- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Immunoprecipitation for Protein-Protein Interactions and for RNA Enrichment in Drosophila melanogaster
Published: Vol 11, Iss 23, Dec 5, 2021 DOI: 10.21769/BioProtoc.4250 Views: 3595
Reviewed by: Alexandros AlexandratosRajesh RanjanMario Ruiz
Original research article
The authors used this protocol in:

Jul 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
To determine the molecular and functional interactions between RNA-binding proteins (RBPs) and their targets RNAs, is of fundamental importance to understand the dynamic organization of the nervous system in health and disease. Nevertheless, this task has remained elusive due to the lack of specific protocols and experimental systems that would allow the combination of biochemical analysis with in vivo functional genetics. In this manuscript, we describe a trustworthy and detailed methodology to establish the molecular organization and intracellular function of RBPs/RNA multimeric complexes in a cell type-defined manner by using the powerful GAL4/UAS system for gene expression in Drosophila melanogaster.
Graphic abstract:

Immunoprecipitation for protein-RNA interaction in Drosophila.
Background
Understanding the molecular and physical interactions between RNA-binding proteins (RBPs) and their RNA targets, especially in in vivo models, can provide important information regarding the functional organization of the eukaryotic nucleus, including RNA metabolism. In this manuscript, we concentrate our analysis on the RNA-binding protein TAR DNA-binding protein 43 (TDP-43) due to its role in the pathogenesis of different neurodegenerative diseases (Prasad et al., 2019; Boer et al., 2021), and describe a protocol to isolate TDP-43-binding mRNA and protein complexes in a cell type (Godena et al., 2011; Romano et al., 2014 and 2020). This methodology is reproducible, relatively costless, and details the passages needed to immunoprecipitate TDP-43 from Drosophila melanogaster neurons and detect the messenger RNA molecules (mRNA), which are able to interact with its RNA-binding domain in vivo. A complementary procedure is also described to separate TDP-43/RBPs protein complexes and identify additional interactors. In addition, we indicate how the powerful genetic tools available in D. melanogaster enable the possibility to study the assembly of RBP complexes in different genetic backgrounds and/or alternative developmental stages, including adulthood and aging. This scope is achieved by utilizing the amenable GAL4/UAS system, which allows the expression of any gene of interest in D. melanogaster, making our approach distinctive and particularly efficient to isolate RBP/mRNA protein combinations in previously inaccessible compartments. In addition, this protocol can be adapted to different experimental models by simply adjusting reagents, concentrations, or proportions accordingly to the ratios described. Finally, we indicate how the material can be safely stored and successively analyzed at whatever time.
Materials and Reagents
RNase-free disposable pellet pestle (Fisher Scientific, catalog number: 12-141-368)
1.5 ml tubes (Eppendorf, catalog number: 0030120086)
2.0 ml tubes (Eppendorf, catalog number: 0030120094)
Glass Petri Dish 100 × 22 mm (Duroplan, catalog number: 217554804)
Drosophila melanogaster strains for research are maintained and distributed from several stock centers, listed here below:
Bloomington Drosophila Stock Center (BDSC) (https://bdsc.indiana.edu/)
Kyoto Stock Center (DGRC) (https://kyotofly.kit.jp/cgi-bin/stocks/index.cgi)
Vienna Drosophila Resource Centre (VDRC) (https://stockcenter.vdrc.at/control/main)
FlyORF (https://www.flyorf.ch/)
Fly Stocks of National Institute of Genetics (NIG-FLY) (https://shigen.nig.ac.jp/fly/nigfly/)
Note: In Figures 1 and 2, the following strains have been used: w1118 (#3605, BDSC) – elav-GAL4/CyO (#8765, BDSC) – UAS-mCD8::GFP/CyO (#30002, BDSC) – UAS-TBPHF/L/TM3, Sb (Godena et al., 2011) – UAS-TBPH/CyO (Feiguin et al., 2009).
qPCR primers [see Romano et al. (2020)]:
Rpl11: forward 5’-ccatcggtatctatggtctgga-3’ reverse 5’-catcgtatttctgctggaacca-3’
Dcr-2: forward 5’gcttttatgtgggtgaacaggg-3’ reverse 5’-ggctgtgccaacaagaactt-3’
Syntaxin: forward 5’-tgttcacgcagggcatcatc-3’ reverse 5’-gccgtctgcacatagtccatag-3’
Liquid nitrogen
Anti-Flag M2 antibody (Sigma-Aldrich, catalog number: F3165)
DynabeadsTM Protein G (Thermo Fisher, catalog number: 10003D)
Complete free protease inhibitors (Roche, catalog number: 11836170001)
Trizol (Invitrogen, catalog number: 15596026)
Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9888)
Potassium chloride (KCl) (Merck, catalog number: P3911)
Magnesium chloride solution (Sigma-Aldrich, catalog number: M1028)
Na2HPO4 (Thermo Fisher, catalog number: 10182863)
KH2PO4 (Thermo Fisher, catalog number: P285-500)
Tris (Thermo Fisher, catalog number: 17926)
Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: 436143)
Sodium deoxycholate (DOC) (ThermoFisher, catalog number: 89904)
Urea (Sigma-Aldrich, catalog number: U5128)
1,4-Dithiothreitol (DTT) (Sigma-Aldrich, catalog number: 10197777001)
Bromophenol Blue (Sigma-Aldrich, catalog number: B0126)
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) HEPES (Sigma-Aldrich, catalog number: H3375)
Ethylenediaminetetraacetic acid (EDTA) (Merck, catalog number: 200-449-4)
Heparin (Sigma-Aldrich, catalog number: H3149)
Glycerol (Sigma-Aldrich, catalog number: G5516)
RNase OUT, RNAse inhibitor (Thermo Fisher, catalog number: 10777019)
cOmpleteTM, EDTA-free Protease Inhibitor Cocktail (Roche, catalog number: 11873580001)
Tween 20 (Merck, catalog number: 9005-64-5)
Triton X-100 (Merck, catalog number: 9002-93-1)
Chloroform (CHCl3) (Merck, catalog number: 67-66-3)
2-Propanol (Merck, catalog number: 67-63-0)
Ethanol (Merck, catalog number: 64-17-5)
SuperScriptTM IV VILOTM Master Mix (ThermoFisher, catalog number: 11756050)
iQTM-SYBR® Green Supermix (Bio-Rad, catalog number: 1708880)
Ultrapure Glycogen (ThermoFisher, catalog number: 10814010)
1× PBS (see Recipes)
1× Protein-mRNA-Lysis Buffer (see Recipes)
1× Protein-Protein-Lysis Buffer (see Recipes)
Bead Binding Buffer (see Recipes)
1× Washing Buffer (see Recipes)
5× Laemmli Buffer (see Recipes)
50 mg/ml Heparin stock solution (see Recipes)
Equipment
Stereomicroscope (Leica, model: M1028)
Glass Dounce Style Tissue Grinder 1 ml (Wheaton, model: 357538)
DynaMag Spin Magnet Stand (ThermoFisher, catalog number: 12320D)
Multi Rotator PTR-35 (Grant-Bio)
Centrifuge 5424 R (Eppendorf)
CFX Connect Real-Time PCR detection system (Bio-Rad)
Tweezers Dumont n.°5 student standard tip 0.1 × 0.06 (FST, catalog number: 91150-20)
Software
ImageJ (Wayne Rasband, NIH, https://imagej.nih.gov/)
Prism (GraphPad, https://www.graphpad.com/scientific-software/prism/)
Procedure
The UAS-GAL4 system (Brand and Perrimon, 1993) allows a targeted tissue specific gene expression by crossing a GAL4-driver stock with a transgenic fly harboring your RBP protein of interest under the control of UAS activating sequences. Protein expression can be targeted to several tissues and animal compartments using different drivers such as GMR-GAL4 or Elav-GAL4, allowing the easy collection of the tissues of interest. In the design of these experiments, it is highly recommended to include a control progeny expressing an unrelated protein (e.g., UAS-GFP).
Collection of heads
Select flies at the stereoscope under anesthesia [exposure to carbon dioxide (CO2)].
Transfer 20 anesthetized flies at the time into an empty 1.5 ml tube.
Immediately flash freeze the tube in liquid nitrogen.
Vortex the tube for 10 seconds at maximum potency.
Pour the contents of the tube on a glass Petri dish laid on ice.
Collect the heads with a tweezer and transfer them into a 1.5 ml tube filled with the appropriate lysis buffer.
During the collection, keep the tubes in ice.
Note: The progeny required for an immunoprecipitation experiment is approximately 100 flies. For this reason, flies can be accumulated and stored at -80°C after flash freezing in liquid nitrogen. Aliquots of 20 flies per tube are the best solution to allow easy detachment of the heads by vortexing at the moment of collection.
Preparation of beads
For each immunoprecipitation sample, 50 μl of bead slurry must be used (approximately 1.5 mg beads). Mix the beads thoroughly by repeated inversion before use.
Transfer the total beads slurry in a single 2 ml tube.
Wash the beads twice with 2 ml of 0.02% Tween 20-PBS, using the magnetic stand to collect the beads and allow buffer changes.
Dilute the primary antibody (a-Flag-M2 in this example) at a final concentration of 2-5 mg/ml in 0.02% Tween 20-PBS, add it to the washed beads, and incubate at room temperature (RT) for a minimum of 10 min on the rotator, to allow coating of the beads.
After the incubation, aliquot the antibody-coated beads in new 1.5 ml tubes (one for each sample).
Discard the buffer with primary antibody just before adding the solution with the homogenized tissue.
Protein-mRNA co-immunoprecipitation
Pile up 100 Drosophila heads in a 1.5 ml tube filled with 200 μl of Protein-mRNA lysis buffer.
Manually squeeze heads with a pestle for 2 min (work on ice).
Transfer the lysate into a dounce (pre wet with lysate buffer).
Collect all the homogenate by adding 500 μl of fresh lysate buffer to the 1.5 ml tube, subsequently add it to the dounce, and proceed to homogenization with 60 strokes.
Mechanically squeeze for another 2 min (work on ice).
Transfer the approximately 700 μl to a clean 1.5 ml tube.
Centrifuge the tubes at 500 × g for 5 min at 4°C.
Collect approximately 600 μl of lysate by avoiding debris, and transfer it to a new clean tube.
Add heparin to the lysate to a final concentration of 5 μg/ml.
Remove 100 μl of lysate and store it as input material (50 μl for protein analysis and 50 μl for RNA extraction). The 50 μl aliquot for protein analysis can be directly stored at -80°C, while 600 μl of Trizol reagent, for RNA extraction, must be added to the 50 μl aliquot before its storage at -80°C.
Transfer the remaining 500 μl lysate into the 1.5 ml tube containing the antibody-coated beads for immunoprecipitation.
Incubate the tubes on a circular rotator for 30 min at 4°C.
After incubation, immobilize the beads on the magnet, and recover 100 μl of each supernatant as "depleted material" (50 μl for protein analysis and 50 μl for RNA extraction). The 50 μl for protein analysis can be stored at -80°C, while 600 μl of Trizol reagent must be added at the 50 μl for RNA extraction before its storage at -80°C.
Remove and discard the remaining supernatant, always keeping the tubes anchored to the magnet to preserve the beads while removing the supernatant.
Wash the beads with 1 ml of washing buffer: add the buffer, transfer the beads to the rotator for 10 min, anchor the tube to the magnet, and remove the washing buffer. Repeat five times, keeping the buffer on ice and the rotator at 4°C.
At the end of the fifth wash, before attaching the tubes to the magnet, transfer 100 μl of the beads in solution to a new tube for protein analysis, and place the remaining 900 μl of the beads in solution in a fresh tube for RNA extraction.
Immobilize the beads and discard the liquid.
Add 50 μl of 1× Laemmli buffer to the tube designated for protein extraction, boil the tube at 95°C for 5 min, immediately transfer to the magnet, and collect the supernatant.
Attach the tube designated for RNA analysis to the magnet, remove the supernatant, and add 600 μl of Trizol to the tube. Leave the tube on the rotator for 5 min at RT, and then collect the supernatant, leaving the beads in the tube (use the magnet).
These materials can be stored at -80°C at this step.
Protein-protein co-immunoprecipitation
Collect 100 heads in a 1.5 ml tube filled with 100 μl of Protein-protein lysis buffer.
Manually squeeze heads with a pestle for 2 min working on ice.
Transfer the lysate into the dounce (pre-wet with lysate buffer), wash the 1.5 ml tube with a further 400 μl of buffer and add it to the lysate in the dounce. Proceed with homogenization with 60 strokes.
Collect the homogenate, transfer it into a new 1.5 ml tube, wash the dounce with 500 μl of fresh Protein-protein lysis buffer, and subsequently add it to the lysate.
Centrifuge the lysate at 500 × g for 5 min at 4°C, and collect the supernatant avoiding the pellet.
Add 100 μl of fresh Protein-protein lysis buffer to the pellet, wash it, and repeat the centrifugation step as previously. Avoiding the pellet, add this supernatant to the lysate already collected, to a final lysate volume of approximately 1 ml.
Take a 100 μl aliquot of the lysate. Use 20 μl of this to proceed with the evaluation of the protein content in the lysate. To the remaining 80 μl of lysate add 20 μl of 5× Laemmli Sample buffer, denature at 95°C for 5 min, and store at -80°C as "Input" for SDS-PAGE analysis.
An equal amount of protein must be used for the immunoprecipitation of your samples of interest and your controls, so add to a new 1.5 ml tube a volume that corresponds to an amount of protein of approximately 1-1.4 mg. Add the protein-protein lysis buffer for equal volumes among samples.
Transfer lysates to tubes with the immune-coated beads and proceed to immunoprecipitation on the rotator at 4°C for 30-60 min.
After incubation, transfer the lysate and beads to a new 1.5 ml tube, anchor the tube to the magnet, and recover 80 μl of supernatant ("depleted material"). To this, add 20 μl of 5× Laemmli Sample buffer, denature at 95°C for 5 min, and store -80°C for the following SDS-PAGE.
Always keeping the tubes anchored to the magnet, remove and discard the remaining supernatant.
Detach the tubes from the magnet, add 1 ml of iced Protein-protein Lysis buffer to the beads, and transfer them to a new 1.5 ml tube. Leave this on the rotator for 10 min at 4°C, then anchor the tubes to the magnet and remove the washing buffer. Repeat to a total of five washes.
At the end of the fifth wash, transfer the beads to a new tube before removing the last wash, then anchor the tube to the magnet, completely remove the liquid while avoiding contact with the beads, add 80 μl of Protein-protein Lysis buffer and 20 μl of 5× Laemmli Sample buffer, transfer to 95 °C for 5 min, then immediately anchor to the magnet and collect the eluate. Store at -80°C for further SDS-PAGE analysis.
Immunoprecipitation validation
Note: To confirm the immunoprecipitation, it is a good practice to run a western blot and verify the presence of your protein of interest in the immunoprecipitated sample. If possible, the best is to perform the western with another antibody that recognizes a different epitope of the immunoprecipitated protein.
Run an SDS-PAGE following the instructions of the pre-cast gel supplier. All the samples collected during the immunoprecipitation procedure: Input (IN), depleted material (DEP), and immunoprecipitated material (IP) must be run on the same gel.
Proceed to western blot, as usual; incubate your membrane with primary antibody, perform washes, and subsequently incubate with the secondary antibody.
Develop the immunoblot following your usual protocols.
If a good immunoprecipitation occurred, loading an equal volume of the IN and DEP material, the signal corresponding to your immunoprecipitated target must be reduced in the DEP fraction if compared to the IN fraction.
The signal of your immunoprecipitated target must be enriched in the IP fraction compared to the mock. In Figure 1, an example of western blot validation is presented.
If any problem occurred, see Table 1. It summarizes the most common problems that can occur, with their respective solutions.
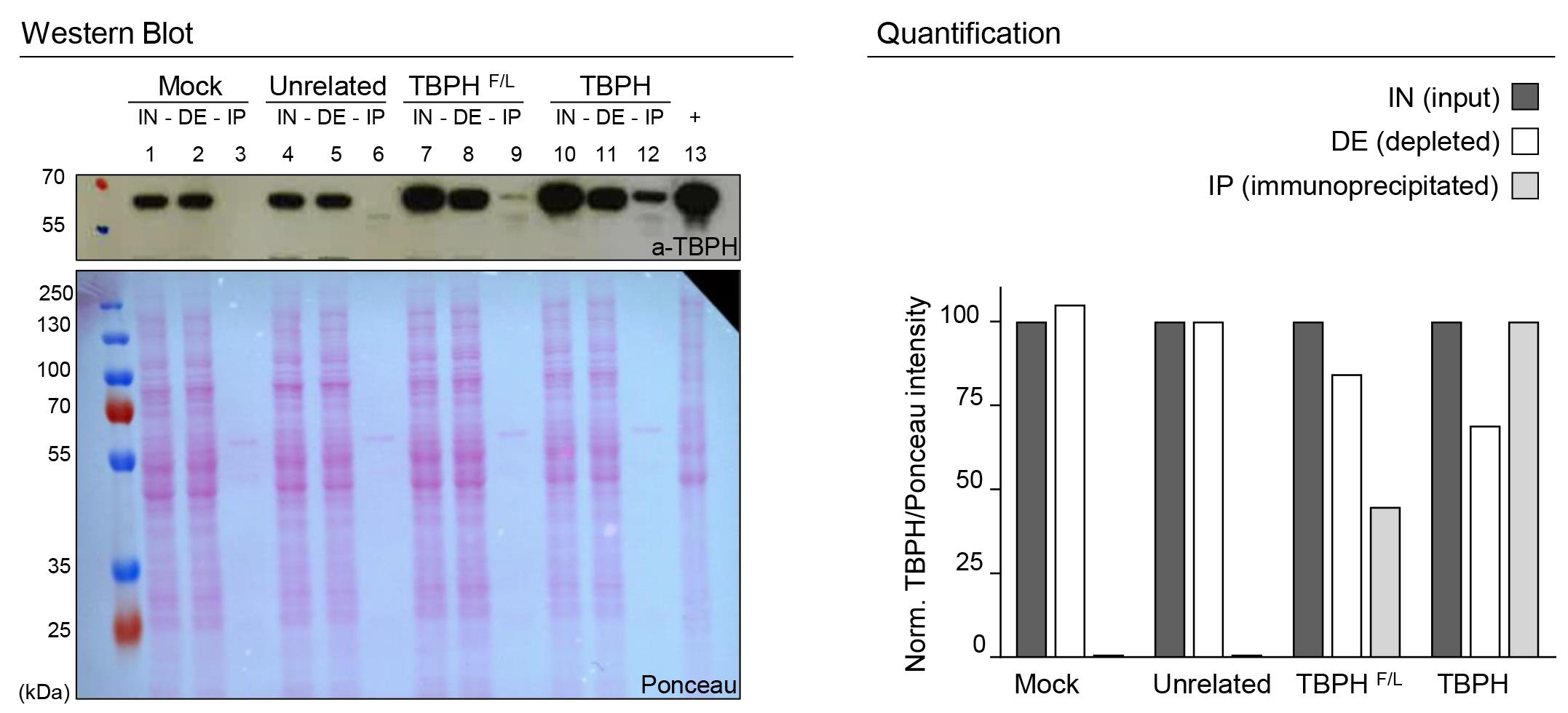
Figure 1. SDS-PAGE IP validation. On the left, western blot analysis of immunoprecipitation with anti FlagM2 from D. melanogaster adult heads: lanes 1, 2, and 3 w1118 (mock- no flag); lanes 4, 5, and 6 elav-GAL4>UAS-unrelated protein (flagged); lanes 7, 8, and 9 elav-GAL4>UAS-TBPHF/L (mutated and flagged); lanes 10, 11, and 12 elav-GAL4>UAS-TBPH (flagged). Respectively, lanes 1, 4, 7, and 10 represent the input fraction (IN), lanes 2, 5, 8, and 11 the depleted fraction (DE), and lanes 3, 6, 9, and 12 the immunoprecipitated fraction (IP); lane 13 is a positive control for the antibody (+). The immunoprecipitation is performed using the anti-FlagM2 antibody, while the immunoblot is developed using an anti-TBPH antibody. The Ponceau of the membrane was used as the loading control. On the right panel, a column graph quantifies the experiment. Results of IN and DE fractions were normalized to the input of each strain, and the IP fraction was normalized to the TBPH strain.
Co-immunoprecipitation controls
Controls are extremely important in immunoprecipitation experiments, and both negative and positive ones are required.
The ones listed below can be used as negative controls for the experiment:
A mock control (Figure 1, lanes 1, 2, 3);
An unrelated protein tagged with the same flag (Figure 1, lanes 4, 5, 6);
The protein of interest harboring a mutation that impaired its RNA binding or protein-protein interactions (Figure 1, lanes 7, 8, 9).
As positive controls for the experiment, if available, an already known target must be confirmed.
RNA extraction
Transfer IN, DEP, and IP samples, stored at -80°C in 600 μl of Trizol reagent to ice.
Once thawed, add 120 μl of chloroform (CHCl3) to each tube, and mix thoroughly for 5 s.
Incubate at RT for 5 min.
Centrifuge at 12,000 × g for 15 min at 4°C.
Recover the transparent aqueous phase (~300 μl) and transfer it to a clean 1.5 ml tube.
Add 1 volume of isopropanol (~300 μl) and glycogen to a final concentration of 0.2 μg/ul
Mildly invert the tubes five times.
To increase yield, leave overnight at -80°C.
Centrifuge at 12,000 × g for 30-45 min at 4°C.
Discard the supernatant, keeping the pellet.
Wash the pellet with 1 ml of 75% ethanol.
Centrifuge at 12,000 × g for 5 min at 4°C, and remove the supernatant.
Repeat the wash.
Centrifuge at 12,000 × g for 5 min at 4°C. Completely remove the supernatant.
Resuspend the pellet in 30 μl of RNase-free water.
Store the RNA at -80°C.
Retrotranscribe 1 μg of RNA into cDNA by using Superscript Vilo Master Mix.
Dilute the cDNA 1:10 in water.
Samples are ready for qPCR with your primers of interest (see Materials and Reagents for primers sequences and references).
For the quantitative PCR, use 2 μl of diluted cDNA with the iQ-Sybr Green Supermix.
The quantitative PCR conditions are: 1 cycle at 95°C for 3 min; 40 cycles at 95°C for 10 s, and at 60°C for 30 s.
Figure 2 shows an example of the outcome (from Romano et al., 2020):
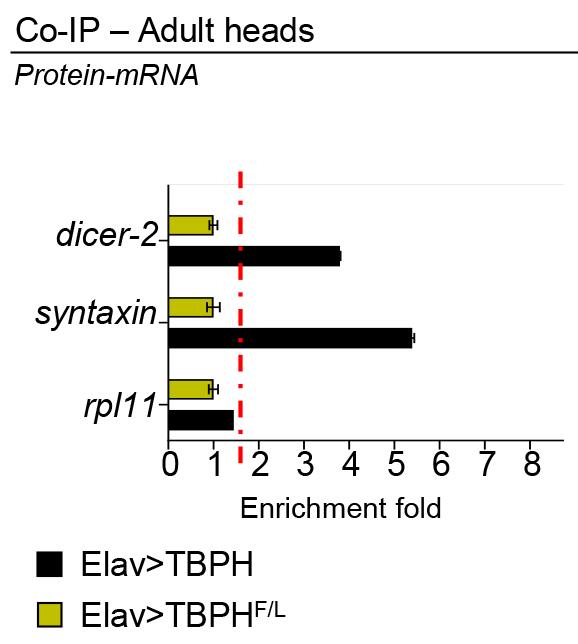
Figure 2. RT-qPCR analysis of mRNAs immunoprecipitated by Flag-M2-tagged TBPH (Elav>TBPH) and its mutant variants TBPH-F/L (Elav>TBPHF/L) in D. melanogaster adult heads. The dicer-2 and syntaxin enrichment folds were referred to rpl11 (housekeeping) (adapted from Romano et al., 2020).
Protein analysis
IN, DEP, and IP samples, all in 1× Laemmli loading dye, stored at -80°C, after a 5 min step of denaturation at 95°C, may be run in precast SDS-PAGE gel upon manufacturer instructions.
After SDS-PAGE, proceed with western blot following your usual protocol. For further information on this sub-protocol, see Romano et al. (2014, 2018, 2020).
Immunoblot must be performed against your immunoprecipitated protein (possibly with an antibody against a different protein epitope) to confirm immunoprecipitation, and with an antibody against your protein of interest, to verify possible interactions.
Data analysis
RT-qPCR Analysis
To calculate the enrichment fold, use the following equation from Livak and Schmittgen (2001): 2-ΔΔCT
Results must be derived from three independent experiments, and differences are determined by t-test (P < 0.05).
Protein Intensity Quantification
Protein bands are quantified using NIH ImageJ.
Briefly, open the western image in ImageJ, select the “Rectangular Selections” tool from the ImageJ toolbar, and select the band of interest.
Press Ctrl+1 and drag the same rectangle selection to all the other bands pressing Ctrl+2, repeating until the last band.
Pressing Ctrl+3, a histogram will open.
Select the “Straight Line” tool from the ImageJ toolbar, and with the line close the bottom of each pick.
Select the “Wand” tool from the ImageJ toolbar, and click inside each histogram.
Area and percentage tables will appear, and they can be copied to Excel.
The intensity of each protein band is divided by the intensity of its loading control.
The result can be plotted in a graph.
Notes
Table 1. Troubleshooting
Problems Suggested Solution Sample degradation -Protease inhibitor has to be freshly prepared; -Ensure all the steps are performed at the correct temperature;
-Check the integrity of all the solutions; for major precaution, filter them with a 0.22 μm membrane;
Antibody has not bound to the beads -Check that the beads are correct for the antibody isotype used (commonly IgG);
-Check for compatibility between the beads and IgG subclasses;
-Ensure the best fit with bead type and antibody, e.g., protein G beads have a greater affinity for human, mouse, rat, and cow, whereas protein A beads have greater affinity for rabbit, dog, pig, and cat;
-Increase the incubation time;
Non-specific proteins are bound to the beads -Add 1% BSA during the bead preparation step; High background/non-specific binding -Prolong washing steps (+ 5-10 minutes);
-Increase buffer volume during the washing steps;
-Test different salt concentration of HEGN buffer (from 150 mM to 400 mM);
Weak binding -Check the expression profile of the target protein and, if low, increase the amount of initial lysate;
-Check for the soluble compatibility between the target protein and the lysis buffer;
-Ensure that beads move freely in the solution during the different steps, allowing a continuous and complete mixing, and that beads are stuck in the tube.
Recipes
1× PBS
137 mM NaCl
10 mM Na2HPO4
1.8 mM KH2PO4
2.7 mM KCl
Adjust to pH 7.4
Autoclave and store at 4°C
1× Protein-mRNA Lysis Buffer
150 mM NaCl
0.5 mM EDTA
20 mM HEPES
10% Glycerol
0.1% Triton X-100
1 mM DTT
RNase OUT (0.2 U/μl)
Protease inhibitors (1×)
Adjust to pH 7.6
Add 0.005 μg/μl Heparin after the squeezing
1× Protein-protein Lysis Buffer
20 mM Tris
pH 7.5
110 mM NaCl
0.5% Triton X-100
Protease inhibitors (1×)
Bead Binding Buffer
PBS 1× pH 7.4
0.02% Tween 20
Washing Buffer
150 mM NaCl
0.5 mM EDTA
20 mM HEPES
10% Glycerol
0.1% Triton X-100
1 mM DTT
RNase OUT (0.2 U/μl)
Protease inhibitors (1×)
0.2% DOC
0.5 M Urea
Adjust to pH 7.5
5× Laemmli Sample Buffer
300 mM Tris-Cl, pH 6.8
50% glycerol
10% SDS
0.05% Bromophenol blue
50 mg/ml Heparin stock solution
Prepare a stock solution of 50 mg/ml by dissolving the heparin powder in distilled water
Aliquots can be stored at -20°C
Acknowledgments
The present work was supported by ICGEB intramural funding. The protocol was adapted and modified from previously published work (Godena et al., 2011; Romano et al., 2014 and 2020).
Competing interests
The authors declare that they have no competing interests.
References
- Brand, A. H. and Perrimon, N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118(2): 401-415.
- de Boer, E. M. J., Orie, V. K., Williams, T., Baker, M. R., De Oliveira, H. M., Polvikoski, T., Silsby, M., Menon, P., van den Bos, M., Halliday, G. M., et al. (2020). TDP-43 proteinopathies: a new wave of neurodegenerative diseases. J Neurol Neurosurg Psychiatry 92(1): 86-95.
- Feiguin, F., l, V.K., Romano, G., D’Ambrogio, A., Klima, R., and Baralle, F.E. (2009). Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett 583: 1586-1592.
- Godena, V. K., Romano, G., Romano, M., Appocher, C., Klima, R., Buratti, E., Baralle, F. E. and Feiguin, F. (2011). TDP-43 regulates Drosophila neuromuscular junctions growth by modulating Futsch/MAP1B levels and synaptic microtubules organization. PLoS One 6(3): e17808.
- Livak, K. J. and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 25(4): 402-408.
- Prasad, A., Bharathi, V., Sivalingam, V., Girdhar, A. and Patel, B. K. (2019). Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front Mol Neurosci 12: 25.
- Romano, G., Klima, R., Buratti, E., Verstreken, P., Baralle, F. E. and Feiguin, F. (2014). Chronological requirements of TDP-43 function in synaptic organization and locomotive control. Neurobiol Dis 71: 95-109.
- Romano, G., Holodkov, N., Klima, R., Grilli, F., Guarnaccia, C., Nizzardo, M., Rizzo, F., Garcia, R., and Feiguin, F. (2018). Downregulation of glutamic acid decarboxylase in Drosophila TDP-43-null brains provokes paralysis by affecting the organization of the neuromuscular synapses.Sci Rep 8: 1809.
- Romano, G., Klima, R. and Feiguin, F. (2020). TDP-43 prevents retrotransposon activation in the Drosophila motor system through regulation of Dicer-2 activity. BMC Biol 18(1): 82.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Romano, G., Klima, R. and Feiguin, F. (2021). Immunoprecipitation for Protein-Protein Interactions and for RNA Enrichment in Drosophila melanogaster. Bio-protocol 11(23): e4250. DOI: 10.21769/BioProtoc.4250.
Category
Neuroscience > Nervous system disorders > Neurodegeneration
Developmental Biology > Cell signaling > Energy homeostasis
Biochemistry > RNA > RNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.