- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

DigiTAG–a RNA Sequencing Approach to Analyze Transcriptomes of Rare Cell Populations in Drosophila melanogaster
(*contributed equally to this work) Published: Vol 10, Iss 21, Nov 5, 2020 DOI: 10.21769/BioProtoc.3809 Views: 4007
Reviewed by: Lijuan DuBenjamin HousdenAnonymous reviewer(s)

Original research article
The authors used this protocol in:

Mar 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Cell-type specific transcriptional programs underlie the development and maintenance of organs. Not only distinct cell types within a tissue, even cells with supposedly identical cell fates show a high degree of transcriptional heterogeneity. Inevitable, low cell numbers are a major hurdle to study transcriptomes of pure cell populations. Here we describe DigiTAG, a high-throughput method that combines transposase fragmentation and molecular barcoding to retrieve high quality transcriptome data of rare cell types in Drosophila melanogaster. The protocol showcases how DigiTAG can be used to analyse the transcriptome of rare neural stem cells (type II neuroblasts) of Drosophila larval brains, but can also be utilized for other cell types or model systems.
Keywords: RNA SequencingBackground
Transitions between different cell types during development and tissue homeostasis are orchestrated by a plethora of transcription factors and their induced transcriptional changes. In the last decade, RNA sequencing (RNA-seq) has become the classical approach to measure transcriptional dynamics across the genome (Stark et al., 2019). Bulk RNA-seq on tissues does not allow to investigate transcriptional networks of different cell populations, specifically those of rare cell types. Thus, RNA-seq protocols that deliver high quality transcriptomes of low input samples are required.
In Drosophila, limited material often constitutes a hurdle to analyse particular tissues or cell types. This is well illustrated by Drosophila neural stem cells, called neuroblasts (Homem and Knoblich, 2012). Several distinct subpopulations of neuroblasts exist. For example, in the larval Drosophila brain only sixteen type II neuroblasts produce neurons innervating brain regions required for locomotion and sensory processing (Walsh and Doe, 2017). Defects in type II neuroblasts do not only result in aberrant neurogenesis but can also induce tumor growth (Knoblich, 2010).
To overcome low cell numbers, mutants displaying tumor-like overgrowth of particular neuroblast subpopulations have been used for bulk RNA-seq to enrich for cells-of-interest (Carney et al., 2012). Although such an approach has given insights into the heterogeneity of neural stem cells, it eliminates the possibility to distinguish between tumor- and cell type-specific changes. Cell isolation by flow cytometry (Berger et al., 2012; Harzer et al., 2013) or by robotic single-cell picking (Yang et al., 2016) have greatly advanced the purity of the isolated cell populations, but manual dissection or the navigation of special equipment still limit cell numbers. Recently, single-cell RNA-seq has been used to map the Drosophila brain (Brunet Avalos et al., 2019), however such high resolution and complex datasets might not be required to answer many scientific questions and is suboptimal due to costs, non-standard analysis, higher technical noise and loss of lowly expressed genes. Therefore, we present here a RNA-seq protocol for Drosophila neuroblasts that can be performed in any standard molecular biology laboratory using commercially available reagents (Figure 1). First, RNA is isolated and polyA-containing RNA molecules are converted into double-stranded cDNA (Figure 1A). Then the simultaneous cDNA fragmentation and ligation of sequencing adapters to each cDNA molecule by the use of transposases (Figure 1B), is time-efficient and highly reproducible. Primers tagged with unique barcodes are subsequently used to amplify the fragmented cDNA molecules and allow the identification of PCR biases, which can be removed during analysis (Figure 3). The combination of transposon-mediated library preparation with molecular barcoding to quantify the original library molecules rather than their amplicons (Figure 1) ensures high quality of the transcriptome data. Recently, DigiTAG has been successfully used to compare tumor cells to their rare cells-of-origin (Landskron et al., 2018), identify growth regulators of differently sized cells (Wissel et al., 2018) and decipher the temporal patterning of transient-amplifying progenitors (Abdusselamoglu et al., 2019).
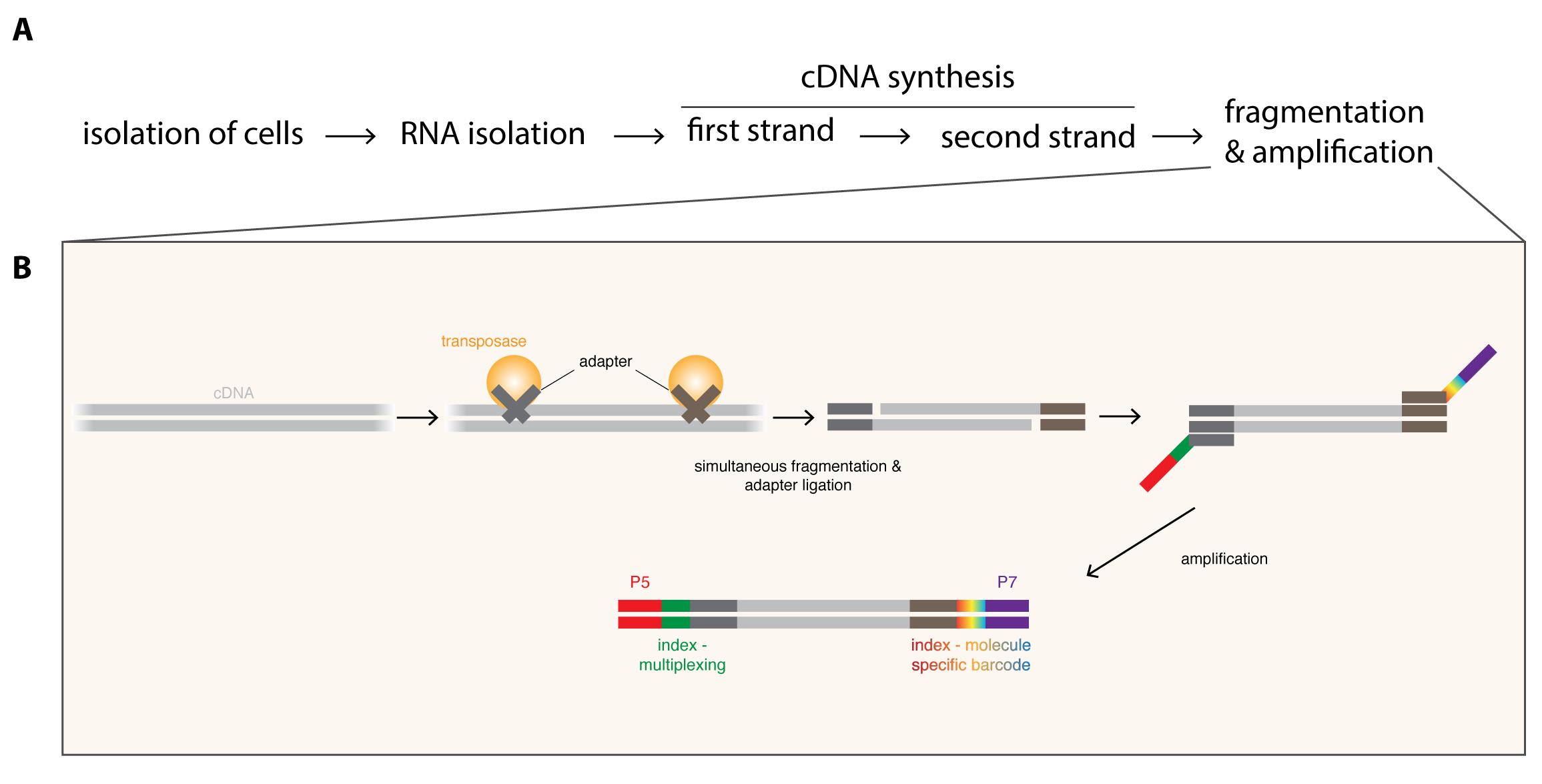
Figure 1. Overview of the DigiTAG workflow. A. RNA is isolated from the purified cells-of-interest and converted into double-stranded cDNA. To generate the final sequencing library, cDNA molecules are fragmented and amplified (for more detail see B). B. Generation of sequencing libraries from cDNA. By using transposases, cDNA is simultaneously fragmented and adapters (dark grey and brown) are ligated. In the following amplification step these adapters serve as binding sites for two types of primers. Forward primers include the Illumina flow cell attachment sequence P5 (red) together with a sample-specific index sequence (green, allowing to multiplex several RNA-seq samples if desired), whereas reverse primers contain the Illumina flow cell attachment sequence P7 (purple) with a molecule-specific index sequence (rainbow-colored, allowing to barcode each amplified molecule uniquely).
Materials and Reagents
Hardshell 96-Well PCR Plate, low profile, thin wall, skirted, white/clear (Bio-Rad, catalog number: HSP9601 )
Microseal ‘B’ PCR Plate Sealing Film, adhesive, optical (Bio-Rad, catalog number: MSB1001 )
Refrigerated centrifuge
DNA LoBind tubes 1.5 ml (Eppendorf, catalog number: 0030108051)
Filter tips for micropipettes
Drosophila
TrizolTM LS (ThermoFisher Scientific, catalog number: 10296028 ) (Storage: 4 °C until expiration date)
Nuclease-free Water (ThermoFisher Scientific, catalog number: AM9932 ) (Storage: Room temperature until expiration date)
InvitrogenTM GlycoBlueTM Coprecipitant (ThermoFisher Scientific, catalog number: AM9515 ) (Storage: -20 °C until expiration date)
Chloroform (Sigma-Aldrich, catalog number: C2432 , handle in fume hood!)
70% and 75% ethanol, prepared from absolute Ethanol (Sigma-Aldrich, catalog number: 32205 , store at room temperature in fireproof cabinet), diluted with Nuclease-free water.
Isopropanol (Sigma-Aldrich, catalog number: 278475 , store at room temperature in fireproof cabinet)
Oligo(dT)20 (50 μM) (ThermoFisher Scientific, catalog number: 18418020 ) (Storage: -20 °C until expiration date)
dNTPs (10 mM each) (ThermoFisher Scientific, catalog number: R0193 ) (Storage: -20 °C until expiration date)
5x First Strand Buffer, DTT (100 mM) and SuperScript III (200 U/μl) are all part of the kit: SuperScriptTM III Reverse Transcriptase (ThermoFisher Scientific, catalog number: 18080093 ) (Storage: -20 °C until expiration date)
MgCl2 (25 mM) (ThermoFisher Scientific, catalog number: R0971 ) (Storage: -20 °C until expiration date)
RNaseOUT (40 U/μl) (ThermoFisher Scientific, catalog number: 10777019 ) (Storage: -20 °C until expiration date)
5x Second Strand Buffer (ThermoFisher Scientific, catalog number: 10812014 ) (Storage: -20 °C until expiration date)
RNase H (5 U/μl) (ThermoFisher Scientific, catalog number: EN0202 ) (Storage: -20 °C until expiration date)
DNA Polymerase I (10 U/μl) (ThermoFisher Scientific, catalog number: 18010017 ) (Storage: -20 °C until expiration date)
AMPure XP Beads (Beckman Coulter, catalog number: A63880 ) (Storage: 4 °C until expiration date)
EB Buffer (Qiagen, catalog number: 19086 ) (Storage: Room temperature until expiration date)
2x Phusion HF master mix (ThermoFisher Scientific, catalog number: F531S ) (Storage: -20 °C until expiration date)
TDE Tagment DNA buffer and TDE1 Tagment DNA enzyme are all part of the kit: Illumina Tagment DNA TDE1 Enzyme and Buffer Kits (Illumina, catalog number: 20034197 ) (Storage: -20 °C until expiration date)
20x Eva Green (Biotum, catalog number: 31000 ) (Storage: -20 °C until expiration date)
Nextera PCR, modified Index one and Index two primers are all part of the kit: Nextera DNA Library Preparation Kit (24 samples) (Illumina, catalog number: FC-121-1030 ) (Storage: -20 °C until expiration date)
Real-Time Standard 1-4 are all part of the kit: KAPA Real-time Library Amplification kit (KAPA Biosystems, catalog number: KK2709 ) (Storage: -20 °C until expiration date)
Equipment
Touch Real-Time PCR Detection System (Bio-Rad, model: CFX96 )
Thermomixer Compact with 1.5 ml block (Eppendorf, model: Thermomixer Compact )
Magnet rack (Fisher Scientific, InvitrogenTM DYNALTM DynaMagTM DynabeadsTM DynaMag-2 Magnet, catalog number: 10723874 )
0.2-2 μl micropipette
10-100 μl micropipette
20-200 μl micropipette
100-1,000 μl micropipette
Software
CFX MaestroTM Software (Bio-Rad)
Procedure
Isolation of cells-of-interest
The cell type-of-interest can be isolated through various different methods. For Drosophila neuroblasts, isolation by flow cytometry is recommended. Briefly, Drosophila brain cells as for example type II neuroblasts cells are labeled using fluorophore-expressing driver lines (e.g., worniu-GAL4, ase-GAL80, UAS-CD8::GFP for type II neuroblasts). Brains are manually dissected and dissociated using enzymatic treatment followed by mechanical disruption. Single-cell suspensions are then subjected to flow cytometry. Neuroblasts identified as a separate cell population, which includes the largest GFP positive cells, are isolated and either stored at -80 °C in TrizolTM or immediately used for RNA isolation. For a step-by-step protocol see Harzer et al. (2013).
RNA isolation
- Collect a minimum of 100 and up to 105 cells of interest with a minimal estimated volume of ~524 μm3 (or ~10μm of diameter) per cell (e.g., type II Neuroblasts) by FACS directly into 750 μl of TrizolTM LS in a 1.5 ml DNA LoBind tube.
Importantly, using less than this amount of initial material will lead to a significantly lowered power to detect transcripts. We advise users experimenting on other cell types to adjust the minimal number of cells proportionally in consideration with their cells-of-interest estimated volume. - Adjust the total volume of each sample to 1 ml with Nuclease-free Water.
- Add 1 μl of GlycoBlueTM Coprecipitant (15 mg/ml) to allow low amount of RNA to be recovered from the RNA extraction.
- Mix by inverting the tubes 4-6 times.
- Optional: store your samples at -80 °C for up to one month.
- Add 200 μl of ~100% pure chloroform (safety note: handle chloroform in fume hood).
- Vortex the tubes for 15 s.
- Incubate for 2 min at room temperature.
- Centrifuge the samples at maximal speed (~24,000 x g) for 15 min at 4 °C in a refrigerated centrifuge.
- Carefully aspirate most of the upper aqueous phase (leave a small fraction to prevent unwanted contamination from the organic phase) preferably with a 20-200 μl micropipette, and transfer it into a new 1.5 ml DNA LoBind tube.
- Add 500 μl of ~100% pure isopropanol to precipitate the RNA.
- Incubate the samples for 15 min at -20 °C to optimize RNA precipitation.
- Optional: keep your samples at -20 °C for up to one week.
- Vortex the tubes for 15 s.
- Centrifuge the samples at maximal speed (~24,000 x g) for 15 min at 4 °C in a refrigerated centrifuge.
- Carefully remove all the supernatant.
- Add 1 ml of ice-cold 75% ethanol to improve the purity of the RNA pellet.
- Vortex the tubes for 15 s.
- Centrifuge the samples at maximal speed (~24,000 x g) for 15 min at 4 °C in a refrigerated centrifuge.
- Carefully remove all the supernatant.
- Remove the very last drop of liquid from each tube with a micropipette but importantly do not let the RNA pellet dry (it will prevent its subsequent solubilization).
- Resuspend the RNA pellet in 7 μl of EB Buffer. Such a low volume is required to enable subsequent reactions to be performed on the whole sample in the situation when the minimal amount of cells was used for the sample’s preparation.
- Collect a minimum of 100 and up to 105 cells of interest with a minimal estimated volume of ~524 μm3 (or ~10μm of diameter) per cell (e.g., type II Neuroblasts) by FACS directly into 750 μl of TrizolTM LS in a 1.5 ml DNA LoBind tube.
Reverse Transcription
- In order to synthesize the first strand of cDNA, add the following reagents to the 7 μl of RNA:
1 μl of oligo(dT)20 (50 μM)
1 μl of dNTPs (10 mM each)
4 μl of 5x First Strand Buffer
4 μl of MgCl2 (25 mM)
2 μl of DTT (100 mM)
0.5 μl of RNaseOUT (40 U/μl)
0.5 μl of SuperScript III (200 U/μl) - Mix well and spin down.
- Incubate 50 min at 50 °C and then 5 min at 85 °C.
- In order to synthesize the first strand of cDNA, add the following reagents to the 7 μl of RNA:
Second Strand Synthesis
- In order to degrade the RNA strand and synthesize a second strand of cDNA instead, add the following reagents to the first strand reaction and mix well:
5 μl of 5x Second Strand Buffer
3 μl of dNTPs (10 mM each)
2 μl of RNase H (5 U/μl)
1 μl of DNA Polymerase I (10 U/μl)
19 μl of Nuclease-free Water - Incubate 2.5 h at 16 °C. The sample contains now double-stranded cDNA.
- Optional: store your samples at 4 °C for up to one day.
- In order to degrade the RNA strand and synthesize a second strand of cDNA instead, add the following reagents to the first strand reaction and mix well:
DNA Purification
- To purify double-stranded cDNA molecules from the previous reaction mixes, add 90 μl of well-suspended AMPure XP Beads.
- Mix by pipetting 10 times. The reaction turns brownish due to the color of the beads. A homogenous color indicates that the reaction is well mixed (Figure 2).
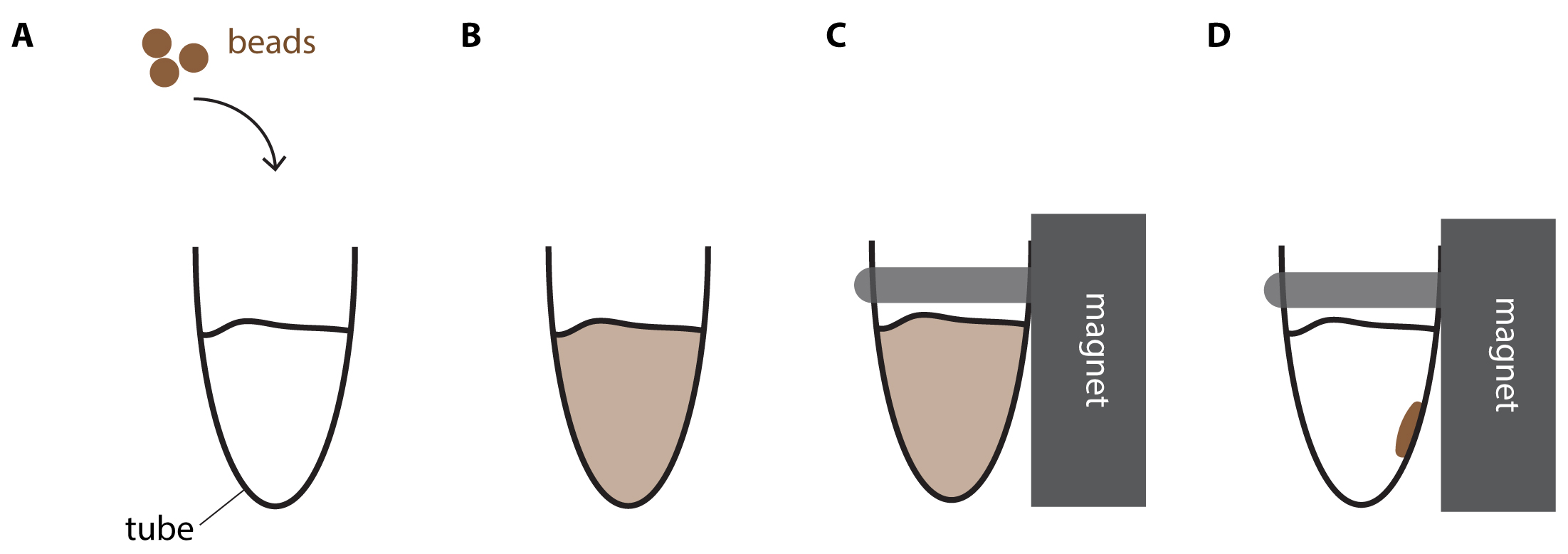
Figure 2. DNA purification using magnetic beads. A. Brownish beads are added to the reaction mix. B. After thoroughly mixing the reaction turns homogenously brown. C. Tube is placed into the magnet rack. D. Shortly after, the reaction mix becomes clear and the brown beads accumulate at the wall closest to the magnet. - Incubate for 5 min at Room-Temperature.
- Put the samples on a magnet rack. Wait 2-3 min or until all beads have migrated to the wall of the tube closest to the magnet (Bead migration is completed when brown beads accumulate clearly visible on the wall of the tube next to the magnet and the reaction is cleared and thus appears transparent, see also Figure 2).
- Wash twice with 70% ethanol while leaving the tube on the magnet rack. Take care not to disturb the beads. If the pellet of beads is disturbed, wait for 1-2 min to let the beads migrate again to the tube’s wall close to the magnet.
- After washing remove all ethanol using a pipette and let all ethanol evaporate for 2 min at Room-Temperature.
- Remove samples from magnet and elute the DNA by resuspending the beads in 15.5 μl of EB buffer by pipetting up and down. Wait one min and place the tubes back on the magnetic rack for 2-3 min to allow all beads to migrate to the wall of the tube closest to the magnet. Transfer 15 μl of the completely cleared eluate to a new DNA low binding 1.5 ml tube.
Tagmentation
- In order to uniquely tag each double-stranded DNA molecule, add on ice the following reagents to the15 μl of double-stranded and purified cDNA from Step E7, and mix:
15 μl of TDE Tagment DNA buffer
0.2 μl of TDE1 Tagment DNA enzyme - Incubate 5 min at 55 °C.
- Purify using 54 μl of AMPure XP beads (see Procedure E. DNA Purification).
- Elute in 20 μl EB Buffer.
- In order to uniquely tag each double-stranded DNA molecule, add on ice the following reagents to the15 μl of double-stranded and purified cDNA from Step E7, and mix:
PCR amplification
- In order to amplify each uniquely tagmented double-stranded DNA molecule to the concentrations required for RNA-sequencing, add the following reagents to the 19.5 μl of tagmented and purified double-stranded cDNA from Step F4 in a single well of a 96-Well qPCR plate and mix well:
25 μl of 2x Phusion HF master mix
2.5 μl 20x Eva Green
1 μl of Nextera PCR primers (10 μM each)
1 μl of Nextera modified Index one primers (“N7XX”; includes a random 8-mer tags for barcoding; 0.5 μM each)
1 μl of Nextera Index two primer (“N501-506”, for multiplexing; 0.5 μM) - Include the four Real-Time fluorescent Standards in duplicates by directly pipetting 50 μl from each Real-Time fluorescent Standards’ tube in separated wells of the qPCR plate.
- Incubate in a CFX96 Touch Real-Time PCR Detection System:
- 3 min at 72 °C
- 30 s at 98 °C
- 10 s at 98 °C
- 30 s at 63 °C
- 3 min at 72 °C (plate read)
- Repeat steps c) to e) up to 25 times (see also Step G5)
- Use the “no baseline subtraction” setting (in User Preference Settings in the Bio-Rad, CFX MaestroTM Software) in order to monitor raw fluorescence amplifications in parallel to the unchanging Real-Time Fluorescent Standards.
- Stop the PCR reaction right after plate read (3.e) and before denaturation (3.c) when an optimal amplification is reached (when the curve reaches a fluorescence level between standard 2 and standard 4; stopping the reaction at this point is key to reach a compromise between sufficient specific amplification of the sample libraries and minimized amplification of non-specific PCR products such as primers dimers) and recover the samples in 1.5 ml DNA LoBind tubes on ice. Optimal amplification is typically reached after 18 to 20 cycles when using a starting material of 100 Neuroblasts or after 14 to 15 cycles when using a starting material of 1,000 Neuroblasts.
- Purify using 90 μl of AMPure XP beads (see Procedure E. DNA Purification).
- Elute in 20 μl EB buffer and send for 50 base-pair Illumina Single-end sequencing. Prior to sending to sequencing, the quality of libraries can be checked by performing quantitative PCR on a house-keeping gene and the resulting amplification should be comparable to an amplification performed in the same settings, using the same DNA amount from a reference cDNA sample (for example, from a whole-tissue extraction).
- In order to amplify each uniquely tagmented double-stranded DNA molecule to the concentrations required for RNA-sequencing, add the following reagents to the 19.5 μl of tagmented and purified double-stranded cDNA from Step F4 in a single well of a 96-Well qPCR plate and mix well:
Data analysis
Summary of the analysis
Most critical analysis steps are the following (see also Figure 3): In a first step, sequencing reads are aligned to the genome. Ribosomal RNA reads are subtracted from all aligned reads. Next, reads that arose due to biased amplification (in the step shown in Figure 1B) are identified using the molecule-specific barcode. At each position in the genome the different barcodes are counted. Reads sharing the same barcode are marked as “duplicated reads”. Only the read with the highest quality (this can be assessed with the PHRED score) is kept while all others are removed. The aligned and deduplicated reads are then counted and polyA-containing transcripts are subjected to a differential expression analysis (for example DESeq, Anders and Huber, 2010).
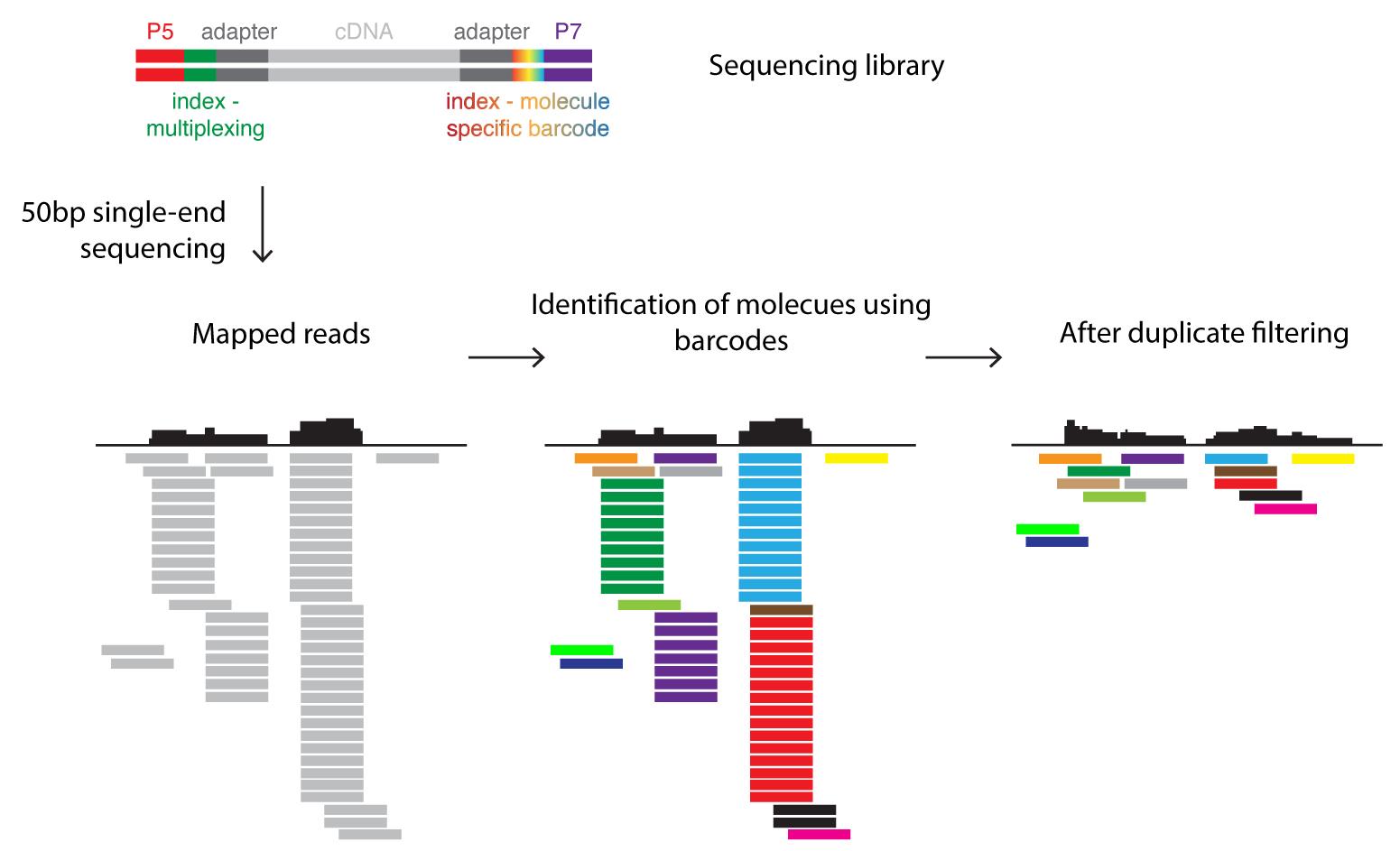
Figure 3. Barcoding strategy for data analysis. The sequencing library (see also Figure 1) contains DNA molecules with sequences allowing the attachment to the flow cell (P5/P7) and indices. The sample-specific index (green) can be used to pool several sequence libraries for a single sequencing run (multiplexing). The molecule-specific barcode is unique for each fragmented, double-stranded DNA molecule. Sequencing reads are mapped to the reference genome. The molecule-specific barcodes identify PCR amplicons derived from the same DNA molecule and thus allow the correction of such PCR bias by filtering duplicates.Step by step procedure of the RNA-seq data analysis
- Demultiplex reads based on “multiplex BC”:
- Create a FASTQ file for each multiplex BC (exact match or edit distance ≤ 2 based)
- Each sequence ID must have following structure: “@readID:multiplexBC:indexMolecule”
- Remove reads of ribosomal RNA origin:
- Align against rRNA sequences derived from RefSeq (BWA, http://bio-bwa.sourceforge.net/bwa.shtml)
- Retrieve un-aligned reads (samtools; http://www.htslib.org/doc/samtools.html)
- Convert un-aligned reads to FASTQ format
- Align reads to the genome with TopHat (https://ccb.jhu.edu/software/tophat/manual.shtml):
- Use the Drosophila melanogaster genome (FlyBase)
- Use min/max intron range of 20-150,000 bp (FlyBase statistics)
- Enable microexon-search
- Provide a gene model as GTF (FlyBase)
- Use our provided custom perl script (digiTag.pl; https://gitlab.com/ii-bioinfo-thomas/ii-digitag) to mark reads arising from duplication events. The index molecule (barcode) must be stored in ID (see 1b). In short the script does following:

- The different barcodes are counted at each genomic position.
- The diversity of barcodes at each position is examined.
- Barcodes are sorted descending by their count.
- If several barcodes have the same occurrence, they are further sorted alphanumerically.
- Reads sharing the same barcode, are sorted by the average PHRED quality.
- If several reads have the same quality, they are further sorted alphanumerically.
- The barcodes are cycled through by their counts.
- Within one barcode, the read with the highest average PHRED quality is the unique correct read and all subsequent reads with the same barcode are marked as duplicates.
- All reads which have barcodes with one mismatch difference compared the pool of valid read barcodes are also marked as duplicates.
- Count with HTSeq (https://htseq.readthedocs.io/en/latest/count.html):
- Set stranded to “no”
- Set mode to “intersection-nonempty”
- use a GTF (FlyBase) without snRNA, rRNA, tRNA, snoRNA and pseudogenes
- Perform standard differential expression analysis with DESeq2 (https://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html)
Further documentation of the analysis can be found in the method section of Landskron et al. (2018) and all data are available on this link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE87085.
- Demultiplex reads based on “multiplex BC”:
Notes
Always use 1.5 ml DNA LoBind tube along the protocol to limit DNA loss.
Always use Nuclease-free Water to prepare ethanol solutions.
Acknowledgments
We would like to particularly thank Jonas Steinman and Heike Harzer for pioneering DigiTAG. This work was funded by the Austrian Academy of Sciences, Austrian science Fund and the European Commission. F.B. was supported by the European Molecular Biology Organization. This protocol is derived from Landskron et al. (2018).
Competing interests
Authors declare no competing interests.
References
- Abdusselamoglu, M. D., Eroglu, E., Burkard, T. R. and Knoblich, J. A. (2019). The transcription factor odd-paired regulates temporal identity in transit-amplifying neural progenitors via an incoherent feed-forward loop. Elife 8: e46566.
- Anders, S. and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol 11(10): R106.
- Brunet Avalos, C., Maier, G. L., Bruggmann, R.and Sprecher, S. G. (2019). Single cell transcriptome atlas of the Drosophila larval brain. Elife 8: e50354.
- Berger, C., Harzer, H., Burkard, T. R., Steinmann, J., van der Horst, S., Laurenson, A. S., Novatchkova, M., Reichert, H. and Knoblich, J. A. (2012). FACS purification and transcriptome analysis of Drosophila neural stem cells reveals a role for Klumpfuss in self-renewal. Cell Rep 2(2): 407-418.
- Carney, T. D., Miller, M. R., Robinson, K. J., Bayraktar, O. A., Osterhout, J. A. and Doe, C. Q. (2012). Functional genomics identifies neural stem cell sub-type expression profiles and genes regulating neuroblast homeostasis. Dev Biol 361(1): 137-146.
- Harzer, H., Berger, C., Conder, R., Schmauss, G. and Knoblich, J. A. (2013). FACS purification of Drosophila larval neuroblasts for next-generation sequencing. Nat Protoc 8(6): 1088-1099.
- Homem, C. C. and Knoblich, J. A. (2012). Drosophila neuroblasts: a model for stem cell biology. Development 139(23): 4297-4310.
- Knoblich, J. A., (2010). Asymmetric cell division: recent developments and their implications for tumour biology. Nat Rev Mol Cell Biol 11(12): 849-860.
- Landskron, L., Steinmann, V., Bonnay, F., Burkard, T. R., Steinmann, J., Reichardt, I., Harzer, H., Laurenson, A. S., Reichert, H. and Knoblich, J. A. (2018). The asymmetrically segregating lncRNA cherub is required for transforming stem cells into malignant cells. Elife 7: e31347.
- Stark, R., Grzelak, M. and Hadfield, J. (2019). RNA sequencing: the teenage years. Nat Rev Genet 20(11): 631-656.
- Walsh, K. T. and Doe, C. Q. (2017). Drosophila embryonic type II neuroblasts: origin, temporal patterning, and contribution to the adult central complex. Development 144(24): 4552-4562.
- Wissel, S., Harzer, H., Bonnay, F., Burkard, T. R., Neumuller, R. A. and Knoblich, J. A. (2018). Time-resolved transcriptomics in neural stem cells identifies a v-ATPase/Notch regulatory loop. J Cell Biol 217(9): 3285-3300.
- Yang, C. P., Fu, C. C., Sugino, K., Liu, Z., Ren, Q., Liu, L. Y., Yao, X., Lee, L. P. and Lee, T. (2016). Transcriptomes of lineage-specific Drosophila neuroblasts profiled by genetic targeting and robotic sorting. Development 143(3): 411-421.
Article Information
Copyright
![]() Landskron et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Landskron et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Landskron, L., Bonnay, F., Burkard, T. R. and Knoblich, J. A. (2020). DigiTAG–a RNA Sequencing Approach to Analyze Transcriptomes of Rare Cell Populations in Drosophila melanogaster. Bio-protocol 10(21): e3809. DOI: 10.21769/BioProtoc.3809.
- Landskron, L., Steinmann, V., Bonnay, F., Burkard, T. R., Steinmann, J., Reichardt, I., Harzer, H., Laurenson, A. S., Reichert, H. and Knoblich, J. A. (2018). The asymmetrically segregating lncRNA cherub is required for transforming stem cells into malignant cells. Elife 7: e31347.
Category
Developmental Biology > Cell growth and fate > Neuron
Developmental Biology > Cell signaling > Fate determination
Molecular Biology > DNA > DNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.