- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A CRISPR-based Strategy for Temporally Controlled Site-Specific Editing of RNA Modifications
(*contributed equally to this work) Published: Vol 13, Iss 3, Feb 5, 2023 DOI: 10.21769/BioProtoc.4607 Views: 2064
Reviewed by: David PaulKenji SugiyamaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2022

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Chemical modifications on RNA play important roles in regulating its fate and various biological activities. However, the impact of RNA modifications varies depending on their locations on different transcripts and cells/tissues contexts; available tools to dissect context-specific RNA modifications are still limited. Herein, we report the detailed protocol for using a chemically inducible and reversible platform to achieve site-specific editing of the chosen RNA modification in a temporally controlled manner by integrating the clustered regularly interspaced short palindromic repeats (CRISPR) technology and the abscisic acid (ABA)-based chemically induced proximity (CIP) system. The procedures were demonstrated using the example of inducible and reversible N6-methyladenosine (m6A) editing and the evaluation of its impact on RNA properties with ABA addition and reversal with the control of ABA or light.
Keywords: CRISPRBackground
More than 170 chemical modifications on RNA have been discovered, which contribute to the regulation of genetic information (Licht and Jantsch, 2016; Ontiveros et al., 2019; Boccaletto et al., 2022). Among all RNA modifications, some are known to be dynamic and reversible, fine-tuned by several endogenous writers, erasers, and reader proteins in cells, which are responsible to generate, remove, and interpret these RNA modifications, respectively (Wang and He, 2014; Xu and Liang, 2022). The most prevalent dynamic RNA modification, N6-methyladenosine (m6A), is deposited by methyltransferase-like 3 (METTL3) and other auxiliary proteins, such as methyltransferase-like 14 (METTL14) and Wilms’ tumor 1associated protein (WTAP) (Wang and He, 2014; Yang et al., 2018). The m6A erasers consist of the fat mass and obesity–associated protein and ALKB homologue 5 (ALKBH5), while heterogenous nuclear ribonucleoproteins and the YTH protein family recognize m6A and facilitate its function (N. Liu and Pan, 2016; Yang et al., 2018). With the rapid advance in the field of RNA epigenetics, consensus has been reached that RNA modifications play important roles in regulating its fate and orchestrating various associated biological processes. Nevertheless, the relationships between a particular RNA modification in specific contexts and the observed phenotypes are still elusive, due to limitations in the current strategies to dissect the functions of RNA modifications in a context-dependent and temporally controlled manner. Strategies to investigate RNA modifications relying on the genetic overexpression and knockdown or knockout of the corresponding enzymes/proteins generate global and irreversible changes, which complicate the functional studies of specific RNA modifications and the understanding of their direct biological consequences.
The last decade has witnessed the flourish of clustered regularly interspaced short palindromic repeats (CRIPSR), which can be reprogrammed for various biological and biomedical applications (Pickar-Oliver and Gersbach, 2019;Lo et al., 2022). There have been different studies using the nuclease inactive Cas (dCas) protein to recruit m6A writers, erasers, and readers to specific sites on RNA that are accessible for m6A editing (Rauch et al., 2018; X. M. Liu et al., 2019; Wilson et al., 2020). These methods achieved site-specific m6A editing, taking the advantage of the targeting ability from the combination of dCas protein and single guide RNA (sgRNA). Unfortunately, the temporal control in these RNA modifications editing processes remains lacking.
To address this critical issue, we recently reported a new strategy by integrating CRISPR with the abscisic acid (ABA)-based chemically induced proximity (CIP) system (Shi et al., 2022). The CRISPR-CIP integrated methods have been used to provide inducible and reversible control of epigenome editing (Chen et al., 2017) and site-specific m6A modification editing (Shi et al., 2022), under the regulation of small molecule ABA. The ABA-controlled inducible platform for m6A writing was designed via individually fusing the dCas13b protein and the m6A writer, METTL3, with PYL and ABI (two inducer-binding domains of ABA). The easy reprogramming nature of this platform makes it a promising tool to target various RNAs of interest by switching the target sequences of sgRNA to achieve site-specific m6A writing. In addition, the m6A writers can be replaced by erasers to achieve site-specific m6A removal, which implies a great potential for achieving editing of other RNA modifications by employing corresponding enzymes. We expect that this versatile inducible tool can be adapted for editing other RNA modifications, which opens a new door for targeted RNA modification editing in a temporally controlled manner.
In this protocol, we describe the experimental details for developing and evaluating the inducible and reversible m6A modification editing platform (Shi et al., 2022). The design of this platform is in the scheme below (Figure 1).
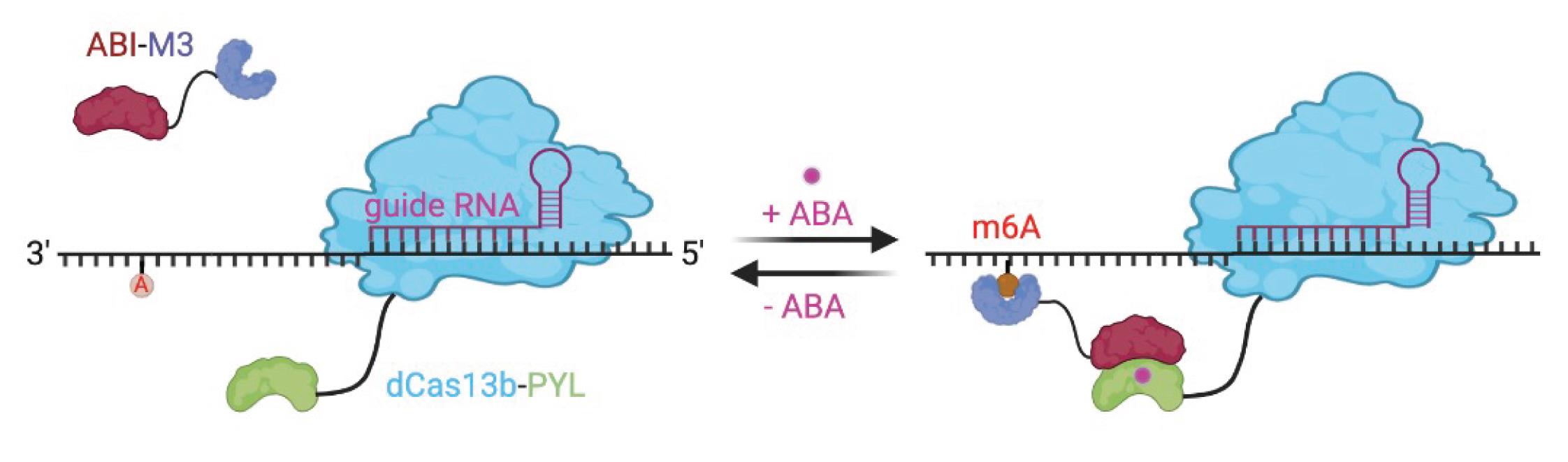
Figure 1. Reversible and inducible m6A editing platform. The targeted m6A editing is triggered by the addition of ABA, which can be reversed by ABA removal. The figure was created with Biorender.com.
Materials and Reagents
Universal pipet tips, 10 µL (VWR, catalog number: 76323-396)
Universal pipet tips, 200 µL (VWR, catalog number: 76323-390)
Universal pipet tips, 1,250 µL (VWR, catalog number: 76323-456)
Microcentrifuge tubes (VWR, catalog number: 87003-294)
1.7 mL culture tubes (VWR, catalog number: 60818-689)
50 mL centrifuge tube (Celltreat, catalog number: 229421)
BioDot pure8-strip PCR tubes (Dot Scientific, catalog number: 415-8PCR)
100 mm TC-treated cell culture dish (Falcon, catalog number: 353003)
Tissue culture plates, 6 well, sterile (VWR, catalog number: 10062-892)
Tissue culture plates, 12 well, sterile (VWR, catalog number: 10062-894)
Tissue culture plates, 24 well, sterile (VWR, catalog number: 10062-896)
500 mL solution bottle, sterile (Celltreat, catalog number: 229784)
500 mL bottle top filter, sterile (Celltreat, catalog number: 229717)
Universal pipet tips, 10 µL, filtered, pre-sterile (VWR, catalog number: 76322-528)
Universal pipet tips, 20 µL, filtered, pre-sterile (VWR, catalog number: 76322-134)
Universal pipet tips, 200 µL, filtered, pre-sterile (VWR, catalog number: 76322-150)
Universal pipet tips, 1,250 µL, filtered, pre-sterile (VWR, catalog number: 76322-528)
Disposable serological pipets, 2 mL (VWR, catalog number: 75816-104)
Disposable serological pipets, 5 mL (VWR, catalog number: 89130-896)
Disposable serological pipets, 10 mL (VWR, catalog number: 89130-898)
Disposable serological pipets, 25 mL (VWR, catalog number: 89130-900)
HEK293T cell (gift from Dr. Gerald R Crabtree at Stanford University Medical School)
HeLa cell (gift from Dr. Gerald R Crabtree at Stanford University Medical School)
Q5 High-Fidelity 2× Master Mix (New England BioLabs, catalog number: M0492S)
Monarch® PCR & DNA Cleanup kit (New England Biolabs, catalog number: T1030S)
NotI-HF restriction enzyme (New England BioLabs, catalog number: R3189)
EcoRI-HF restriction enzyme (New England BioLabs, catalog number: R3101S)
PmeI restriction enzyme (New England BioLabs, catalog number: R0560S)
BbsI restriction enzyme (New England BioLabs, catalog number: R0539S)
HindIII-HF restriction enzyme (New England BioLabs, catalog number: R3104S)
KpnI-HF (New England BioLabs, catalog number: R3142)
CutSmart buffer (New England BioLabs, catalog number: B7204)
Bst 2.0 DNA polymerases (New England BioLabs, catalog number: M0537S)
SplintR ligase (New England BioLabs, catalog number: M0375S)
ATP solution (100 mM) (Thermo Scientific, catalog number: R0441)
pCMV-dCas13-M3nls (Addgene, catalog number: 155366)
pC0043-PspCas13b-crRNA (Addgene, catalog number: 103854)
QIAquick Gel Extraction kit (QIAGEN, catalog number: 28704)
Agarose I (Thermo Fisher Scientific, catalog number: 17852)
T4 DNA ligase reaction buffer (New England BioLabs, catalog number: B0202A)
T4 DNA ligase (New England Biolabs, catalog number: M0202L)
In-Fusion Snap Assembly bundles for seamless DNA cloning kit (Takara, catalog number: 638952)
1 kb plus DNA ladder (New England BioLabs, catalog number: N3200)
T4 polynucleotide kinase (New England BioLabs, catalog number:M0201S)
TAE buffer (Tris-acetate-EDTA) (50×) (Thermo Fisher Scientific, catalog number: B49)
Ampicillin sodium salt (Sigma, catalog number: A9518)
Nuclease-free water (Sigma, catalog number: BCCG1748)
Dulbecco’s modified Eagle’s medium, high glucose (Sigma, catalog number: RNBK9791)
Opti-MEM reduced serum medium (OMEM), GlutaMax supplement (Gibco, catalog number: 51985034)
Fetal bovine serum, premium selected (R&D Systems, catalog number: S11550)
Lipofectamine 2000 transfection reagent (Invitrogen, catalog number: 11668019)
TRIzol reagent (Invitrogen, catalog number: 15596026)
Choloroform:isoamyl alcohol 24:1 (Sigma, catalog number: MKCM1807)
2-propanol (Sigma, catalog number: 19516)
Ethanol (Fisher Bioreagents, catalog number: BP28184)
RIPA buffer (Thermo Fisher Scientific, catalog number: 89901)
Protease inhibitor cocktail (Thermo Fisher Scientific, catalog number: 78430)
Dulbecco’s phosphate buffered saline (PBS) (Sigma, catalog number: D8537)
PierceTM 660 nm protein assay reagent (Thermo Fisher Scientific, catalog number: 22660)
2-mercaptoethanol (Sigma, catalog number: M6250)
4× Laemmli sample buffer (Bio-Rad, catalog number: 1610747)
EZ-Run prestained protein (Fisher Bioreagents, catalog number: BP36011)
4%–15% precast polyacrylamide gel (Bio-Rad, catalog number: 4561084)
Methanol (Fisher Scientific, catalog number: A412-4)
Non-fat milk (Bio-Rad, catalog number: 1705016)
Tween-20 Surfact-AmpsTM detergent solution (Thermo Scientific, catalog number: PI85113)
Anti-HA Tag monoclonal antibody (2-2.2.14) (Invitrogen, catalog number: 26183)
Anti-Flag Tag monoclonal antibody (M2) (Sigma, catalog number: F1804)
Anti-Gapdh antibody GAPDH (D16H11) XP® Rabbit mAb (Cell Signaling Technology, catalog number: 5174)
Anti-FOXM1(A-11) (Santa Cruz, catalog number: sc-271746)
Anti-SOX2 (E-4) (Santa Cruz, catalog number: sc-365823)
Anti-vinculin (E1E9V, 1:1,000) (Cell Signaling Technology, catalog number: 13901)
Anti-rabbit IgG HRP-linked antibody (Cell Signaling Technology, catalog number: 7074)
Anti-mouse IgG HRP-linked antibody (Cell Signaling Technology, catalog number: 7076)
ClarityTM Western ECL substrate (Bio-Rad, catalog number: 1705060)
Immuno-Blot PVDF membrane (Bio-Rad, catalog number: 1620177)
4% paraformaldehyde (Thermo Fisher Scientific, catalog number: J19943.K2)
TritonTM X-100 surfact-AmpTM detergent solution (Thermo Fisher Scientific, catalog number: 85111)
Alexa Fluor Plus 488 (Thermo Fisher Scientific, catalog number: 32723)
DAPI (Fisher Scientific, catalog number: NBP2311561)
Actinomycin D (Thermo Fisher Scientific, catalog number: 11805017)
RNA clean & concentrator-5 (Zymo Research, catalog number: R1014)
N6-Methyladenosine (m6A) (D9D9W) rabbit mAb (Cell Signaling Technology, catalog number: 56593)
PierceTM Protein G magnetic beads (Thermo Fisher Scientific, catalog number: 88848)
N6-Methyladenosine 5′-monophosphate sodium salt (m6A salt) (Sigma, catalog number: M2780-10MG)
Protease K solution, RNA grade (Invitrogen, catalog number: 25530049)
RNasin Plus ribonuclease inhibitor (Promega, catalog number: N2611)
Ultrapure bovine serum albumin (BSA), 50 mg/mL (Invitrogen, catalog number: AM2616)
Zinc chloride (ZnCl2) (Sigma, catalog number: Z0152)
0.5 M EDTA pH 8.0 (Invitrogen, catalog number: AM9260G)
Formaldehyde solution (Sigma, catalog number: F8775-500ML)
Anti-METTL3 antibody (Invitrogen, catalog number: 15073-1-AP)
Glycine (Fisher bioreagents, catalog number: BP181-1)
Magnesium chloride solution, 1 M (MgCl2) (Sigma, catalog number: M1028-100ML)
Sodium dodecyl sulfate (SDS) (Sigma, catalog number: L3771-500G)
5 M sodium chloride (NaCl) (Invitrogen, catalog number: AM9759)
1 M Tris-HCl, pH 7.0 (Sigma, catalog number: T1819-1L)
1 M Tris-HCl, pH 7.4 (Sigma, catalog number: T2663-1L)
IGEPAL-CA-630 (Sigma, catalog number: I8896)
Sodium acetate (3 M), pH 5.5, RNase-free (Invitrogen, catalog number: AM9740)
Pierce RIPA buffer (Thermo Fisher Scientific, catalog number: 89900)
Halt protease inhibitor cocktail (Thermo Scientific, catalog number: 1861279)
PowerUpTM SYBRTM Green Master Mix (Thermo Fisher, catalog number: A25778)
iScript cDNA Synthesis kit (Bio-Rad, catalog number: 1708891)
Abscisic acid (ABA) (Gold Biotechnology, catalog number: A-050-5)
Dimethyl sulfoxide (DMSO) (Sigma, catalog number: D8418-100 mL)
Tris base (Sigma, catalog number: T1503-500 g)
Agar (Sigma, catalog number: A6686)
LB media (see Recipes)
LB plate with ampicillin (see Recipes)
1× gel running buffer (see Recipes)
1× membrane transfer buffer (see Recipes)
1× TBST buffer (see Recipes)
Fragmentation buffer, 10× (see Recipes)
5× IP buffer for m6A-IP study (see Recipes)
m6A elution buffer (see Recipes)
Wash buffer for RNA-crosslink immunoprecipitation (see Recipes)
NT-2 buffer, 5× (see Recipes)
Protease K digestion buffer (see Recipes)
Equipment
Centrifuge 5424/5424R (Eppendorf, catalog number: EP5404000537)
New Brunswick I26/I26R stackable incubator shakers (Eppendorf, catalog number: VWR-89173-938)
Sorvall LYNX 6000 superspeed centrifuge (Thermo Scientific, catalog number: 75006590)
7900HT Fast Real-Time PCR system (Applied Biosystems, catalog number: 4329001)
ChemiDoc MP imaging system (Bio-Rad, catalog number: 17001402)
C1000 touch thermal cycler (Bio-Rad, catalog number: 1851148)
NanoDrop One spectrophotometer (Thermo Scientific, catalog number: ND-ONE-W4)
Bioruptor Pico sonication device (Diagenode, catalog number: B01060010)
Fluorescence microscopy (Agilent BioTek LFXSN, catalog number: BTFLX)
CriterionTM Blotter with wire electrodes (Bio-Rad, catalog number: 1704071)
Procedure
Cloning of dCas13b-PYL-HA plasmid
Amplify the dCas13b fragment with the template of pCMV-dCas13b-M3nls, using the designed primers via Q5 High-Fidelity 2× Master Mix according to the manufacturer’s protocol.
Amplify the PYL [pyrabactin resistance (PYR)/PYR1-like] fragment with the template of dCas9-PYL (a plasmid cloned by our former group member), using the designed primers for In-Fusion cloning via Q5 High-Fidelity 2× Master Mix according to the manufacturer’s protocol.
Run a 0.8% DNA agarose gel to purify the DNA fragments amplified above with the QIAquick Gel Extraction kit. The size of amplified fragment dCas13b is 3,147 bp and the size of PYL fragment is 560 bp.
Linearize 1 µg backbone plasmids dCas13b-ALKBH5-HA with the HindIII-HF and KpnI-HF restriction enzymes by incubating at 37°C for 2 h and purify the linearized vector by running a 0.8% DNA agarose gel. The size of the linearized backbone is 5,452 bp.
Use the In-Fusion master mix from the In-Fusion Snap Assembly bundles for seamless DNA cloning kit to insert the dCas13b and PYL fragments into the linearized vector in the step A4 to get the dCas13b-PYL-HA plasmids. The primers used for PCR and sequencing are listed in Table 1 and the scheme of cloning is shown in Figure 2.
Notes:
Set up the PCR reaction in 50 µL of reaction. Briefly, gently mix 10 µL of 5× Q5 reaction buffer, 1 µL of 10 mM dNTPs, 2.5 µL of forward primer (10 µM), 2.5 of µL reverse primer, 100 ng of template (1 µL), 0.5 µL of forward primer Q5 high-fidelity DNA polymerase, and 32.5 µL of nuclease-free water. Run the PCR reaction at 98°C for 30 s, 34 cycles of 98°C for 10 s + 60 °C for 30 s + 72°C for 90 s, and finally 72°C for 120 s.
There are many plasmids containing PYL fragments that can be obtained from Addgene for the purpose of amplification. For example: PYL1-nTEVp (catalog number: 119213).
Table 1. Information on primers used to clone and sequence the dCas13b-PYL-HA plasmid
Primers used for cloning and sequencing Sequences (5′-3′) Forward primer to amplify the dCas13b fragment cgtttaaacttaagcttGCCACCatgaaacggacagccgacgga Reverse primer to amplify the dCas13b fragment cttgagtagcggccgcgctctctggtgttgctgactc Forward primer to amplify the PYL fragment gagcgcggccgctactcaagacgaattcacccaa Reverse primer to amplify the PYL fragment CGTATGGGTAGGTACCgttcatagcttcagtgatcga Sequencing primer for the dCas13b-PYL-HA aagcagagctctctggctaac Sequencing primer for the dCas13b-PYL-HA tgcgagttcctgaccagcaca Sequencing primer for the dCas13b-PYL-HA gattacggcaagctgttcgac Sequencing primer for the dCas13b-PYL-HA agcggcaatgcccacggcaag Sequencing primer for the dCas13b-PYL-HA cccagacagatgttcgacaat Sequencing primer for the dCas13b-PYL-HA atcaccagcgagggcatgaag
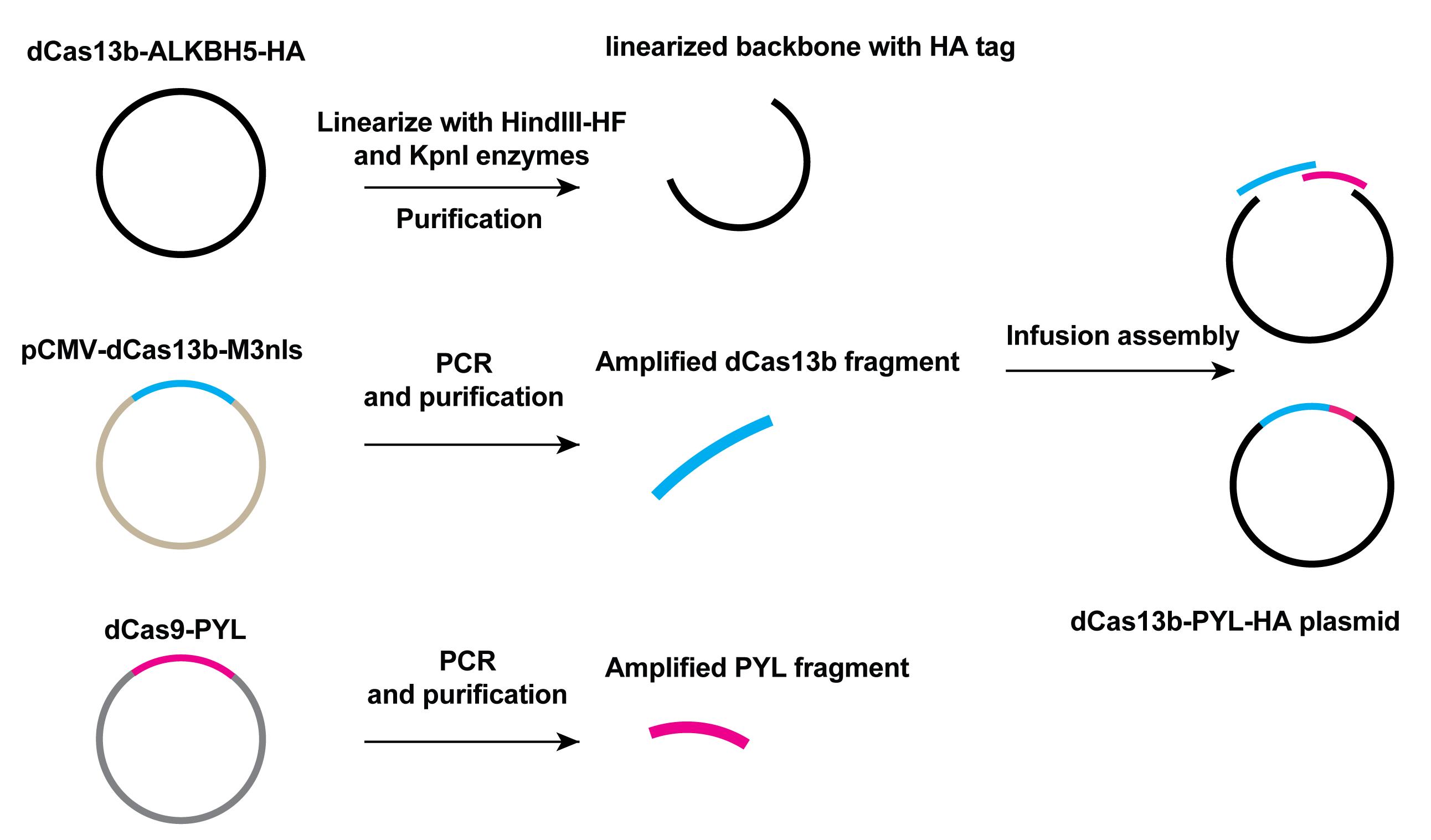
Figure 2. Scheme of cloning of the dCas13b-PYL-HA plasmid. The dCas13b-ALKBH5-HA plasmid was linearized to give the backbone of target plasmid. Amplified dCas13b and PYL fragments, as well as the linear backbone, were mixed together with In-fusion assembly master mix from the In-Fusion Snap Assembly bundles for seamless DNA cloning kit to generate dCas13b-PYL-HA plasmid.Cloning of ABI (abscisic acid insensitive 1)-M3nls plasmid
Amplify the ABI (abscisic acid insensitive 1) fragments with the designed primers and template of ABI-P300 plasmid (P300 is a lysine acetyltransferase and ABI-P300 plasmid is constructed by former members in our lab).
Amplify SV40 NLS (nuclear localization sequence) fragment with the template of dCas13b-M3nls using designed primers listed in Table 2 for In-Fusion cloning method.
Purify the amplified ABI fragment and SV40 NLS fragments by running a 0.8% DNA agarose gel. The size of the ABI fragment is 1,013 bp and the SV40 NLS is 122 bp.
Linearize the 1 µg backbone plasmids pCMV-dCas13-M3nls with EcoRI and NotI restriction enzymes by incubating at 37°C for 2 h to get the linearized vector containing M3nls fragment.
Purify the linearized vector by 0.8% DNA agarose gel electrophoresis. The linearized vector size is 4,365 bp. Use 1 kb plus DNA ladder as the size marker.
Use the In-Fusion master mix from the In-Fusion Snap Assembly bundles for seamless DNA cloning kit to insert the SV40 NLS and ABI fragments into the linearized vector according to the manufacturer’s protocol, to generate ABI-M3nls plasmid construct. The primers used for PCR and sequencing are listed in Table 2 and the scheme of cloning is shown in Figure 3.
Notes:
The PCR setup condition is the same as described in section A.
There are many plasmids containing ABI fragments that can be obtained from Addgene for the purpose of amplification. For example: ABI-cTEVp (catalog number: 19214).
Table 2. Information of primers used to clone and sequence the ABI-M3nls plasmid
Primers used for cloning and sequencing Sequences (5′-3′) Forward primer to amplify the SV40 NLS fragment tagagatccgcggccgctaatacgactcactataggga Reverse primer to amplify the SV40 NLS fragment ctttgtagtcgcgcgcgactttccgcttcttctttgg Forward primer to amplify the ABI fragment Agtcgcgcgcgactacaaagaccatgacggtgattataaagatcatgacatcg attacaaggatgacgatgacaaggtccccctgtatgggttcacc
Forward primer to amplify the ABI fragment tcttgggctcgaattcgctgccgtcggcggttcttttggatcccttcaggtccacgacgacgac Sequencing primer for the ABI-M3nls caactccgccccattgacgca Sequencing primer for the ABI-M3nls ctcctgagaccgtgggctcta
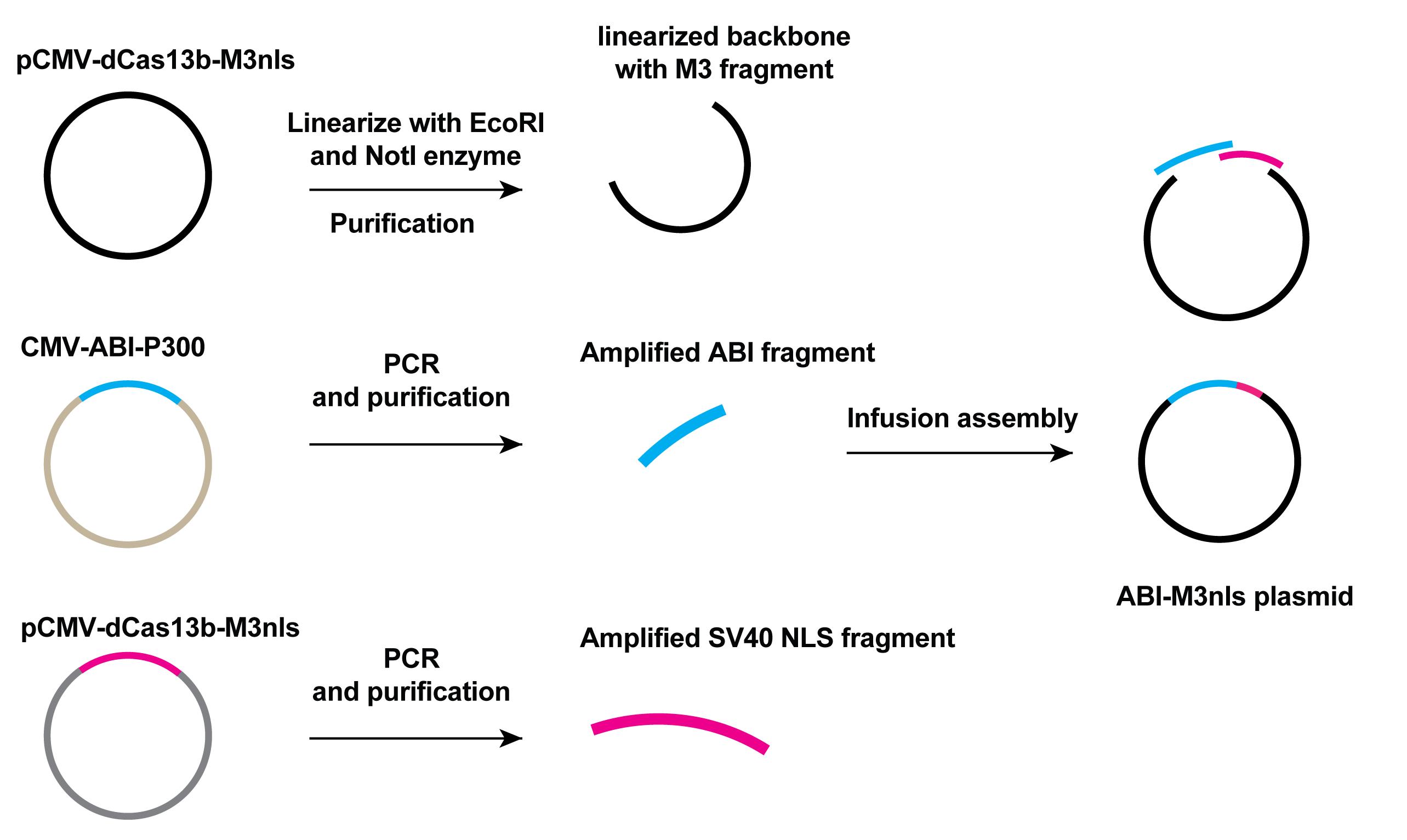
Figure 3. Scheme of cloning of the ABI-M3nls plasmid. The pCMV-dCas13b-M3nls plasmid was linearized to give the backbone of target plasmid. Amplified ABI and NLS fragments as well as the linear backbone were mixed together with In-fusion master mix from the In-Fusion Snap Assembly bundles for seamless DNA cloning kit to generate ABI-M3nls plasmid.
Cloning of sgRNA
For sgRNA cloning, anneal the forward and reverse spacer sequences with overhang to obtain the sgRNA spacer DNA fragments in an 18 µL reaction. Then, phosphorylate the annealed insert with T4 polynucleotide kinase according to the manufacturer’s protocol.
Linearize 1 µg of pC0043-PspCas13b-crRNA backbone vector with BbsI restriction enzyme by incubating at 55°C for 2 h and purify the linearized vector with Monarch® PCR & DNA Cleanup kit.
Ligate the sgRNA spacer inserts with the linearized sgRNA backbone with T4 ligase according to the manufacturer’s protocol to obtain the sgRNA targeting the RNA of interest. The spacer sequences of sgRNA are listed in Table 3 and the scheme of sgRNA cloning is shown in Figure 4.
Note: The distance of sgRNA spacer from the targeting site can impact the editing efficiency; we chose the sgRNA with its’ 3’-end two nucleotides (nts) away from the targeting site after screening. We suggest the audience to screen the optimal sgRNA for different targets.
Table 3. Information of spacers of targeting sgRNA
Spacer of sgRNA targeting RNA of interest Sequence (5′-3′) Actb gRNA-2 TCCATCGTCCACCGCAAATGCTTCTAGGCG Actb gRNA-8 GGCCCCTCCATCGTCCACCGCAAATGCTTC Actb gRNA-22 AGTATGACGAGTCCGGCCCCTCCATCGTCC Gapdh gRNA-8 GGGAAACTGTGGCGTGATGGCCGCGGGGCT Foxm1 gRNA ATGTTTCTCTGATAATGTCCCCAATCATAC Sox2 gRNA AACGGCACACTGCCCCTCTCACACATGTGA MALAT gRNA-1 AACGGAAGTAATTCAAGATCAAGAGTAATT MALAT gRNA-50 GAAGGCCTTAAATATAGTAGCTTAGTTTGA MALAT gRNA-100 ATTTAAAAAAAACTAAGGCAGAAGGCTTTT 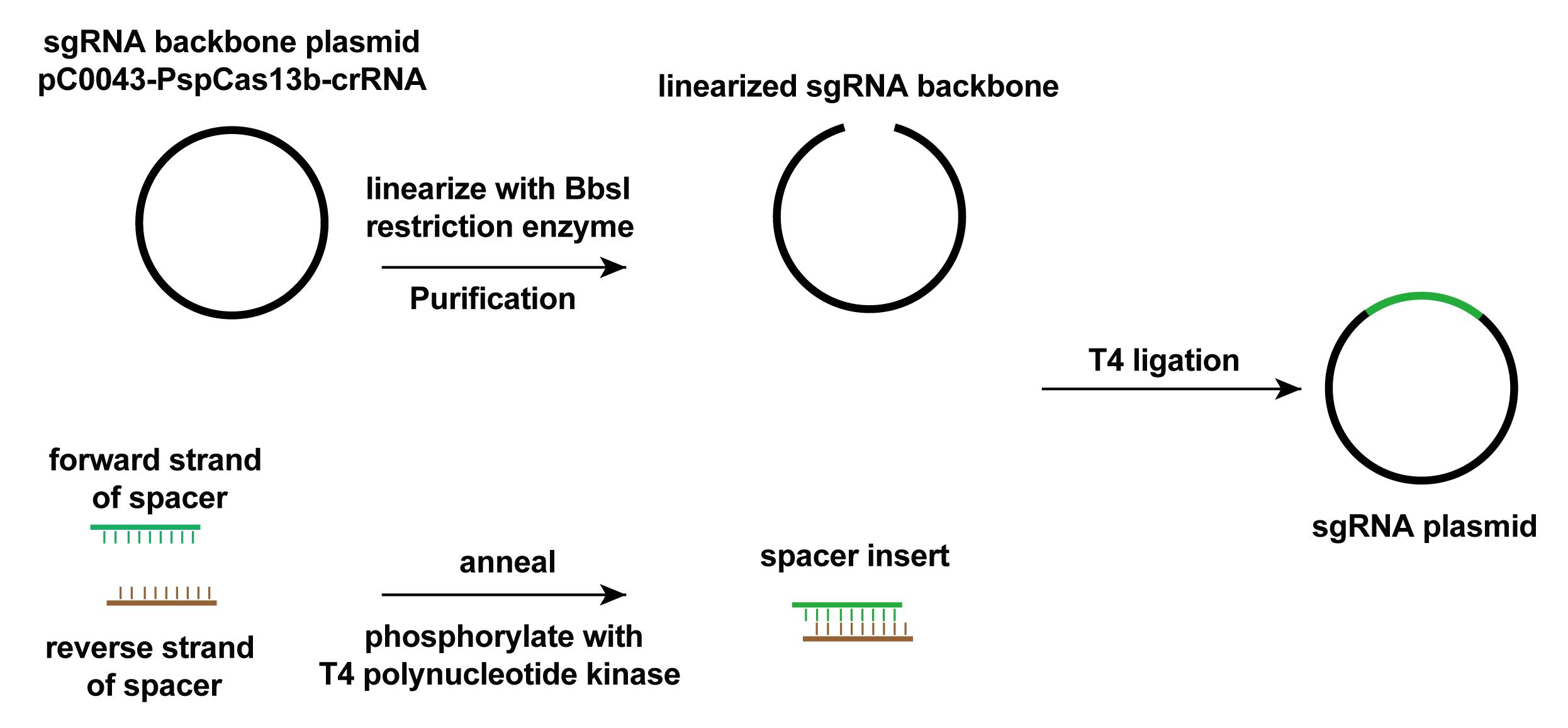
Figure 4. Scheme of sgRNA cloning. The pC0043-PspCas13b-crRNA plasmid was linearized to give the backbone of target plasmid. Annealed and phosphorylated spacer inserts and the linear backbone were mixed together with In-fusion master mix from the In-Fusion Snap Assembly bundles for seamless DNA cloning kit to generate sgRNA plasmid.
Study the expression of plasmids by western blot
Seed 250,000 HEK293T or HeLa cells in each well of a 6-well plate and incubate for 16 h to reach a confluency of 75%–80%.
Transfect cells with 1.5 µg of each plasmid (dCas13b-PYL-HA, Flag-ABI-METTL3, or Flag-ABI-ALKBH5) with 3 µL of lipofectamine 2000 and incubate for 24 h.
Remove the cell culture media and wash cells with DPBS.
Lyse cells with 200 µL of ice-cold RIPA buffer with 2 µL of protease inhibitor cocktail for 30 min on rotation at 4°C.
Spin down the cell debris via centrifugation at 12,000 × g for 15 min at 4°C.
Save the supernatant and measure the protein concentration with the PierceTM 660 nm protein assay reagent according to the manufacturer’s protocol.
Denature 20 µg of protein extracted from cells transfected with the different plasmids described in step D2 by incubation at 95°C for 5 min with the addition of 4× Laemmli sample buffer supplemented with 2-mercaptoethanol according to the manufacturer’s protocol.
Prepare the gel electrophoresis setup and load the samples or EZ-Run prestained protein (marker) into the wells of a 4%–15% precast polyacrylamide gel.
Run the gel electrophoresis in 1× gel running buffer at 60 V for 5 min, then change the voltage to 120 V and run for another 60 min.
Prepare the Immuno-Blot PVDF membrane transfer setup and run the transfer process with 10 mA current for 16 h.
Block the membrane with 5% non-fat milk in TBST buffer for 1 h.
Dilute the primary antibodies (anti-HA, anti-Flag, anti-GAPDH, anti-FOXM1, anti-SOX2, and anti-vinculin) in 1% TBST buffer (1:1,000 dilution) and incubate with the membrane at room temperature for 2 h.
Wash the membrane three times with TBST buffer.
Dilute the secondary antibody in 1% TBST buffer (anti-rabbit, anti-mouse) (1:2,000 dilution) and incubate with the membrane at room temperature for 1 h.
Wash the membrane three times with TBST buffer.
Prepare the ECL substrate by combining the two components in a 1:1 ratio.
Image each membrane with 200 µL of prepared ECL substrate using the ChemidocTM touching imaging system.
Study the localization of inducible m6A editors in cells via immunofluorescence imaging
Seed 250,000 HEK293T or HeLa cells in a 6-well plate and incubate for approximately 16 h to reach 75%–80% confluency.
Transfect cells with 1.5 µg of dCas13b-PYL and ABI-METTL3 or 1.5 µg of dCas13b-PYL and ABI-ALKBH5 and incubate for 24 h. The ratio of plasmids to lipofectamine 2000 is 100 ng:0.2 µL.
Fix cells with 4% paraformaldehyde for 10 min.
Treat the above fixed cells with 0.1% TritonTM X-100 for 15 min for permeabilization.
Block the cells with 1% ultrapure bovine serum albumin for 1 h at room temperature.
Incubate cells with anti-HA or anti-Flag antibody (1:1,000 dilution) at 4°C overnight.
Incubate cells with Alexa Fluor Plus 488 for 1 h at room temperature. In addition, add DAPI into fixed cells and incubate for 30 min.
Wash the cells three times with PBS and obtain fluorescence images using the fluorescence microscope with the objective lens (10×).
RNA extraction for m6A-immunoprecipitation
Seed 2,000,000 cells into 100 mm dishes and incubate for approximately 16 h to reach 75%–80% confluency.
Transfect cells with 6 µg of dCas13b-PYL plasmid and 6 µg of ABI-X (X refers to M3, M3*, ALK, or ALK*) plasmid and 4 µg of PspCas13b guide RNA plasmid using lipofectamine 2000. (Set condition of 6 µg of dCas13b-PYL, dCas13b-M3, or dCas13b-ALK plasmid with 4 µg of sgRNA as negative and positive control.) The ratio of plasmids to lipofectamine 2000 is 100 ng:0.2 µL.
Add 100 µM ABA or DMSO into cells transfected with dCas13b-PYL+ABI-M3+gRNA and incubate for 24 h.
Remove cell culture media in each group with different transfection and treatment and add 2 mL of TRIzol reagent to the 100 mm culture dish to lyse the cells.
Pipette the lysate up and down several times to homogenize.
Incubate for 5 min on ice to allow complete dissociation of nucleoprotein complexes.
Add 0.2 mL of choloroform:isoamyl alcohol 24:1 per 1 mL of TRIzol reagent used for lysis, thoroughly mix by shaking, and incubate for 2–3 min.
Centrifuge the sample for 15 min at 12,000 × g and 4°C (the mixture separates into a lower red phenol-chloroform, a white interphase, and a colorless upper aqueous phase)
Transfer the aqueous phase containing RNA by angling the tube 45o and gently transferring the upper layer solution into a new centrifuge tube.
Add 0.5 mL of 2-propanol to the aqueous phase per 1 mL of TRIzol Reagent used for lysis and incubate on ice for 10 min.
Centrifuge for 10 min at 12,000 × g and 4°C. (Total RNA precipitate forms a white gel-like pellet at the bottom of the tube.)
Discard the supernatant with a pipette.
Resuspend the pellet in 1 mL of 75% ethanol per 1 mL of TRIzol reagent used for lysis.
Centrifuge for 10 min at 10,000 × g and 4°C.
Discard the supernatant with a pipette.
Vacuum or air dry the RNA pellet for 5–10 min.
Resuspend the pellet in 200 µL of nuclease-free water by pipetting up and down and incubate for 5–10 min at room temperature for RNA to dissolve.
Investigation of m6A level by m6A-immunoprecipitation
Seed 2,000,000 cells into 100 mm dishes and incubate for approximately 16 h to reach 75%–80% confluency.
Transfect cells with 6 µg of dCas13b-PYL plasmid and 6 µg of ABI-X (X refers to M3, M3*, ALK, or ALK*) plasmid and 4 µg of PspCas13b guide RNA plasmid using lipofectamine 2000. (Set condition of 6 µg of dCas13b-PYL, dCas13b-M3, or dCas13b-ALK plasmid with 4 µg sgRNA as negative and positive control.) The ratio of plasmids to lipofectamine 2000 is 100 ng:0.2 µL.
Incubate cells for 24 h and replace cell culture media with ABA at different concentrations for another 24 h of incubation before harvesting for RNA extraction with TRIzol reagent. (For studying the reversible m6A writing, this step was done after gently washing the cell once with 10 mL of PBS, replacing the ABA-containing media with ABA-free fresh media, and incubating for another 24 h.)
Extract RNA following the procedures described above.
Dispense RNA into 1 µg/µL concentration for fragmentation.
Fragment 180 µg RNA with 20 µL of fragmentation buffer (see Recipes) in 10 PCR tubes equally at 94°C for 1 min. (Proceed as quickly as possible.)
Add 2 µL of 0.5 M EDTA to each reaction (18 µL) immediately to terminate fragmentation on ice. (The addition of EDTA should be quick to avoid excessive fragmentation, which will generate improper size of the RNA fragments.)
Collect fragmented RNA into a new 1.7 mL tube.
Add 22 µL of sodium acetate (3 M, pH 5.5) and 550 µL of 100% ethanol, then incubate at -80°C overnight.
Pellet RNA with centrifugation at 15,000 × g for 30 min at 4°C and discard the supernatant.
Wash the RNA pellet with 1 mL of 75% ethanol.
Centrifuge again at 15,000 × g for 15 min at 4°C.
Carefully remove the supernatant and let the RNA air-dry; use 150 µL of nuclease-free water to dissolve RNA.
Prepare the m6A-immunoprecipitation (m6A-IP) reaction as below.
Component Volume (µL) Fragmented RNA 120 RNasin Plus ribonuclease inhibitor (40 U/µL) 4 IP buffer, 5× 80 m6A-specific antibody (~0.5 mg/mL)
Nuclease-free water
10
186
Total volume (µL) 400 Incubate the m6A-IP reaction on a rotator at 4°C for 2 h.
While the samples are incubating, wash 40 µL of recombinant protein G beads twice with 1× IP buffer (diluted five times from the 5× IP buffer, see Recipes) for each IP reaction.
Resuspend the beads in 1 mL of 1× IP buffer supplemented with BSA (0.5 mg/mL) at 4°C for 2 h.
Incubate the IP reaction with beads at 4°C overnight on rotation.
Wash beads with 1 mL of 1× IP buffer three times and remove the buffer.
Add 100 µL of m6A elution buffer (with RNasin, see Recipes) to the sedimented beads and incubate the mixture for 1 h with continuous shaking at 4°C.
Spin down the beads carefully and save the supernatant containing eluted RNA fragments.
Repeat the elution step again and combine the 200 µL eluted RNA together.
Purify RNA with RNA clean & concentrator-5 and dissolve RNA in 15 µL of nuclease-free water according to the manufacturer’s protocol.
Perform reverse-transcription PCR (RT-PCR) with 12 μL of RNA after IP or 2 μL of input RNA (RNA before IP) to synthesize the cDNA using the iScript cDNA Synthesis kit according to the manufacturer’s protocol. Analyze the RNA level with PowerUpTM SYBRTM Green Master Mix according to the manufacturer’s protocol. The primers used for qPCR are listed in Table 4 and the scheme for the investigation of m6A level on target RNA is in Figure 5.
Table 4. Information of primers used in the RT-qPCR assay to study the m6A enrichment and enzyme recruitment
Primers used for RT-qPCR in this study Sequences (5′-3′) Actb-m6A-Foward AGATGTGGATCAGCAAGC Actb-m6A-Reverse TCATCTTGTTTTCTGCGC Gapdh-m6A-Forward CATCACTGCCACCCAGAAGA Gapdh-m6A-Reverse CAGTAGAGGCAGGGATGATGTT Foxm1-m6A-Forward TGCCCAGATGTGCGCTATTA Foxm1-m6A- Reverse CTTCTCAAGCCTCCACCTGA Sox2-m6A-Forward GGCCATTAACGGCACACTG Sox2-m6A-Reverse TCTTTTGCACCCCTCCCATT MALAT1-m6A- Forward CGTAACGGAAGTAATTCAAG MALAT1-m6A- Reverse GTCAATTAATGCTAGTCCTC CYB5A-m6A- Forward GTTTTAAGGGAACAAGCTGGAG CYB5A-m6A- Reverse TCCACCAACTGGAACTAGAATC CTNNB1-m6A- Forward TGGATTGATTCGAAATCTTGCC CTNNB1-m6A- Reverse GAACAAGCAACTGAACTAGTCG MYC-m6A- Forward CAGCTGCTTAGACGCTGGATT MYC-m6A- Reverse GTAGAAATACGGCTGCACCGA 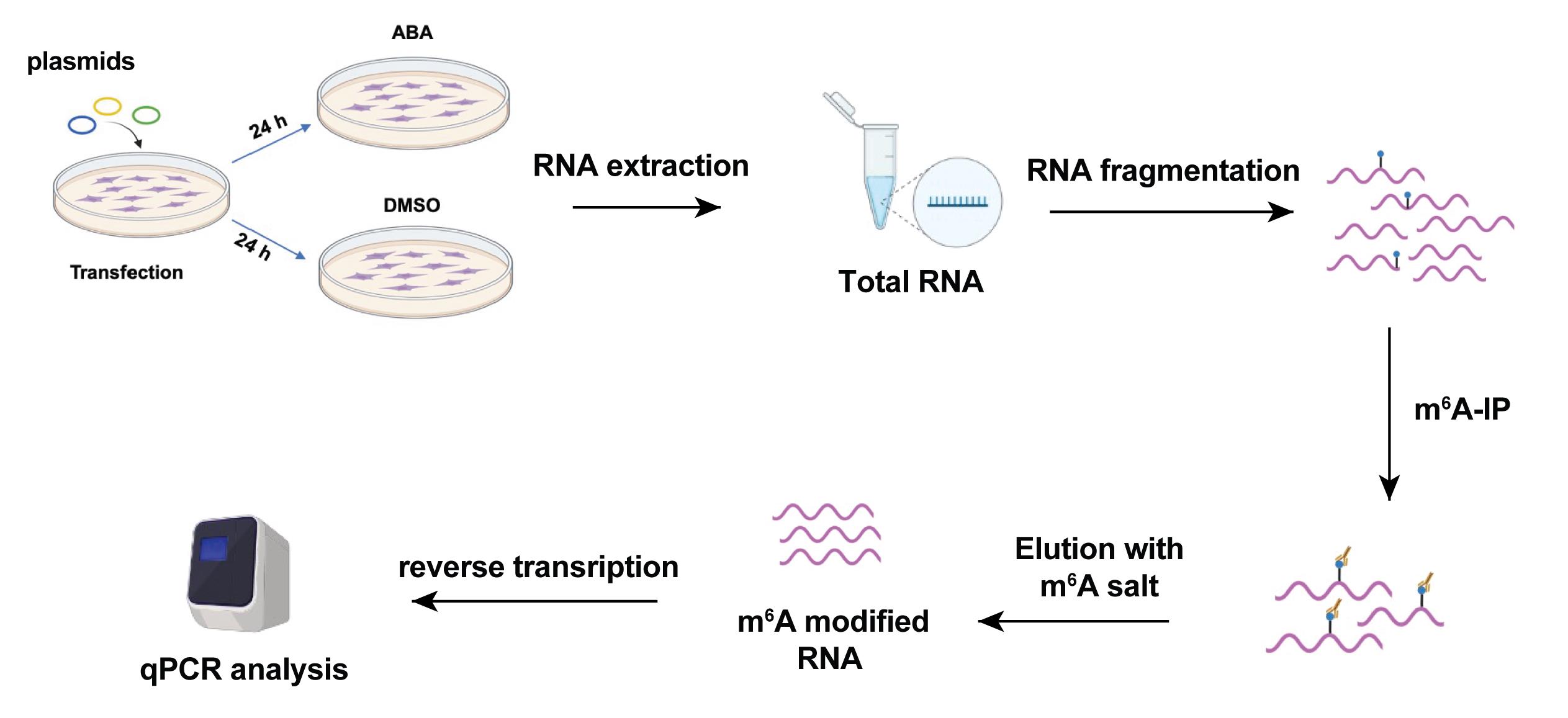
Figure 5. Scheme of the study of m6A investigation assay. The cartoon was created with Biorender.com.
mRNA stability assay
Seed 50,000 HEK293T or HeLa cells into a 24-well plate and incubate for approximately 16 h to reach 80% confluency.
Transfect the cells with DNA plasmids expressing different components of ABA-induced m6A modification system with lipofectamine 2000 for 24 h (0.9 μg of dCas13b-PYL + 0.6 μg of gRNA, 0.9 μg of dCas13b-M3 + 0.6 μg of gRNA, 0.9 μg of dCas13b-PYL + 0.9 μg of ABI-M3 + 0.6 μg of gRNA). The ratio of plasmids to lipofectamine 2000 is 100 ng:0.2 µL.
Add 100 µM ABA or DMSO into cells transfected with dCas13b-PYL+ABI-M3+gRNA and incubate for 24 h.
Treat cells with 5 µg/mL actinomycin D and harvest cells at specific time points (e.g., 0, 1, 3, and 6 h). (For studying the reversible effect on RNA stability, this step was done after gently washing the cell once with 5 mL of PBS, replacing the ABA-containing media with ABA-free fresh media, and incubating for another 24 h.)
Extract total RNA with TRIzol reagent and purify according to the manufacturer’s protocol.
Reverse-transcribe the RNA into cDNA with the iScript cDNA Synthesis kit.
Perform qPCR assays to study the mRNA levels of target transcripts (ACTB, GAPDH, SOX2, and FOXM1) with the PowerUpTM SYBRTM Green Master Mix according to the manufacturer’s protocol. The primers used for qPCR are listed in Table 5.
Table 5. Information of primers used in RT-qPCR to study mRNA level
Primers used in this study Sequences (5′-3′) Actb- Forward TCCCAAGTCCACACAGG Actb- Reverse CACGAAGGCTCATCATTCAAA Gapdh- Forward GGTGTGAACCATGAGAAGTATGA Gapdh- Reverse GAGTCCTTCCACGATACCAAAG Foxm1- Forward CAATGGCAAGGTCTCCTTCT Foxm1- Reverse GGTAGCAGTGGCTTCATCTT Sox2- Forward AGACGCTCATGAAGAAGGATAAG Sox2- Reverse TCATGTGCGCGTAACTGT
Note: The impact of installed m6A modification is in a context-dependent manner, which means that different results of stabilization or destabilization can be observed depending on the target RNA. In the cases studied in Shi’s work, the deposited m6A destabilize the ACTB mRNA, FOXM1 mRNA, and SOX2 mRNA, but no destabilization was found of GAPDH.
Enrichment of METTL3 on targeted A1216 of ACTB mRNA
Seed 250,000 cells into a 6-well plate and incubate for approximately 16 h to reach 75% confluency.
Transfect cells with 1.5 µg of dCas13b-PYL plasmid and 1.5 µg of ABI-M3 plasmid and 1 µg of PspCas13b guide RNA plasmid using lipofectamine 2000. (Set condition of 1.5 µg of dCas13b-PYL with 1 µg sgRNA as negative control.) The ratio of plasmids to lipofectamine 2000 is 100 ng:0.2 µL.
Incubate cells for 24 h and replace cell culture media with 100 µM of ABA for another 24 h incubation.
Add 35 µL formaldehyde to each well and incubate for 10 min for crosslinking.
Add 167 µL glycine (0.9 mg/mL) to each well and incubate for 5 min.
Remove the media in each well and wash cell with 1 mL of ice-cold DPBS twice.
Add another 167 µL of DPBS supplemented with protease inhibitor cocktail and collect all the cells into a new tube using a scraper.
Centrifuge cells at 5,000 × g for 5 min at 4°C.
Discard supernatant, resuspend cells with 220 µL of RIPA buffer (with protease inhibitor cocktail and RNasin) and incubate for 10 min on ice.
Sonicate cells in RIPA buffer for 2 min (30 s on/30 s off for one repeat) with the Bioruptor Pico sonication device.
Centrifuge at 16, 000 × g for 20 min at 4°C.
Save supernatant for immunoprecipitation or input and store at -80°C.
Wash 10 µL of Protein G magnetic beads with 1 mL of wash buffer (see Recipes) twice for each IP reaction in a 1.7 mL centrifuge tube.
Resuspended the beads in 200 µL of wash buffer with 1 µg of anti-METTL3 antibody and 3 µL of RNasin Plus ribonuclease inhibitor and incubate at room temperature for 2 h on rotation for the RNA-protein immunoprecipitation.
Remove the supernatant on a magnetic rack and wash the beads three times with 200 µL of wash buffer.
Add 200 µL of the supernatant saved in step I12 into the beads and incubate at 4°C overnight on rotation.
Add the remaining lysate supernatant to each antibody bead reaction.
Incubate all tubes on a rotating wheel overnight at 4°C on rotation.
Remove the supernatant on a magnetic rack and wash beads with 200 µL of wash buffer.
Add 100 µL of protease K digestion buffer to the beads after IP or 10 µL input.
Incubate tubes at 65°C for 3 h with shaking to digest the proteins.
Spin down all tubes and collect the supernatants on a magnetic rack.
Use RNA clean & concentrator-5 kit for RNA purification and dissolve RNA in 15 µL of nuclease-free water according to the manufacturer’s protocol.
Reverse-transcribe 12 μL of RNA after IP or input into cDNA and analyze the RNA level with PowerUpTM SYBRTM Green Master Mix according to the manufacturer’s protocol. The primers used in the qPCR assay are listed in Table 4 and the scheme for the study of enrichment of the METTL3 enzyme is in Figure 6.
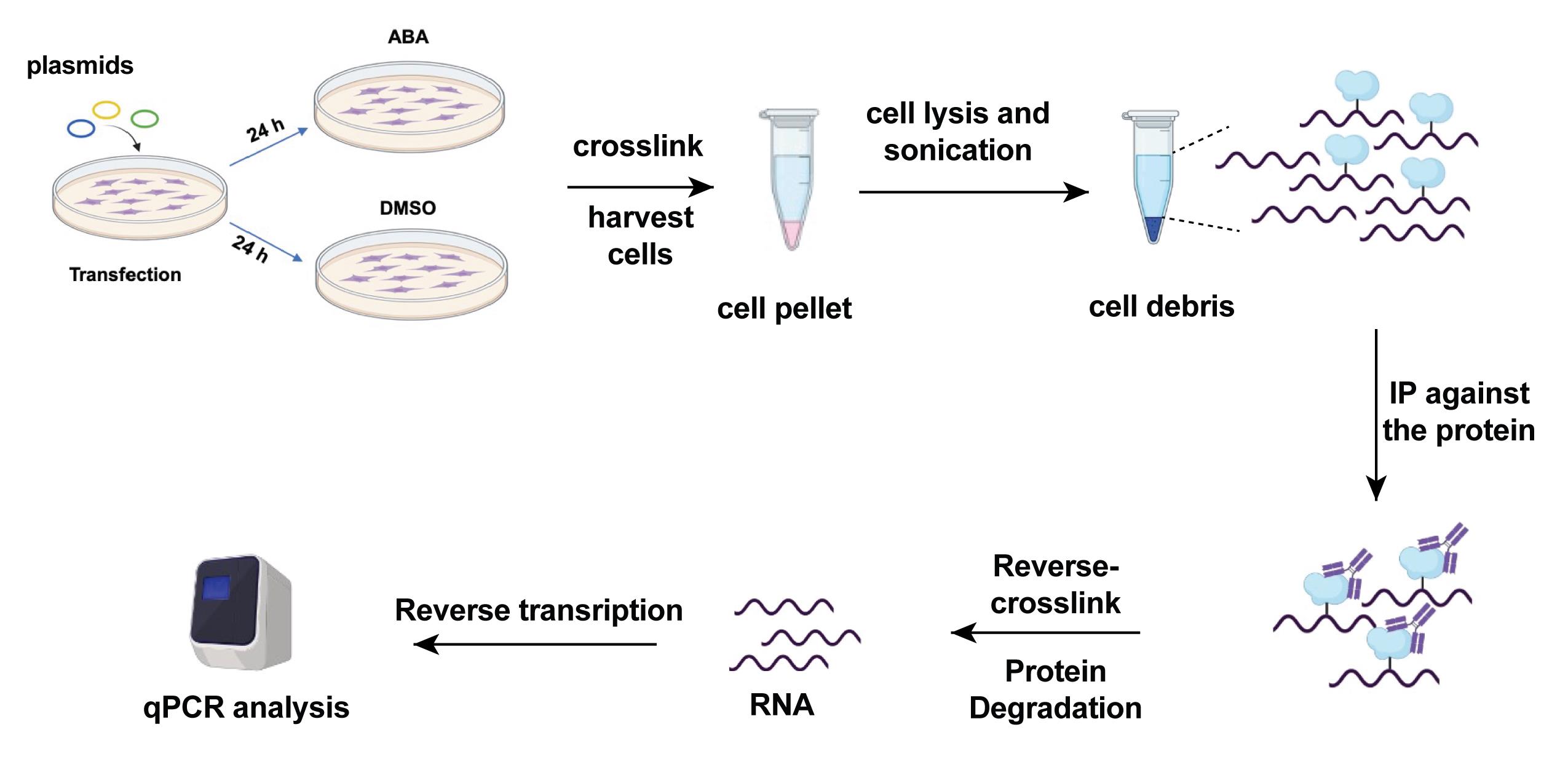
Figure 6. Scheme of investigation of enrichment of METTL3 on target RNA of interest. The cartoon was created with Biorender.com
Specificity of targeted m6A editing by SELECT
Seed 100,000 cells in a 12-well plate and incubate for approximately 16 h to reach 80% confluency.
Transfect cells with different components of ABA-induced m6A modification system for 24 h (1.2 µg of dCas13b-PYL + 0.8 µg of gRNA, 1.2 µg of dCas13b-M3 + 0.8 µg of gRNA, 1.2 µg of dCas13b-PYL + 1.2 µg of ABI-M3 + 0.8 µg of gRNA). The ratio of plasmids to lipofectamine 2000 is 100 ng:0.2 µL.
Add 100 µM ABA or DMSO into cells transfected with dCas13b-PYL+ABI-M3+gRNA and incubate for 24 h.
Harvest cells for RNA extraction as described above.
Mix 1.5 µg of total RNA with 40 nM Up primer, 40 nM Down primer, and 5 µM dNTP in 17 µL of 1× CutSmart buffer.
Run the following PCR program to anneal the RNA mixture: 90°C (1 min), 80°C (1 min), 70°C (1 min), 60°C (1 min), 50°C (1 min), and 40°C (6 min).
Incubate 17 µL of annealing products with 3 µL of enzyme mixture containing 0.01 U Bst 2.0 DNA polymerases, 0.5 U SplintR ligase, and 10 nmol ATP.
Incubate the final mixture from above (total 20 µL) at 40°C for 20 min and denature at 80°C for 20 min.
Analyze the DNA level via qPCR with PowerUpTM SYBRTM Green Master Mix. Briefly, mix 2 µL of the product from step J8, 5 µL of master mix, 1 µL of forward primer, and 1 µL of reverse primer, as well as 1 µL of nuclease-free water to prepare the qPCR reaction. Run qPCR with the setup of 95°C for 2 min and 40 cycles of 95°C for 15 s and 60°C for 1 min, followed by a dissociation step of 95°C for 15 s, then 60°C for 1 min and 95 °C for 15 s.
Light-inducible m6A writing
Seed 2,000,000 HEK293T cells in 100 mm dishes and incubate for approximately 16 h to reach 75%–80% confluency.
Transfect cells with different components of ABA-induced m6A modification system for 24 h (6 µg of dCas13b-PYL + 4 µg of gRNA, 6 µg of dCas13b-M3 + 4 µg of gRNA, 6 µg of dCas13b-PYL + 6 µg of ABI-M3 + 4 µg of gRNA). The ratio of plasmids to lipofectamine 2000 is 100 ng:0.2 µL.
Treat cells with 100 µM ABA or ABA-DMNB (see note below) with and without exposure to 405 nm light for 2 min.
Incubate cells for another 24 h.
Harvest cells for RNA extraction, m6A-IP, and RT-PCR as described above.
Note: The photo-caged ABA, ABA-DMNB, was synthesized by lab members by caging the ABA with 4,5-Dimethoxy-2-nitrobenzyl (DMNB), which can be removed by 405 nm light. The light was emitted from a fluorescence microscopy (model information provided in the Equipment section). Additionally, the primer information can be found in section G. Investigation of m6A level by m6A-immunoprecipitation.
Exemplary results
Here are some exemplary results (Figure 7) adapted from the original article “Inducible and reversible RNA N6-methyladenosine editing” (Shi et al., 2022).
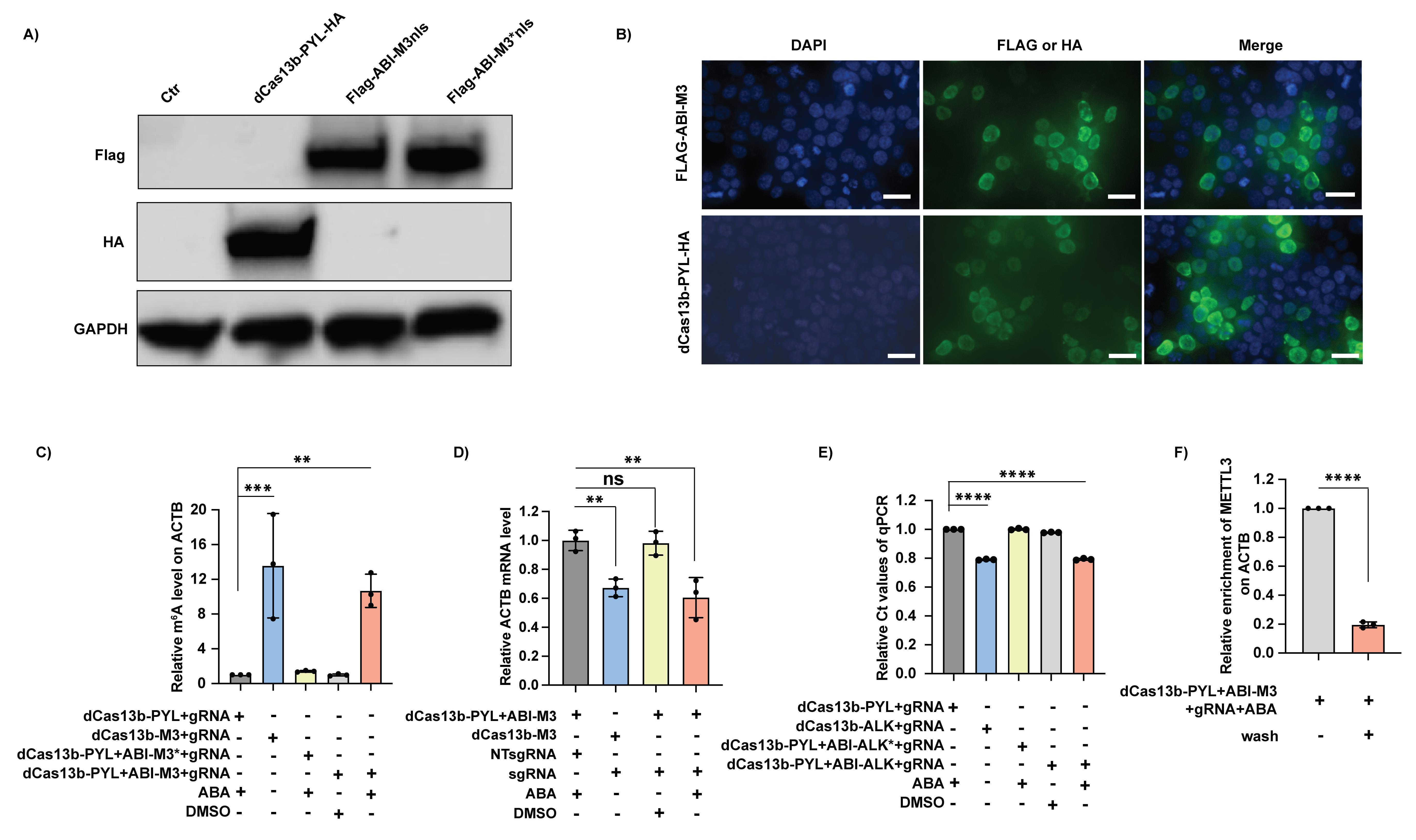
Figure 7. Exemplary results of main assays. A) Protein expression of the constructs of dCas13b-PYL with HA tag and ABI-M3nls with Flag tag. B) Localization of the expressed dCas13b-PYL and ABI-M3nls. Both proteins were localized in the nucleus. C) Targeted m6A deposition by the inducible m6A writing system on the targeted ACTB mRNA. M3 refers to the m6A writer, METTL3, and M3* refers to the mutant version of METTL3 without the catalytic function. The m6A level can only be elevated by the inducible m6A writing platform (dCas13b-PYL + ABI-M3 + gRNA) in the presence of ABA to a comparable level as the positive control (dCas13b-M3 + gRNA), but no increase can be observed without ABA or by using the catalytic inactive M3*. Results are normalized to the group of dCas13b-PYL + sgRNA + ABA. D) Impact of installed m6A on the stability of target ACTB mRNA. The installed m6A on the ACTB mRNA by the inducible m6A writing platform destabilized the mRNA just like the reported dCas13b-M3 + sgRNA. Results are normalized to the group of dCas13b-PYL + ABI-M3 + NT-sgRNA + ABA. E) Targeted m6A level removal by the inducible m6A erasing system on the target Malat1 lncRNA by SELECT assay. The smaller Ct value reflects the higher amounts of full-length product of SELECT-PCR, indicating the lower enrichment of m6A on the target. ALK refers to the m6A eraser, ALKBH5, and ALK* refers to the mutant version of ALKBH5 without the catalytic function. The m6A level can only be reduced by the inducible m6A erasing platform (dCas13b-PYL + ABI-ALK + gRNA) in the presence of ABA to a comparable level as the positive control (dCas13b-ALK + gRNA), but no increase can be observed without ABA or by using the catalytic inactive ALK*. Results are normalized to the group of dCas13b-PYL + sgRNA + ABA. F) Recruitment of METTL3 on the target ACTB mRNA after ABA removal. The METTL3 enzyme of inducible m6A writing platform can be recruited to target site by the treatment of ABA, which will be released after the ABA removal by wash. Results are normalized to the group of dCas13b-PYL + sgRNA + ABA.
Data analysis
For each assay, three independent trials were performed to obtain data and no exclusion of data was performed. Data and statistics were analyzed with one-way ANOVA using GraphPad Prism 8.0. The p value lower than 0.05 was marked as *, lower than 0.01 as **, lower than 0.001 as ***, and lower than 0.0001 as ****.
Notes
RNA extraction and other RNA-related experiments should be performed on ice or at 4°C, unless specifically noted otherwise, to avoid RNA degradation, as RNA integrity is important.
We suggest investigators to optimize the time of the fragmentation process in the m6A-IP by themselves, as the speed of taking the PCR tubes out of the thermo-cycler and addition of EDTA will impact the size of fragmented RNA. Usually, a size between 100 and 300 nt is optimal.
For all transfection procedures mentioned in this protocol, mix the plasmid and Lipofectamine 2000 transfection reagent separately in OMEM, then combine and mix them according to the manufacturer’s protocol.
For the amplification of the plasmids that we cloned, LB media and LB plates with ampicillin sodium salt are needed, because all plasmids have ampicillin resistance.
Recipes
LB plates with ampicillin
Reagents Final concentration Amount LB broth 25 g/L 25 g Agar 15 g/L 15 g 100 mg/mL ampicillin sodium salt 100 μg/mL 1 mL ddH2O N/A 980 mL Total volume N/A 1,000 mL Dissolve 25 g of LB broth and 15 g of agar in 980 mL of ddH2O in a glass bottle of 1 L volume.
Autoclave the LB broth liquid at 121°C for 15 min.
Add 1 mL of 100 mg/mL ampicillin solution to the autoclaved liquid when it cools down to 30–40°C.
Transfer 15 mL of the prepared solution to 10 cm Petri dishes using a serological pipette.
Allow the agar plates to cool and dry in a laminar flow hood for 20 min.
Store the plates at 4°C in a plastic bag.
LB media
Reagents Final concentration Amount LB broth 25 g/L 25 g ddH2O N/A 985 mL Total volume N/A 1,000 mL Dissolve 25 g of LB broth in 985 mL of ddH2O in a glass bottle of 1 L volume.
Autoclave the LB broth liquid at 121°C for 15 min.
Store the LB media at 4°C when it cools down. The shelf life for LB media is three months at 4°C.
1× TBST buffer
Reagents Final concentration Amount Tris base 2.42 g/L 2.42 g NaCl 8 g/L 8 g Tween-20 0.1% 1 mL ddH2O N/A 995 mL Total volume N/A 1,000 mL Dissolve 2.42 g of Tris base and 8 g of NaCl in 995 mL of ddH2O in a glass bottle of 1 L volume.
Add 1 mL of Tween-20 into the bottle and mix well.
Store the buffer at room temperature before use.
1× membrane transfer buffer
Reagents Final concentration Amount Glycine 1.44 g/L 1.44 g Tris base 3.03 g/L 3.03 g Methanol 20% 200 mL ddH2O N/A 800 mL Total volume N/A 1,000 mL Dissolve 1.44 g of glycine and 3.03 g of Tris base in 800 mL of ddH2O in a glass bottle of 1 L volume.
Add 200 mL of methanol into the bottle and mix well. Store at 4°C before use.
1× gel running buffer
Reagents Final concentration Amount Tris base 3.04 g/L 3.04 g Glycine 1.44 g/L 1.44 g SDS 1 g/L 1 g ddH2O N/A 995 mL Total volume N/A 1,000 mL Dissolve 1.44 g of glycine, 3.04 g of Tris base, and 1 g of SDS in 995 mL of ddH2O in a glass bottle of 1 L volume.
Store the buffer at room temperature before use.
Fragmentation buffer, 10×
Reagents Final concentration Amount Tris-HCl, pH 7.0 (1 M) 100 mM 100 µL ZnCl2 (1 M) 100 mM 100 µL ddH2O N/A 800 µL Total volume N/A 1,000 µL Mix 800 µL of nuclease-free water with 100 µL (1 M stock) of Tris-HCl (pH 7.0) and 100 µL of ZnCl2 (1 M stock).
Freshly prepare the buffer.
5× IP buffer for m6A-IP study
Reagents Final concentration Amount Tric-HCl, pH 7.4 (1 M) 50 mM 0.5 mL NaCl (5 M) 750 mM 1.5 mL IGEPAL-CA-630 (10% vol/vol) 0.5% vol/vol 0.5 mL ddH2O N/A 10 mL Total volume N/A 10 mL Mix 0.5 mL of Tris-HCl (1 M, pH 7.4), 1.5 mL of NaCl (5 M), and 0.5 mL of IGEPAL-CA-630 (10% vol/vol) in a total volume of 10 mL nuclease-free water.
Freshly prepare the buffer for use.
m6A elution buffer
Reagents Final concentration Amount 5× IP buffer 1× 90 µL 20 mM m6A salt 2.67 mM 60 µL RNasin Plus ribonuclease inhibitor N/A 7 µL Nuclease-free water N/A 293 µL Total volume N/A 450 µL Wash buffer for RNA-crosslink immunoprecipitation
Reagents Final concentration Amount DPBS N/A 50 mL Tween-20 0.02% 10 μL NT-2 buffer, 5×
Reagents Final concentration Amount Tric-HCl, pH 7.4 (1 M) 250 mM 12.5 mL MgCl2, 1 M 5 mM 0.25 mL NaCl (5 M) 750 mM 7.6 mL IGEPAL-CA-630 (10% vol/vol) 0.25% vol/vol 1.25 mL ddH2O N/A 50 mL Total volume N/A 50 mL Protease K digestion buffer
Reagents Final concentration Amount Protease K N/A 5 µL SDS (10%) 1% 15 µL NT-2 buffer, 5× N/A 30 µL Nuclease-free water N/A 100 µL Total volume N/A 150 µL
Acknowledgments
This work was supported by National Institutes of Health (R21CA247638).
We acknowledge our previous work “Inducible and reversible RNA N6-methyladenosine editing” published in Nature Communication. DOI: https://doi.org/10.1038/s41467-022-29665-y.
Competing interests
The authors declare no competing interests.
References
- Boccaletto, P., Stefaniak, F., Ray, A., Cappannini, A., Mukherjee, S., Purta, E., Kurkowska, M., Shirvanizadeh, N., Destefanis, E. and Groza, P. (2022). MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic Acids Res 50(D1): D231-D235.
- Chen, T., Gao, D., Zhang, R., Zeng, G., Yan, H., Lim, E. and Liang, F.-S. (2017). Chemically controlled epigenome editing through an inducible dCas9 system. J Am Chem Soc 139(33): 11337-11340.
- Licht, K. and Jantsch, M. F. (2016). Rapid and dynamic transcriptome regulation by RNA editing and RNA modifications. J Cell Biol 213(1): 15-22.
- Liu, N. and Pan, T. (2016). N6-methyladenosine-encoded epitranscriptomics. Nat Struct Mol Biol 23(2): 98-102.
- Liu, X. M., Zhou, J., Mao, Y., Ji, Q. and Qian, S. B. (2019). Programmable RNA N(6)-methyladenosine editing by CRISPR-Cas9 conjugates. Nat Chem Biol 15(9): 865-871.
- Lo, N., Xu, X., Soares, F. and He, H. H. (2022). The Basis and Promise of Programmable RNA Editing and Modification. Front Genet 13: 834413.
- Ontiveros, R. J., Stoute, J. and Liu, K. F. (2019). The chemical diversity of RNA modifications. Biochem J 476(8): 1227-1245.
- Pickar-Oliver, A. and Gersbach, C. A. (2019). The next generation of CRISPR–Cas technologies and applications. Nat Rev Mol Cell Biol 20(8): 490-507.
- Rauch, S., He, C. and Dickinson, B. C. (2018). Targeted m6A Reader Proteins To Study Epitranscriptomic Regulation of Single RNAs. J Am Chem Soc 140(38): 11974-11981.
- Shi, H., Xu, Y., Tian, N., Yang, M. and Liang, F.-S. (2022). Inducible and reversible RNA N6-methyladenosine editing. Nature Commun 13(1): 1958.
- Wang, X. and He, C. (2014). Dynamic RNA modifications in posttranscriptional regulation. Mol Cell 56(1): 5-12.
- Wilson, C., Chen, P. J., Miao, Z. and Liu, D. R. (2020). Programmable m6A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat Biotechnol 38(12): 1431-1440.
- Xu, Y. and Liang, F.-S. (2022). On demand CRISPR-mediated RNA N6-methyladenosine editing. Genes Dis 9(6): 1389-1390.
- Yang, Y., Hsu, P. J., Chen, Y. S. and Yang, Y. G. (2018). Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res 28(6): 616-624.
Article Information
Copyright
© 2023 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Xu, Y., Wang, Y. and Liang, F. S. (2023). A CRISPR-based Strategy for Temporally Controlled Site-Specific Editing of RNA Modifications. Bio-protocol 13(3): e4607. DOI: 10.21769/BioProtoc.4607.
Category
Cell Biology > Cell engineering > CRISPR-cas9
Molecular Biology > RNA > Epitranscriptome
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.