs to be separately confirmed on selected clones.
We typically conduct a first screen using a dot blot. For this, take 3 μL of the supernatant of each single cell clone and spot on a nitrocellulose membrane using a 96-well plate as a guide. Let the nitrocellulose membrane dry for 30 min and proceed with the blocking step and secondary staining of a general western blot protocol, using the antibodies indicated in Table 4 to stain the desired part of the produced antibodies (isotype, tag, etc.).
For a second screen, use diluted supernatant of positive clones to confirm production of the correct protein using a traditional western blot. Here, we analyzed the supernatant of NLDC-145 engineered cell lines to produce Fab’OTI and Fab’OTII using anti-rIgG2a (H+L) and anti-His-tag (Figure 8H). The results show production of a HC of 25 kDa bearing a His-tag over the wild-type (WT) HC of 50 kDa, confirming HDR insertion.
Alternatively, SDS-PAGE analysis can confirm production of the right product (Figure 8I). Here, we sequentially modified NKI.B20/1 parental cell line into a cell line producing a Fab’ fragment bearing two site-specific modification sites, on its heavy and light chains. Following the first and second step of modification, we respectively detected a shift downwards of the HC from 50 to 25 kDa (HC WT > HC Fab’-srt-his), and a shift upwards of the LC (LC WT > LC-srt-myc) (Figure 8I).
Table 4. Antibodies used to characterize presence of the right insert
Target Clone Fluorophore Company Cat. number Dilution rabbit anti 6×His-tag RM146 unconjugated Abcam AB14923 1,000 goat anti-rabbit IgG (H + L) polyclonal IRD800 LI-COR 926-32211 5,000 goat anti-rat IgG (H + L) polyclonal AF680 Thermo Fisher A-21096 5,000
Confirming production of a functional protein
Confirmation that the selected clone produces functional antibodies is paramount to validate the selected cell line.
Production of high levels of modified antibody
Based on genomic screen and confirmation at the protein level, one clone can be selected based on its production and growth rate and used to produce high levels of the modified antibody. For this, expand the selected clone to a 5-layer flask or CELLineCL 1000 bioreactors, following the manufacturer’s instructions.
Isolation of modified antibody
Isolation methods depend on the nature of the insert. The HDR template can be designed to include a purification tag such as His-tag or Myc-tag, for which affinity resins are available (e.g., Ni-NTA agarose). Antibodies that do not contain such tags can be isolated using e.g., protein G resin, in which case the media needs to be serum-free (CD hybridoma medium).
Functional assays
Assays needed to confirm antibody functionality typically depend on the nature of the modification, target, and intended use. Here, we provide examples of functional assays we conducted to validate the cell lines presented. These results illustrate that CRISPR/Cas9 knock in in the immunoglobulin locus conserves the specificity of the antibodies, and that the inserts are functional.
We generated MIH5 cell lines that produce mAbs of the m2a and m2a(silent) isotypes. To confirm functionality of the isotype, we conducted surface plasmon resonance analysis of purified mAbs on immobilized murine fragment crystallizable gamma receptors (FcyR) (Figure 8J). As expected, m2a antibodies bind to murine FcyR, and binding is abrogated with the mutation introduced in m2a(silent). In addition, we performed in vitro killing of PD-L1-expressing cancer cells opsonized with the different isotypes (Figure 8K). Incubation with whole blood from C57BL6/J mice showed specific lysis of target cells for m2a, but not r2a (WT) and m2a(silent). Similarly, in vivo killing of splenocytes opsonized with m2a or m2a(silent) (Figure 8L) reveals specific killing of m2a-treated cells over m2a(silent). Together, these results validate the functionality of the novel chimeric antibodies (van der Schoot et al., 2019).
The engineered NLDC-145 cell lines produce Fab’OTI and Fab’OTII that can be used to specifically deliver antigens to DEC-205-expressing dendritic cells (DCs). OTI and OTII antigens are respectively recognized by CD8 OTI and CD4 OTII primary T cells upon presentation on MHC molecules by DCs. We incubated DEC-205-expressing primary murine dendritic cells for 2 h with the constructs, short peptides (positive control, OVA257-264 or OVA323-339) that do not need processing, or OVA protein. We then exposed DCs to antigen-specific CD8 or CD4 T cells (Figure 8M-N) that were labeled with Cell Trace Violet (CTV). CD8 (Figure 8M) and CD4 (Figure 8N) T-cell activation was measured by means of proliferation (CTV dilution by flow cytometry), activation maker expression, and pro-inflammatory cytokine production in the supernatant [interleukin-2 (IL-2), interferon gamma (IFNγ) by ELISA]. CD8 and CD4 T cells proliferated and produced pro-inflammatory cytokines upon recognition of their cognate antigen by DCs exposed to short peptide and to the Fab’ antigen fusions. This confirmed that the Fab’ antigen fusion proteins retain their ability to be processed and presented by DCs, and potently induce T cell activation (unpublished data by Fennemann et al).
Lastly, we generated a NKI.B20/1 cell line that produces a Fab’ fragment bearing orthogonal site-selective modification sites on both HC and LC at their C-terminus (Le Gall et al., 2021). To confirm functionality of the orthogonal sites, we sequentially modified the HC to introduce fluorescein isothiocyanate and the LC to introduce an azide group, as confirmed on fluorescent SDS-PAGE (Figure 8O). We finally confirmed that site-specific modification did not alter binding to the target by performing a competitive binding assay on CD20-expressing target cells (Figure 8P).
Notes
We have successfully applied this protocol to various hybridoma cell lines. We advise operators to get a good sense of the growth rate, production levels, and viability of the target cell lines before starting genetic engineering. We found that the following details can help refining the procedure:
Ensuring quality of the parental cell line
Hybridoma may lose production capacity over time or have a low viability, which severely impairs the efficacy of the procedure. To address this, we advise operators to subclone the parental line by performing a limiting dilution (as described in section E) and to quantify viability and WT antibody production of SCCs to select a new monoclonal line. This ensures selection of a highly viable parental clone with high production. Performing subcloning on the parental cell line also confirms that SCC can grow. If viability of the parental line does not improve, adjust culture conditions, for instance by removing β-ME (ensure that antibody production is not impaired) or increasing FBS concentration in the medium, spinning down cells at 800 rpm for 10 min instead of 1,500 rpm for 5 min, or removing dead cells using a density gradient such as Lymphoprep.
Confirm sensitivity to antibiotic pressure of the parental line
Normally, hybridoma lines do not have resistance to selection antibiotics, and we do not need to perform antibiotic titration of mock-transfected hybridoma. It might not be the case for every cell line, and in some situations, titration needs to be performed on mock-transfected hybridoma. If this is the case, follow section D to mock-transfect the cell line, then perform titration as indicated in section C.
Adjusting concentration of the selection drug during the procedure
Cell death starts around day 2–3. For puromycin, cells should be dead by day 7, and for blasticidin by day 10. Upon antibiotic pressure, we typically observe massive cell death in both GFP and HDR-transfected plates. If this is not the case, the concentration of the selection drug is too low. This might happen even after titration, and operators should increase the concentration.
If cells do not grow out following antibiotic pressure
If transfected cells do not sustain antibiotic pressure, try the following solutions. Check hybridoma viability prior to transfection and GFP transfection efficacy. Sequence the target region to ensure that no mutations are present that could affect the gRNA/HDR efficiency. Perform a T7 assay to determine gRNA ability to cut the target locus and try other gRNAs if necessary. Test for mycoplasma contamination. Synchronize the cells prior to transfection by starving (low/no FBS). Incorporate a non-homologous end joining inhibitor such as nocodazole to favor HDR.
If SCCs do not grow out after limiting dilution
While we have never encountered this issue, some cell lines might not grow out after limiting dilution. To solve this, doubling the FBS concentration in the culture medium can rescue single cells. Additionally, using conditioned medium to perform limiting dilution can help single cells to survive. To generate conditioned medium, plate ~1 × 106 cells from the parental line in 10 cm Petri dishes. Twenty-four or forty-eight hours later, depending on the growth rate, centrifuge the supernatant and pass it through a 0.45 µm filter.
If resistant cells do not produce (only) modified antibodies
Among resistant clones, we usually find a high fraction of correctly edited clones producing high amounts of antibodies (50%–90%). We have found sporadic SCC that resist antibiotic pressure and show correct integration of the insert, but produce WT antibodies, which we attribute to exceptions in allelic exclusion. Typically, other SCCs will produce the right protein and should be selected instead.
However, if all SCCs surviving antibiotic pressure show integration of the insert in the target region by PCR but do not secrete modified antibodies, the HDR template might require adjustments. For instance, due to alternative splicing, SCCs can resist antibiotic pressure but produce both modified and WT antibodies, or only WT antibodies. To address this, remove splice acceptor sequences for exons downstream of the insert in 3′ Has, and insert a synthetic splice acceptor sequence (such as GCTAGCGATCGCAGGCGCAATCTTCGCATTTCTTTTTTCCAG) upstream of the synthetic exon. Especially if knocked in upstream of an exon, make sure that the insert is in frame with the variable sequence after splicing.
Table 5. Homology arms used in this protocol. Bold: mutated PAM sequences to inactivate Cas9 after successful HDR recombination. Alternatively, complete excision in the donor sequence is possible (e.g., Fab r2a_HC). Underlined: original splice acceptor site upstream of rIgG2a CH1 exon: remove or mutate in HDR template to prevent alternative splicing of variable region with the original isotype.
Homology arms 5′-3′ Sequence 5′ HA r2a_iso (rIgG2a WT > mIgG2a) AGAAAGATCTGAGTAGAACCAAGGTAAAAAGTGTGGGTAAAAACACATGTTCACAGGCCTGGCTGACATGATGCTGGGCACGTATGGAGGCAAAGTCAAGAGGGCAGTGTAAGGGCCAGAAGTGAATCCTGACCCAAGAATAGAGAGTGCTAAACCTACGTAGATCGAAGCCAACTAAAAAGACAAGCTACAAAACGAAGCTAAGGCCAGAGATCTTGGACTGTGAAGAGTTCAGAGAACCTAGGATCAGGAACCATTAGTAACAGGCCAAGGAAGATAGAAGCTGCCTAGGACTTGGCAAGAGCCAACATGGTTGGACTGGAAAAGAAAGGAGGAGACAGAAGACAGGAGAGATGTGCCAACTTGATTTTGGGCTTCACTGTTGTCCATACTGTGTGCAGCCATATGGCCCACAGATAACAGGTTTAGCCGAGGAACACAGATACCCACATTGGACAATGGTGGGGGAACACAGATACCCATACTACAGGGCTCTTTAGGGCATTTCCTGAAAGTGTACTAGGAGTGGGACTGGGCTCAAAGGGATTAGGTGTGATCTGGCCTGGTGAGGCTGACATTGGCAAGCCCAATGGTTGGGTGTTGCCTCCTCCATGT 3′ HA r2a_iso (rIgG2a WT > mIgG2a) TGTACAACTTGGGGAGGGTACAAAATGGAGGACTTGTAGGAGCTTGGGTCCAGACCTGTCAGACAAAATGATCACGCATACTTATTCTTGTAGCTGAAACAACAGCCCCATCTGTCTATCCACTGGCTCCTGGAACTGCTCTCAAAAGTAACTCCATGGTGACCCTGGGATGCCTGGTCAAGGGCTATTTCCCTGAGCCAGTCACCGTGACCTGGAACTCTGGAGCCCTGTCCAGCGGTGTGCACACCTTCCCAGCTGTCCTGCAGTCTGGACTCTACACTCTCACCAGCTCAGTGACTGTACCCTCCAGCACCTGGTCCAGCCAGGCCGTCACCTGCAACGTAGCCCACCCGGCCAGCAGCACCAAGGTGGACAAGAAAATTGGTGAGAGAACAACCAGGGGATGAGGGGCTCACTAGAGGTGAGGATAAGGCATTAGATTGCCTACACCAACCAGGGTGGGCAGACATCACCAGGGAGGGGGCCTCAGCCCAGGAGACCAAAAATTCTCCTTTGTCTCCCTTCTGGAGATTTCTATGTCCTTTACACCCATTTATTAATATTCT 5′ HA r2a_HC (rIgG2a WT > Fab’ fragment) CCTGGAACTCTGGAGCCCTGTCCAGCGGTGTGCACACCTTCCCAGCTGTCCTGCAGTCTGGACTCTACACTCTCACCAGCTCAGTGACTGTACCCTCCAGCACCTGGTCCAGCCAGGCCGTCACCTGCAACGTAGCCCACCCGGCCAGCAGCACCAAGGTGGACAAGAAAATTGGTGAGAGAACAACCAGGGGATGAGGGGCTCACTAGAGGTGAGGATAAGGCATTAGATTGCCTACACCAACCAGGGTGGGCAGACATCACCAGGGAGGGGGCCTCAGCCCAGGAGACCAAAAATTCTCCTTTGTCTCCCTTCTGGAGATTTCTATGTCCTTTACACCCATTTATTAATATTCTGGGTAAGATGCCCTTGCATCATGACATACAGAGGCAGACTAGAGTATCAACCTGCAAAAGGTCATACCCAGGAAGAGCCTGCCATGATCCCACACCAGAACCAACCTGGGGCCTTCTCACCTATAGACCATACTAACACACAGCCTTCTCTCTGCA 3′ HA r2a_HC (rIgG2a WT > Fab’ fragment) GGTAAGTCACTAGGACTATTACTCCAGCCCCAGATTCAAAAAATATCCTCAGAGGCCCATGTTAGAGGATGACACAGCTATTGACCTATTTCTACCTTTCTTCTTCATCTACAGGCTCAGAAGTATCATCTGTCTTCATCTTCCCCCCAAAGACCAAAGATGTGCTCACCATCACTCTGACTCCTAAGGTCACGTGTGTTGTGGTAGACATTAGCCAGAATGATCCCGAGGTCCGGTTCAGCTGGTTTATAGATGACGTGGAAGTCCACACAGCTCAGACTCATGCCCCGGAGAAGCAGTCCAACAGCACTTTACGCTCAGTCAGTGAACTCCCCATCGTGCACCGGGACTGGCTCAATGGCAAGACGTTCAAATGCAAAGTCAACAGTGGAGCATTCCCTGCCCCCATCGAGAAAAGCATCTCCAAACCCGAAGGTGGGAGCAGCAGGGTGTGTGGTGTAGAAGCTGCAGTAGGCCATAGACAGAGCTTGACTTAACTAGACTT 5′ HA m1_HC (mIgG1 WT > Fab’ fragment) CAGTATTGTCCAGATTGTGTGCAGCCATATGGCCCAGGTATAAGAGGTTTAACAGTGGAACACAGATGCCCACATCAGACAGCTGGGGGGCGGGGGTGAACACAGATACCCATACTGGAAAGCAGGTGGGGCATTTTCCTAGGAACGGGACTGGGCTCAATGGCCTCAGGTCTCATCTGGTCTGGTGATCCTGACATTGATAGGCCCAAATGTTGGATATCACCTACTCCATGTAGAGAGTCGGGGACATGGGAAGGGTGCAAAAGAGCGGCCTTCTAGAAGGTTTGGTCCTGTCCTGTCCTGTCTGACAGTGTAATCACATATACTTTTTCTTGTAGCCAAAACGACACCCCCATCTGTCTATCCACTGGCCCCTGGATCTGCTGCCCAAACTAACTCCATGGTGACCCTGGGATGCCTGGTCAAGGGCTATTTCCCTGAGCCAGTGACAGTGACCTGGAACTCTGGATCCCTGTCCAGCGGTGTGCACACCTTCCCAGCTGTCCTGCAGTCTGACCTCTACACTCTGAGCAGCTCAGTGACTGTCCCCTCCAGCACCTGGCCCAGCCAGACCGTCACCTGCAACGTTGCTCATCCGGCATGTAGCACCAAGGTAGACAAAAAGATA 3′ HA m1_HC (mIgG1 WT> Fab’ fragment) GGTGAGAGGACGTATAGGGAGGAGGGGTTCACTAGAGGTGAGGCTCAAGCCATTAGCCTGCCTAAACCAACCAGGCTGGACAGCCATCACCAGGAAATGGATCTCAGCCCAGAAGATCGAAAGTTGTTCTTCTCCCTTCTGGAGATTTCTATGTCCTTTACACTCATTGGTTAATATCCTGGGTTGGATTCCCACACATCTTGACAAACAGAGACAATTGAGTATCACCAGCCAAAAGTCATACCCAAAAACAGCCTGGCATGACCTCACACCAGACTCAAACTTACCCTACCTTTATCCTGGTGGCTTCTCATCTCCAGACCCCAGTAACACATAGCTTTCTCTCCACAGTGCCCAGGGATTGTGGTTGTAAGCCTTGCATATGTACAGGTAAGTCAGTAGGCCTTTCACCCTGACCCCAGATGCAACAAGTGGCCATGTTAGAGGGTGGCCCAGGTATTGACCTATTTCCACCTTTCTTCTTCATCCTTAGTCCCAGAAGTATCATCTGTCTTCATCTTCCCCCCAAAGCCCAAGGATGTGCTCACCATTACTCTGACTCCTAAGGTCACGTGTGTTGTGGTAGACATCAGCAAGGATGATCCCGAGGTCCAGTTCAGCTGGTTTGT 5′ HA mκ_LC (mκ LC > mκLC-tag) TCTGTCTGAAGCATGGAACTGAAAAGAATGTAGTTTCAGGGAAGAAAGGCAATAGAAGGAAGCCTGAGAATATCTTCAAAGGGTCAGACTCAATTTACTTTCTAAAGAAGTAGCTAGGAACTAGGGAATAACTTAGAAACAACAAGATTGTATATATGTGCATCCTGGCCCCATTGTTCCTTATCTGTAGGGATAAGCGTGCTTTTTTGTGTGTCTGTATATAACATAACTGTTTACACATAATACACTGAAATGGAGCCCTTCCTTGTTACTTCATACCATCCTCTGTGCTTCCTTCCTCAGGGGCTGATGCTGCACCAACTGTATCCATCTTCCCACCATCCAGTGAGCAGTTAACATCTGGAGGTGCCTCAGTCGTGTGCTTCTTGAACAACTTCTACCCCAAAGACATCAATGTCAAGTGGAAGATTGATGGCAGTGAACGACAAAATGGCGTCCTGAACAGTTGGACTGATCAGGACAGCAAAGACAGCACCTACAGCATGAGCAGCACCCTCACGTTGACCAAGGACGAGTATGAACGACATAACAGCTATACCTGTGAGGCCACTCACAAGACATCAACTTCACCGATTGTAAAGAGTTTTAATAGCAATGAATGCGGATCC 3′ HA mκ_LC (mκ LC > mκLC-tag) TGAAGACAAAGATCCTGAGACGCCACCACCAGCTCCCCAGCTCCATCCTATCTTCCCTTCTAAGGTCTTGGAGGCTTCCCCACAAGCGACCTACCACTGTTGCGGTGCTCCAAACCTCCTCCCCACCTCCTTCTCCTCCTCCTCCCTTTCCTTGGCTTTTATCATGCTAATATTTGCAGAAAATATTCAATAAAGTGAGTCTTTGCACTTGAGATCTCTGTCTTTCTTACTAAATGGTAGTAATCAGTTGTTTTTCCAGTTACCTGGGTTTCTCTTCTAAAGAAGTTAAATGTTTAGTTGCCCTGAAATCCACCACACTTAAAGGATAAATAAAACCCTCCACTTGCCCTGGTTGGCTGTCCACTACATGGCAGTCCTTTCTAAGGTTCACGAGTACTATTCATGGCTTATTTCTCTGGGCCATGGTAGGTTTGAGGAGGCATACTTCCTAGTTTTCTTCCCCTAAGTCGTCAAAGTCCTGAAGGGGGACAGTCTTTACAAGCACATGTTCTGTAATCTGATTCAACCTACCCAGTAAACTTGGCGAAGCAAAGTAGAATCATTATCACAGGAAGCAAAGGCAACCTAAATGTGCA
Recipes
Wash buffer
PBS + 0.5% BSA
PBA
PBS + 5% BSA + 0.03% NaN3
Culture medium
RPMI-1640 (formulation with HEPES) + 10% heat-inactivated FBS + 2 mM ultraglutamine-1 + 1% (v/v) antibiotic-antimycotic + freshly added 50 μM β-ME.
1× selection medium
RPMI-1640 (formulation with HEPES) + 10% heat-inactivated FBS + 2 mM ultraglutamine + 1% (v/v) antibiotic-antimycotic + freshly added 50 μM β-ME + freshly added selection antibiotic at pre-determined concentration.
Acknowledgments
M.V. was the recipient of ERC Starting Grant CHEMCHECK (679921) and a Gravity Program Institute for Chemical Immunology tenure track grant by NWO. F.A.S. was the recipient of an LUMC Strategic Fund (#049–19). This work was derived from two papers from our research group previously published by van der Schoot et al. (2019) and Le Gall et al. (2021). We thank Arthur E. H. Bentlage and Gestur Vidarsson (Sanquin, Amsterdam, The Netherlands) for collecting the surface plasmon resonance data (van der Schoot et al., 2019).
Competing interests
The authors declare no competing interest.
Ethics
Work described in this protocol was partially performed in animals, which was approved by the local authority for the Ethical Evaluation of Animal Experiments and Animal Welfare (Instantie voor Dierenwelzijn Radboudumc). Mice were kept in accordance with federal and state policies on animal research, and Annex III of the EU Directive (Directive 2010-63-EU).
References
- Adhikari, P., Zacharias, N., Ohri, R. and Sadowsky, J. (2020). Site-Specific Conjugation to Cys-Engineered THIOMABTM Antibodies. In: Tumey, L. (Ed.). Antibody-Drug Conjugates. Methods in Molecular Biology. pp. 51-69.
- Beers, S. A., Glennie, M. J. and White, A. L. (2016). Influence of immunoglobulin isotype on therapeutic antibody function. Blood 127(9): 1097-1101.
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A., et al. (2013). Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 339(6121): 819-823.
- Evers, M., Stip, M., Keller, K., Willemen, H., Nederend, M., Jansen, M., Chan, C., Budding, K., Nierkens, S., Valerius, T., et al. (2021). Anti-GD2 IgA kills tumors by neutrophils without antibody-associated pain in the preclinical treatment of high-risk neuroblastoma. J Immunother Cancer 9(10): e003163.
- Fennemann, F. L., Le Gall, C. M., van der Schoot, J. M. S., Ramos-Tomillero, I., Figdor, C. G., Scheeren, F. A., and Verdoes, M. (2021). Generation of dendritic cell targeted anti-cancer vaccines by CRISPR- Cas9-editing of the hybridoma immunoglobulin locus. In: Molecularly Defined Dendritic Cell-Targeted Vaccines for Cancer Immunotherapy. pp. 42-63.
- Le Gall, C. M., van der Schoot, J. M. S., Ramos-Tomillero, I., Khalily, M. P., van Dalen, F. J., Wijfjes, Z., Smeding, L., van Dalen, D., Cammarata, A., Bonger, K. M., et al. (2021). Dual Site-Specific Chemoenzymatic Antibody Fragment Conjugation Using CRISPR-Based Hybridoma Engineering. Bioconjug Chem 32(2): 301-310.
- Khoshnejad, M., Brenner, J. S., Motley, W., Parhiz, H., Greineder, C. F., Villa, C. H., Marcos-Contreras, O. A., Tsourkas, A. and Muzykantov, V. R. (2018). Molecular engineering of antibodies for site-specific covalent conjugation using CRISPR/Cas9. Sci Rep 8(1): 1760.
- Kim, H. K., Lee, S., Kim, Y., Park, J., Min, S., Choi, J. W., Huang, T. P., Yoon, S., Liu, D. R. and Kim, H. H. (2020). High-throughput analysis of the activities of xCas9, SpCas9-NG and SpCas9 at matched and mismatched target sequences in human cells. Nat Biomed Eng 4(1): 111-124.
- Kirkling, M. E., Cytlak, U., Lau, C. M., Lewis, K. L., Resteu, A., Khodadadi-Jamayran, A., Siebel, C. W., Salmon, H., Merad, M., Tsirigos, A., et al. (2018). Notch Signaling Facilitates In Vitro Generation of Cross-Presenting Classical Dendritic Cells. Cell Rep 23(12): 3658-3672 e3656.
- Köhler, G. and Milstein, C. (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256(5517): 495-497.
- Nanda, J.S. and Lorsch, J.R. (2014). Labeling a protein with fluorophores using NHS ester derivitization. Methods Enzymol 536: 87-94.
- van der Schoot, J. M. S., Fennemann, F. L., Valente, M., Dolen, Y., Hagemans, I. M., Becker, A. M. D., Le Gall, C. M., van Dalen, D., Cevirgel, A., van Bruggen, J. A. C., et al. (2019). Functional diversification of hybridoma-produced antibodies by CRISPR/HDR genomic engineering. Sci Adv 5(8): eaaw1822.
- Sharma, A., Subudhi, S. K., Blando, J., Scutti, J., Vence, L., Wargo, J., Allison, J. P., Ribas, A., and Sharma, P. (2019). Anti-CTLA-4 immunotherapy does not deplete FOXP3+ regulatory T cells (Tregs) in human cancers.Clin Cancer Res 25(4): 1233-1238.
- Waldor, M. K., Mitchell, D., Kipps, T. J., Herzenberg, L. A., and Steinman, L. (1987). Importance of immunoglobulin isotype in therapy of experimental autoimmune encephalomyelitis with monoclonal anti-CD4 antibody. J Immunol 139(11): 360-3664.
- Yamada, K., Shikida, N., Shimbo, K., Ito, Y., Khedri, Z., Matsuda, Y. and Mendelsohn, B. A. (2019). AJICAP: Affinity Peptide Mediated Regiodivergent Functionalization of Native Antibodies. Angew Chem Int Ed Engl 58(17): 5592-5597.
- Zhao, Y., Gutshall, L., Jiang, H., Baker, A., Beil, E., Obmolova, G., Carton, J., Taudte, S. and Amegadzie, B. (2009). Two routes for production and purification of Fab fragments in biopharmaceutical discovery research: Papain digestion of mAb and transient expression in mammalian cells. Protein Expr Purif 67(2): 182-189.
Article Information
Copyright
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Le Gall, C. M., Fennemann, F. L., van der Schoot, J. M. S., Scheeren, F. A. and Verdoes, M. (2023). CRISPR/Cas9-based Engineering of Immunoglobulin Loci in Hybridoma Cells. Bio-protocol 13(4): e4613. DOI: 10.21769/BioProtoc.4613.
- van der Schoot, J. M. S., Fennemann, F. L., Valente, M., Dolen, Y., Hagemans, I. M., Becker, A. M. D., Le Gall, C. M., van Dalen, D., Cevirgel, A., van Bruggen, J. A. C., et al. (2019). Functional diversification of hybridoma-produced antibodies by CRISPR/HDR genomic engineering. Sci Adv 5(8): eaaw1822.
Category
Immunology > Antibody analysis > Antibody modification
Cell Biology > Cell engineering > CRISPR-cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.