- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Fe-NTA Microcolumn Purification of Phosphopeptides from Immunoprecipitation (IP) Eluates for Mass Spectrometry Analysis
Published: Vol 11, Iss 15, Aug 5, 2021 DOI: 10.21769/BioProtoc.4113 Views: 4072
Reviewed by: Khyati Hitesh ShahYu JinAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Protein phosphorylation is a nearly universal signaling mechanism. To date, a number of proteomics tools have been developed to analyze phosphorylation. Phosphoproteome-wide analyses using whole cell extracts suffer from incomplete coverage, often missing phosphorylation events from low-abundance proteins. In order to increase coverage of phosphorylation sites on individual proteins of interest (“phospho-mapping”), immunoprecipitation (IP) followed by phosphoenrichment is necessary. Unfortunately, most commercially available phosphoenrichment kits are not readily scalable to the low-microgram quantities of protein present in IP eluates. Here, we describe a simple method specifically optimized for the enrichment of phosphopeptides from IP samples using an Fe-NTA based method. This method can be added downstream of any standard immunoprecipitation protocol and upstream of any MS analysis pipeline. The protocol described herein is cost effective, uses commonly available laboratory reagents, and can be used to obtain deep coverage of individual protein phosphorylation patterns, supplementary to phosphoproteomics data.
Graphical abstract:
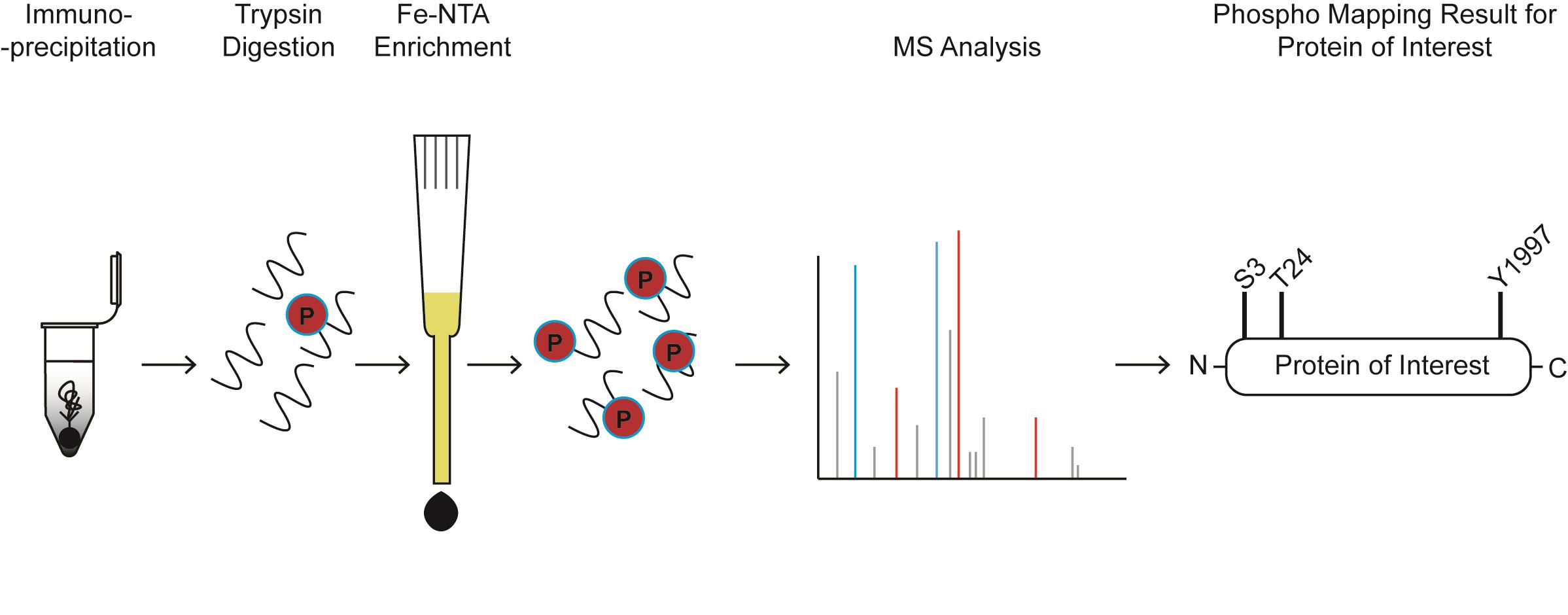
Phospho-mapping workflow for a hypothetical protein of interest
Background
Phosphorylation regulates key cellular processes such as transcription, translation, nutrient sensing, and the DNA damage response (Gingras et al., 2001; Chapman et al., 2007; Ohouo et al., 2010; Kim et al., 2011; Cussiol et al., 2015). Given the ubiquity of protein phosphorylation, its importance in normal cellular function, and its potential for dysregulation in disease, a number of proteomics methods have been developed to identify and quantitate phosphorylation events (Dephoure et al., 2013; Humphrey et al., 2018; Li et al., 2019; Faca et al., 2020). Many of these methods for phosphopeptide purification and analysis are performed on the proteome-wide scale and may suffer from relatively low coverage of individual proteins, especially those with limited abundance. High coverage of phosphorylation sites in individual proteins is often necessary to comprehensively study a given protein and to increase the chances of generating mutations of phospho-acceptor residues that elicit a phenotype. One strategy of increasing coverage of individual proteins is to first conduct an immunoprecipitation (IP) of the protein of interest using either an antibody raised against the protein itself or a common recombinant tag such as FLAG, HA, or V5. Phosphopeptides can then be isolated from the IP eluate. Optionally, differences in phosphorylation patterns between mutants or treatments can be analyzed using quantitative mass spectrometry methods such as SILAC or TMT (Ong et al., 2002; Zhang and Elias, 2017).
There are a number of commercially available kits for phosphopeptide enrichment, although these are difficult to scale down to the low-microgram quantities of protein yielded by immunopurification. There is a paucity of existing protocols for phosphopeptide enrichment following immunopurification, and those that do exist often require costly titanium dioxide (TiO2) reagents, making it difficult to scale up to multiple IPs across a panel of bait proteins (Breitkopf and Asara, 2012). Iron-based phospho-enrichment takes advantage of the affinity of negatively charged phosphate groups toward Fe3+ cations and is a cheaper alternative to TiO2; indeed, even where TiO2 is feasible, iron-based resin can be used to collect additional phospho-site data since the two methods display different phosphopeptide specificities (Bodenmiller et al., 2007).
The protocol described herein uses readily available Ni-NTA silica-based resin columns. The NTA-coupled resin is stripped of nickel, loaded with iron, and finally, the IP eluate is passed over the Fe-NTA resin to isolate phosphorylated peptides and remove unphosphorylated peptides. The procedure is performed using a home-made column (a gel-loading tip). The amount of resin generated from one Ni-NTA column is sufficient for 4-5 enrichments, making this protocol highly cost effective. Reproducibility is internally monitored via the inclusion of α/β casein phosphopeptides as controls for phosphopeptide enrichment. While this protocol has been primarily used for immunopurifications from yeast and mammalian cells, it is usable for any organism for which antibodies or recombinant tagging are available.
Materials and Reagents
C18 Sep-Pak column (Waters, catalog number: WAT043395)
Capillary tubing, 125 µm inner diameter (Polymicro, catalog number: 1068151718)
Centrifuge tubes, 50 ml (VWR, catalog number: 21008-178 or similar)
Glass fiber (Corning, catalog number: UX-34552-01)
Gel-loading pipette tips 1-200 µl (Fisher, catalog number: 02-707-138)
Low-protein binding microcentrifuge tubes, 1.5 ml (Eppendorf, catalog number: 022431081)
Microcentrifuge tubes, 1.5 ml (Eppendorf, catalog number: 0030108442)
Micro-spin columns (Thermo, catalog number: 89879)
Kimwipes (Fisher, catalog number: 06-666-1A)
Parafilm (VWR, catalog number: 52858-076)
Qiagen Ni-NTA spin columns (Qiagen, catalog number: 31014)
Razor blades (VWR, catalog number: 55411-055)
Sample vials (Sun SRI, catalog number: 501-300)
Sample vial snap caps, 11 mm (Fisher, catalog number: 14-823-379)
Acetic acid (Fisher, catalog number: A38S-500)
Acetone (Fisher, catalog number: A11-1)
Acetonitrile (Fisher, catalog number: A998-1)
Alpha casein (Sigma, catalog number: C6780-250MG)
Ammonium hydroxide solution (Sigma, catalog number: 205840010)
Angiotensin II peptide (Sigma, catalog number: A9525-5MG)
Beta casein (Sigma, catalog number: C6905-250MG)
Dithiothreitol (Sigma, catalog number: 10197777001)
Ethanol (VWR, catalog number: 89125-186)
Ethylenediamine tetraacetic acid (VWR, catalog number: MK258012)
Formic acid (Sigma, catalog number: 33015-500ML)
Iodoacetamide (Sigma, catalog number: I1149-5G)
Iron (III) chloride hexahydrate (Honeywell, catalog number: 236489-500G)
NaCl (Promega, catalog number: PRH5273)
Sodium dodecyl sulfate (Sigma, catalog number: L3771-100G)
Target polypropylene conical insert (Thermo, catalog number: C4010-629P)
Trifluoroacetic acid (Thermo, catalog number: 28904)
Tris base (Fisher, catalog number: BP152-500)
Trypsin Gold, mass spectrometry grade (Promega, catalog number: V5280)
Urea (Fisher, catalog number: U15-500)
Urea/Tris solution (see Recipes)
α/β casein peptide digest (see Recipes)
0.1 P solution (see Recipes)
Trypsin Gold solution (see Recipes)
C18 Buffer E (see Recipes)
C18 Buffer W1 (see Recipes)
C18 Buffer W2 (see Recipes)
C18 Buffer W3 (see Recipes)
FeCl3 solution (see Recipes)
IMAC elution solution (see Recipes)
IMAC stripping solution (see Recipes)
IMAC wash I (see Recipes)
Iodoacetamide (IAc) solution (see Recipes)
IP elution buffer (see Recipes)
PPT solution (see Recipes)
Equipment
1 ml BD syringes with slip-tip (BD, catalog number: 309659)
2× water bath or heat block (Thermo, model: TSGP02 or similar)
Tweezers (VWR, catalog number: 89259-984)
Desktop centrifuge (Beckman, model: A99469 or similar)
Desktop microcentrifuge (Beckman, model: A46471 or similar)
Micropipettes (Gilson, catalog number: F167380 or similar kit with P2, P20, P200, P1000 types)
Note: One water bath/heat block should be set to 42°C and the other to 65°C.Orbitrap mass spectrometer; for example, Q-Exactive HF (Thermo, IQLAAEGAAPFALGMBFZ)
Scissors (VWR, catalog number: 82027-596 or similar)
UHPLC system coupled to a mass spectrometer; for example, UltiMate 3000 (Thermo, model: IQLAAAGABHFAPBMBEX)
Vacuum concentrator centrifuge (Thermo, model: SPD131DDA)
Vacuum concentrator filter (Thermo, model: VPOF110)
Vacuum concentrator pump (Thermo, model: VLP120)
Vacuum concentrator vapor trap (Thermo, model: RVT5105)
Note: Items 9-12 are individual parts of what is referred to as a “speedvac” in the later steps of the protocol.
Software
XCalibur Qual Browser (Thermo Scientific, https://www.thermofisher.com/order/catalog/product/OPTON-30965?SID=s rch-srp-OPTON-30965#/OPTON-30965?SID=srch-srp-OPTON-30965)
Comet (http://comet-ms.sourceforge.net/)
Note: Any other commonly available mass spectrometry analysis software can be used.
Procedure
Reduction, denaturation, alkylation, and trypsin digestion of IP eluate
Most standard IP protocols are adaptable to this procedure. It is recommended to immunoprecipitate bait protein from 2-10 mg protein extract for this procedure (the amount of protein present in the sample following immunoprecipitation will be in the low-microgram range). Following lysis, immunoprecipitation, and washes, add 3× resin volume of IP elution buffer (see Recipes section for info on bolded solutions) to the sample. Incubate at 65°C for 15 min.
Note: Resin volume refers to the quantity of beads used in the IP. For example, if 30 μl of a 50:50 agarose resin:buffer slurry is used, the resin volume is 15 μl, and the volume of elution buffer used would be 45 μl.
Spin down the sample at 1,000 × g for 1 min. Take the supernatant (= volume of IP elution buffer used in the previous step) and transfer to a new tube.
Note: If the sample has been metabolically labeled for SILAC analysis, light and heavy IP eluates should be pooled prior to iodoacetamide treatment.
Add iodoacetamide (IAc) solution to a final concentration of 25 mM. Incubate in the dark at room temperature for 15 min.
Add 4 volumes PPT solution (see Recipes) to the sample. Vortex the tube for 10 s to wash the entire interior, and incubate on ice for 30 min-1 h. Longer incubation times may marginally improve the yield, but 30 min should be sufficient to precipitate most proteins. PPT contains flammable acetone and should be handled with care.
Spin the sample at max speed (~14,000 × g) in a benchtop centrifuge for 10 min.
Pour off PPT solution. You should see a small white pellet at the bottom of the tube. Alternatively, there may be small granules of protein spattered along the side of the tube.
Add 1 ml PPT solution to the tube and vortex to wash the top and sides. Spin down at max speed for 1 min. Pour off PPT.
Gently tap the open tube against a kimwipe to remove residual PPT, taking care not to dislodge the pellet at the bottom. Leave the tube open at room temperature for ~5 min, or until the acetone smell is nearly gone.
Add 20 µl urea/tris solution to the tube. Vortex the sample 30 s, then add 60 µl ddH2O. Vortex again.
Add 1 µl Trypsin Gold solution to the tube. Parafilm the top of the tube and nutate at 37°C overnight.
Desalting the sample
The next day, acidify the sample by adding formic acid (FA) at a final concentration of 0.25% and trifluoroacetic acid (TFA) at a final concentration of 0.25%. Additionally, add 2 µl α/β casein peptide digest (see Recipes section for details on how to prepare this solution).
Note: It is recommended to make 10% working stocks of TFA and FA in water for use.
Prepare a micro-spin desalting column by adding 20 µl 100 mg/ml C18 resin in acetonitrile.
First, remove the luer plug from the micro-spin column.
Obtain C18 resin by cutting open a Waters C18 Sep-Pak column of any size. Weigh out a small amount of C18 powder and resuspend in pure acetonitrile at a concentration of 100 mg/ml.
Spin at 1,500 × g for 30 s. If C18 resin is not yet level at the bottom of the micro-spin column, add 40 µl acetonitrile to the column and spin again.
Condition the column by adding 100 µl C18 Buffer E and spin at 1,500 × g for 30 s.
Equilibrate the column by adding 100 µl C18 Buffer W1 and spin at 1,500 × g for 30 s.
Load the acidified sample onto the column; approx. 100 µl. Spin at 1,500 × g for 30 s.
Wash the column by adding 100 µl C18 Buffer W1 and spin at 1,500 × g for 30 s.
Wash the column by adding 100 µl C18 Buffer W2 and spin at 1,500 × g for 30 s.
Wash the column by adding 100 µl C18 Buffer W3 and spin at 1,500 × g for 30 s.
Elute the sample into a low protein binding Eppendorf tube by adding 100 µl C18 Buffer E and spin at 1,500 × g for 30 s.
Place the sample in a speedvac until dry; approx. 40 min.
Note: It is important to ensure that the sample is completely dry. If some liquid persists after 40 min of drying, continue drying in the speedvac until all the liquid is gone.
Reconstitute the sample in 20 µl 0.1% acetic acid in ddH2O.
Preparation of Fe-NTA resin
Add 25 ml IMAC stripping solution to a 50-ml tube.
Remove a Qiagen Ni-NTA spin column from its package (Figure 1a) and associated collection tube. Make sure that the cap is open and hold the column over the open 50-ml tube containing 25 ml IMAC stripping solution (Figures 1b and 1c). With the tip of tweezers or similar thin, pointy object, poke the bottom aperture of the column to dislodge the two frits and the resin in between (Figure 1c). The frits and resin will fall into the 50-ml tube (Figure 1d). Nutate for 1 h to strip the nickel from the NTA resin.
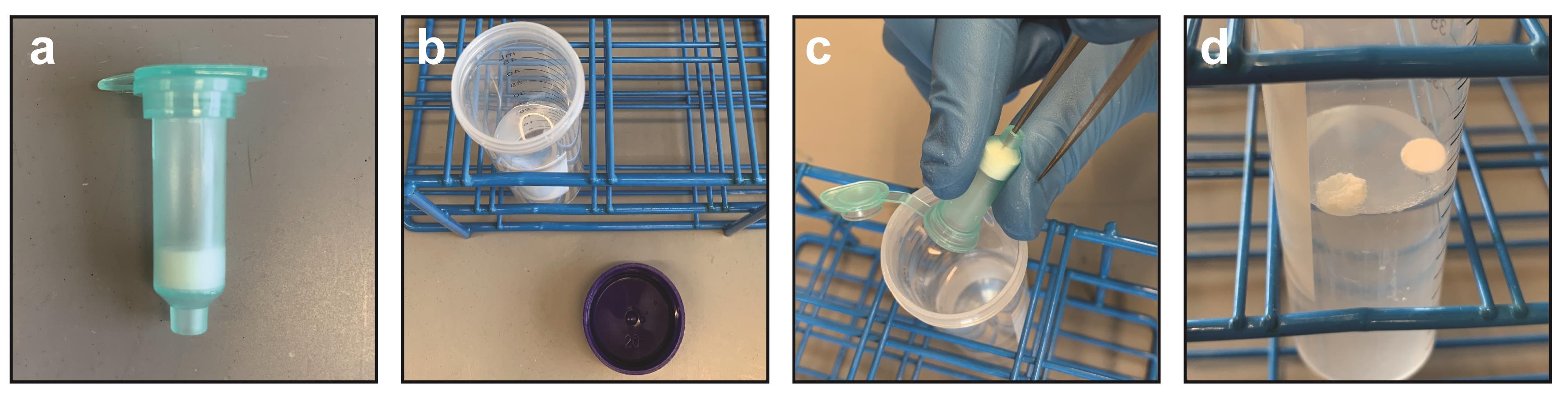
Figure 1. Harvesting Ni-NTA resin from spin column. (a) Ni-NTA column removed from collection tube. (b) 25 ml IMAC stripping solution in 50-ml conical tube. (c) Tweezer method for poking frit and dry resin into IMAC stripping solution. (d) Frits from Ni-NTA column afloat in stripping solution. Resin will sink to the bottom. After a 50-min nutation in stripping solution, frits can be discarded.Spin at 1,000 × g for 1 min. Discard the supernatant. This first supernatant will include the two frits from the Ni-NTA column.
Wash once with 25 ml ddH2O.
Wash once with 25 ml 0.6% acetic acid in ddH2O.
Resuspend the resin in 18 ml 0.3% acetic acid in ddH2O. Add 2 ml FeCl3 solution. Nutate for 2-3 h to bind the iron to the NTA resin.
Spin at 1,000 × g for 1 min. Discard the supernatant.
Wash once with 25 ml 0.6% acetic acid in ddH2O.
Wash once with 25 ml IMAC wash I.
Note: For this step, nutate for 1 min before spinning down.
Wash once with 25 ml 0.1% acetic acid in ddH2O.
Resuspend the resin in 0.1 ml 0.1% acetic acid in ddH2O and transfer to a microcentrifuge tube. The Fe-NTA resin should be prepared fresh every time.
Preparation of gel-loader tip column.
Clean a small area of bench surface with ethanol. Have glass fiber (4), gel-loader tips (3), BD syringes with slip tip (2), and a small section of 125-µm capillary tubing ready (1) (see Figure 2a).
Fit the gel loader tip onto a micropipette. Take up 10 µl ddH2O and make a mark at the 10 µl level with a permanent marker.
Remove the gel-loader tip from the pipette and cut the top of it with scissors so that it will fit snugly onto a 1-ml BD syringe.
Roll a tiny wisp of glass fiber between gloved fingers. Cut into a small 5-10 mm segment using a razor blade.
Insert the glass fiber segment into the top end of the gel-loader tip with the 10-µl mark (Figure 2b). Use the section of 125-μm capillary tubing to force the glass fiber segment into the tip of the gel-loader tip. Crimp the end of the gel loader tip with tweezers. Some glass fiber should be protruding from the end of the gel-loader tip at this point (Figure 2b, note wisp at end of tip). Use a razor blade to cut off any protruding glass fiber (Figure 2c).

Figure 2. Preparation of gel-loader tip micro-column. (a) Materials needed for column assembly: (1) 125-μm capillary tubing; (2) 1-ml BD syringe with slip tip; (3) gel-loader tip; and (4) glass fiber, rolled and cut into small sections. (b) Gel-loader tip (note 10-μl mark in blue ink) with top cut to adapt to BD syringe and glass fiber wisp protruding slightly from tip. (c) Final resin-filled gel-loader tip ready for phosphopeptide enrichment. Inset shows where end has been crimped and wisp removed to form a flat tip.
Phosphopeptide enrichment.
Prepare the column (see Figures 2 and 3 for column assembly) by adding 20 µl 0.1% acetic acid:resin mixture from Step C11 to the gel-loader tip. Use the BD syringe to apply backpressure to the column, forcing the resin to settle. Keep the column vertical at all times to ensure that the resin bed is even and undisturbed. Keep adding 0.1% acetic acid:resin mixture until the resin level reaches the 10-µl mark on the gel-loader tip. Once the settled resin has reached the 10-µl mark, use the BD syringe to force the remaining 0.1% acetic acid solution through the column.
Note: It is critically important to not allow the column to dry out. This is challenging due to the small volumes being worked with. When initially preparing the column and when performing washes, allow the liquid to reach the very edge of the resin bed (meniscus should nearly disappear). At this point, the column must be quickly removed from the BD syringe to prevent drying. In between washes, place the resin-filled gel-loader tip upright in a 1.5-ml microcentrifuge tube.

Figure 3. BD syringe with phosphopeptide enrichment column. Figure shows a completed, assembled phospho-peptide enrichment column.Once the phosphopeptide enrichment column has been assembled, wash once with 25 µl IMAC wash II to equilibrate the column.
Note: When passing solutions over the column, great care must be taken not to disturb the resin bed. Pipette carefully!
Apply the acidified, trypsinized sample to the column (20 µl reconstituted in 0.1% acetic acid, from protocol Step B12). Apply consistent pressure using the BD syringe and discard the flowthrough.
Note: Optionally, the flowthrough can be saved for a second attempt at phospho-enrichment if the first attempt fails.
Once the sample has been fully loaded onto the resin, pipette 100 µl IMAC wash I onto the column, then remove 90 µl, leaving 10 µl. Push 10 µl through the column using the BD syringe.
Pipette 100 µl ddH2O onto the column, then remove 90 µl, leaving 10 µl. Push 10 µl through the column using the BD syringe. While the ddH2O elutes, use a kimwipe to wipe the very tip of the gel-loader column. This will help to remove any residual contaminants.
Elute the phosphopeptides with 30 µl IMAC elution solution into a target polypropylene conical insert tube inside a microcentrifuge tube labeled “90%”. Then transfer 3 µl into another target polypropylene conical insert tube inside a microcentrifuge tube labeled “10%.”
Place both the samples in a speedvac until fully dry.
Resuspend both samples in 5 µl 0.1 P solution.
Proceed to MS analysis. Due to the low abundance and hydrophobicity of phosphopeptides, it is recommended to avoid using a trapping column. First run 5% of the total sample. If the MS chromatogram is relatively free of polymers and other contaminants, load increasing amounts of sample (for example: 10%, 20%, and 50%). Proceed to the “Data analysis” section.
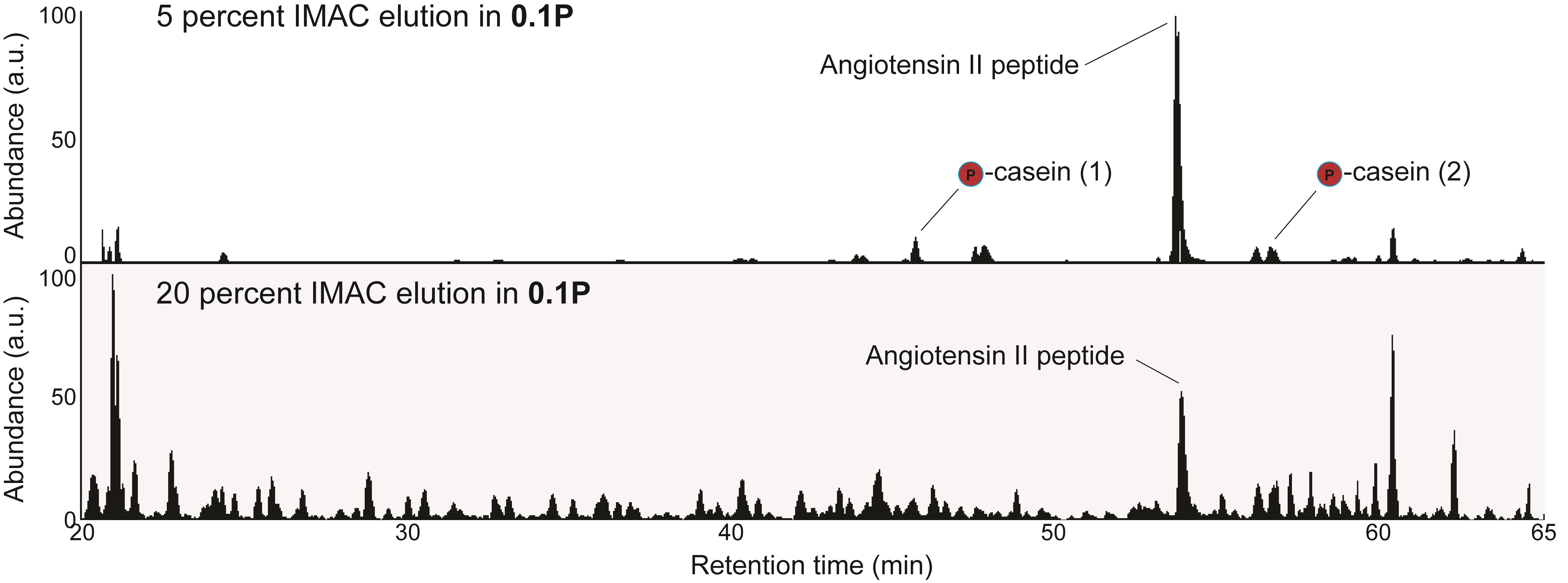
Figure 4. Representative IP-IMAC chromatogram. Representative chromatogram from 5% sample in 0.1P solution (top panel). Proceed to an injection of 20% sample (bottom panel) if chromatography from the 5% sample shows symmetric peaks with few contaminants. Contaminants are identifiable as any +1 charge state ions present in the sample. The same quantity of angiotensin II peptide is present in both samples (~200 fmoles). Casein peptides are also indicated. Casein peptide (1): β casein, m/z 1031.41, FQSphosEEQQQTEDELQDK; casein peptide (2): α casein, m/z 830.90, VPQLEIVPNSphosAEER.
Data analysis
Internal control for phosphopeptide recovery (monitoring α/β casein phosphopeptides)
Trypsinized α/β casein should be spiked into the sample prior to desalting as an internal control to verify that the phospho-enrichment portion of the protocol worked properly. Once 5% of the sample has been run on a mass spectrometer, add the following MS1 m/z ranges in Qual Browser (Thermo Scientific): 830.5-831.5; 1030.5-1031.5. Retention times may vary between users depending on the HPLC gradient profile that is used. These ranges encompass two diagnostic α/β casein phosphopeptides (see Figure 4 legend for details). The spectral intensity of the phosphorylated casein peptides should be greater than 1.00E6 in the 5% sample. Lower intensities indicate that phospho-enrichment troubleshooting is needed. Often, inefficient phospho-enrichment is due to excessive agitation of the Fe-NTA resin or column drying between washes.
Mass spectrometry data analysis
Mass spectrometry data can be analyzed using any existing software such as MaxQuant, Sorcerer, Comet, or Proteome Discoverer. Parameters for the identification of phosphorylated peptides by mass spectrometry have been exhaustively described elsewhere (Mann and Jensen, 2003; Mueller et al., 2008; Deutsch et al., 2010; Bastos de Oliveira et al., 2018; Faca et al., 2020). Typical mass spectrometry data analysis parameters will include a semi-tryptic requirement, a precursor mass tolerance of 10-20 ppm, a differential mass modification of 79.966331 Da for the phosphorylation of serine, threonine, or tyrosine residues, and a static mass modification of 57.021465 for carbamidomethylcysteine.
There are several ways to display the data generated from an IP phosphoenrichment experiment. Phosphopeptides with 2 or more peptide-spectral matches (PSMs) may be displayed in the absence of quantitative information along with a stick model of the protein of interest. In the presence of quantitative MS information, such as that obtained from SILAC or TMT analysis, scatterplots or histograms can be generated that indicate quantitative differences in bait protein phosphorylation between two genetic or pharmacological conditions. For example, we refer to the following work containing IMAC data generated in our lab (Lanz et al., 2018; Baile et al., 2019).
Recipes
0.1 P solution
1 pmol/µl angiotensin II peptide, 0.1% trifluoroacetic acid in ddH2O
Store at -20°C in 500-µl aliquots
C18 Buffer E
80% acetonitrile, 0.1% acetic acid in ddH2O
Store at room temperature
C18 Buffer W1
0.1% trifluoroacetic acid in ddH2O
Store at room temperature
C18 Buffer W2
3% acetonitrile, 0.1% formic acid in ddH2O
Store at room temperature
C18 Buffer W3
0.1% acetic acid in ddH2O
Store at room temperature
DTT solution (for IP elution buffer)
1 M DTT in ddH2O
Freeze in small aliquots and minimize freeze-thaw cycles
FeCl3 solution
1 M FeCl3, 0.3% acetic acid in ddH2O
Store at room temperature. Prepare a few days in advance of the experiment and do not agitate. Stable for several years at room temp.
IMAC elution solution
12% ammonium hydroxide, 10% acetonitrile in ddH2O
Store at room temperature. Keep tightly capped to prevent evaporation of ammonium hydroxide.
IMAC stripping solution
50 mM EDTA, 1 M NaCl in ddH2O
Store at room temperature
IMAC wash I
25% acetonitrile, 100 mM NaCl, 0.1% acetic acid in ddH2O
Store at room temperature. Make sure that this solution is tightly capped to prevent evaporation of acetonitrile.
IMAC wash II
1% acetic acid in ddH2O
Store at room temperature
Iodoacetamide (IAc) solution
500 mM iodoacetamide, 1 M Tris pH 8.0 in ddH2O
Prepare fresh
IP elution buffer
1% sodium dodecyl sulfate, 100 mM Tris pH 8.0 in ddH2O, 10 mM dithiothreitol (DTT).
IP elution buffer minus DTT may be stored long-term at room temperature; 10 mM DTT should be added fresh every time.
PPT solution
50% acetone
49.9% ethanol
0.1% acetic acid
Store at room temperature. Make sure that this solution is tightly capped to prevent evaporation of acetone and ethanol.
Trypsin Gold solution
1 µg/µl Trypsin Gold, 0.3% acetic acid in ddH2O
Store at -80°C in 1-µl aliquots
Urea/Tris solution
8 M urea, 50 mM Tris pH 8.0 in ddH2O
Prepare fresh
α/β casein peptide digest
5 pmol/µl trypsin-digested α-casein, 5 pmol/µl trypsin-digested β-casein, 1% acetic acid in ddH2O
Store at -20°C in 500-µl aliquots
Acknowledgments
The authors thank members of the Smolka Lab for valuable insights related to this work – particularly William Comstock for his careful testing of this protocol. We thank Beatriz S.V. Almeida for technical support. This work was supported by grants from the National Institutes of Health to M.B.S. (R35GM141159, R01GM097272, R01GM123018, and R01HD095296; Equipment Supplement R01GM097272‐07S1). The protocol was derived from experiments appearing in (Lanz et al., 2018; Sanford et al., 2021).
Competing interests
The authors declare no competing interests.
Ethics
No human or animal ethics are relevant to this protocol.
References
- Baile, M. G., Guiney, E. L., Sanford, E. J., MacGurn, J. A., Smolka, M. B. and Emr, S. D. (2019). Activity of a ubiquitin ligase adaptor is regulated by disordered insertions in its arrestin domain. Mol Biol Cell 30(25): 3057-3072.
- Bastos de Oliveira, F. M., Kim, D., Lanz, M. and Smolka, M. B. (2018). Quantitative Analysis of DNA Damage Signaling Responses to Chemical and Genetic Perturbations. Methods Mol Biol 1672: 645-660.
- Bodenmiller, B., Mueller, L. N., Mueller, M., Domon, B. and Aebersold, R. (2007). Reproducible isolation of distinct, overlapping segments of the phosphoproteome. Nat Methods 4(3): 231-237.
- Breitkopf, S. B. and Asara, J. M. (2012). Determining in vivo phosphorylation sites using mass spectrometry. Curr Protoc Mol Biol Chapter 18: Unit18 19 11-27.
- Chapman, R. D., Heidemann, M., Albert, T. K., Mailhammer, R., Flatley, A., Meisterernst, M., Kremmer, E. and Eick, D. (2007). Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science 318(5857): 1780-1782.
- Cussiol, J. R., Jablonowski, C. M., Yimit, A., Brown, G. W. and Smolka, M. B. (2015). Dampening DNA damage checkpoint signalling via coordinated BRCT domain interactions. EMBO J 34(12): 1704-1717.
- Dephoure, N., Gould, K. L., Gygi, S. P. and Kellogg, D. R. (2013). Mapping and analysis of phosphorylation sites: a quick guide for cell biologists. Mol Biol Cell 24(5): 535-542.
- Deutsch, E. W., Mendoza, L., Shteynberg, D., Farrah, T., Lam, H., Tasman, N., Sun, Z., Nilsson, E., Pratt, B., Prazen, B., Eng, J. K., Martin, D. B., Nesvizhskii, A. I. and Aebersold, R. (2010). A guided tour of the Trans-Proteomic Pipeline. Proteomics 10(6): 1150-1159.
- Faca, V. M., Sanford, E.J., Tieu, J., Comstock, W., Gupta, S., Marshall, S., Yu, H., and Smolka, M.B. (2020). Maximized quantitative phosphoproteomics allows high confidence dissection of the DNA damage signaling network. Scientific Reports: 10
- Gingras, A. C., Raught, B., Gygi, S. P., Niedzwiecka, A., Miron, M., Burley, S. K., Polakiewicz, R. D., Wyslouch-Cieszynska, A., Aebersold, R. and Sonenberg, N. (2001). Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev 15(21): 2852-2864.
- Humphrey, S. J., Karayel, O., James, D. E. and Mann, M. (2018). High-throughput and high-sensitivity phosphoproteomics with the EasyPhos platform. Nat Protoc 13(9): 1897-1916.
- Kim, J., Kundu, M., Viollet, B. and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13(2): 132-141.
- Lanz, M. C., Oberly, S., Sanford, E. J., Sharma, S., Chabes, A. and Smolka, M. B. (2018). Separable roles for Mec1/ATR in genome maintenance, DNA replication, and checkpoint signaling. Genes Dev 32(11-12): 822-835.
- Li, J., Paulo, J. A., Nusinow, D. P., Huttlin, E. L. and Gygi, S. P. (2019). Investigation of Proteomic and Phosphoproteomic Responses to Signaling Network Perturbations Reveals Functional Pathway Organizations in Yeast. Cell Rep 29(7): 2092-2104 e2094.
- Mann, M. and Jensen, O. N. (2003). Proteomic analysis of post-translational modifications. Nat Biotechnol 21(3): 255-261.
- Mueller, L. N., Brusniak, M. Y., Mani, D. R. and Aebersold, R. (2008). An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J Proteome Res 7(1): 51-61.
- Ohouo, P. Y., Bastos de Oliveira, F. M., Almeida, B. S. and Smolka, M. B. (2010). DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol Cell 39(2): 300-306.
- Ong, S. E., Blagoev, B., Kratchmarova, I., Kristensen, D. B., Steen, H., Pandey, A. and Mann, M. (2002). Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1(5): 376-386.
- Sanford, E. J., Comstock, W.J., Faça, V. M., Vega, S. C., Gnugge, R., Symington, L.S., and Smolka, M. B. (2021) Phosphoproteomics reveals a distinctive Mec1/ATR signaling response upon DNA end hyper-resection. EMBO J 40(10):e104566. doi: 10.15252/embj.2020104566.
- Zhang, L. and Elias, J. E. (2017). Relative Protein Quantification Using Tandem Mass Tag Mass Spectrometry. Methods Mol Biol 1550: 185-198.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sanford, E. J. and Smolka, M. B. (2021). Fe-NTA Microcolumn Purification of Phosphopeptides from Immunoprecipitation (IP) Eluates for Mass Spectrometry Analysis. Bio-protocol 11(15): e4113. DOI: 10.21769/BioProtoc.4113.
Category
Molecular Biology > Protein > Phosphorylation
Developmental Biology > Cell signaling
Cancer Biology > Genome instability & mutation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.