- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantitative Kinetic Analyses of Histone Turnover Using Imaging and Flow Cytometry
Published: Vol 10, Iss 17, Sep 5, 2020 DOI: 10.21769/BioProtoc.3738 Views: 5721
Reviewed by: Imre GáspárYong-Yu LiuPooja Verma
Original research article
The authors used this protocol in:

Jun 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Dynamic histone changes occur as a central part of chromatin regulation. Deposition of histone variants and post-translational modifications of histones are strongly associated with properties of chromatin status. Characterizing the kinetics of histone variants allows important insights into transcription regulation, chromatin maintenance and other chromatin properties. Here we provide a protocol of quantitative and sensitive approaches to test the timing of incorporation and dissociation of histones using a two-color SNAP-labeling system, labelling pre-existing and newly-incorporated histones distinctly. Together with cell cycle synchronization methods and cell cycle markers, this approach enables a pulse-chase analysis to determine the turnover of histone variants during the cell cycle, detected using imaging or flow cytometry methods at single cell resolution. As well as testing global histone turnover, cell cycle-dependent cellular localization of histone variants can be also addressed using imaging approaches.
Keywords: Chromatin dynamicsBackground
Chromatin remodeling is part of the numerous fundamental cellular activities in eukaryotic cells (Geiman and Robertson, 2002; Clapier and Cairns, 2009). Accessibility by transcription factors and RNA polymerases are generally associated with changes in DNA methylation and chromatin states, including accessibility, post-translational histone modifications and deposition of histone variants. Histone variants differentially coordinate the gene expression that regulates development, cell differentiation or other physiological activities (Banaszynski et al., 2010). They also play diverse roles in DNA repair, telomere maintenance, heterochromatin formation and chromatin segregation (Henikoff and Smith, 2015; Zink and Hake, 2016). Moreover, dysregulation of a histone variant’s incorporation is associated with cancer (Vardabasso et al., 2014), indicating a significant role in human disease. To understand the kinetics of histone variants, including incorporation into and dissociation from specific chromatin regions during the cell cycle, is crucial since it tightly links regulatory properties with the mitotic maintenance of epigenetic (heritability from parent to daughter cells). DNA replication involves major chromatin remodeling to duplicate the entire chromatin structure following mitosis. Histones that associate with chromatin prior to DNA replication are transiently dissociated from DNA by the access of the DNA polymerase complex. Release of pre-existing histones randomly re-associate at newly synthesizing replication forks together with newly synthesized histones (Balhorn et al., 1975; Alabert and Groth, 2012; Annunziato, 2012). Pre-existing post-translational modifications on histones and some histone variants also re-associate on the newly synthesized DNA at the replication fork, explaining re-association of pre-existing histone at the replication fork is one part of the maintenance of mitotic inheritance of chromatin states. To determine the timing of post-translational modifications or incorporation of histone variants, a sensitive pulse-chase system that can distinguish the detection of newly incorporated histone from pre-existing histones is required. Newly synthesized canonical histones are unmodified when they are incorporated into chromatin during DNA replication. Unlike de novo DNA methylation, which occurs together with DNA replication (Tillo et al., 2016), most histone marks are not established on newly incorporated histones on the replicating fork. Proteomic based pulse-chase approaches (see in Alternative methods section below) have been used to determine the global kinetics of histone post-translational modifications by detecting the pre-existing and new deposition of histone acetylation and methylation at lysine residues (Pesavento et al., 2008; Scharf et al., 2009; Martinez-Garcia et al., 2011; Xu et al., 2011; Zee et al., 2012; Alabert et al., 2015). This approach identified two distinctive kinetic patterns of histone modifications. One group, such as histone H3 acetyl-lysine at 27 (H3 K27 ac), exhibits rapid turnover to equalize, and notably they are not maintained through the cell cycle (Scharf et al., 2009). This rapid acetylation kinetics probably represent temporally active transcription dynamics (Stasevich et al., 2014). While another group including H3 lysine tri-methylation at 9 (K9me3) or H3 K27me3 is acquired more slowly and step-wise from mono and di to tri-methylation, established during G1 phase following the cell cycle instead of before mitosis (Pesavento et al., 2008; Scharf et al., 2009; Martinez-Garcia et al., 2011; Xu et al., 2011; Zee et al., 2012; Alabert et al., 2015). Importantly, this group has the property of mitotic chromosome memory. The potential mechanism of maintenance of histone marks over cell division has been addressed by imaging approaches. In situ proximity ligation assays using specific antibodies against histone lysine methylations and their methyltransferases detected that histone modifiers continuously associate with the replicating DNA component in Drosophila embryos, suggesting the association of modifiers on replicating DNA may provide a “tag” to be methylated (Petruk et al., 2012).
Recent advances in chemical protein labeling technologies provide us with a powerful tool for protein tagging applications in living cells (e.g., SNAP (New England Biolabs), CLIP (New England Biolabs), Halo (Promega) and TMP (Active Motif) tag). These technologies are based on the covalent labeling of genetically encoded tags that bind with specific ligands conjugated to cell permeable substrates such as synthetic fluorescent dyes or biotin, which can mediate affinity purification in biochemical applications. In contrast with common genetically encoded tags, this chemical labeling of protein can be utilized in timing-dependent labeling with many choices of fluorophores, which allows the pulse-chase labeling of specific protein. This labeling technology has revealed the deposition timing of histone variants at specific chromatin architectures using imaging detection (e.g., CENP-A at centromeres [Dunleavy et al., 2009] and macroH2A at heterochromatin [Sato et al., 2019]).
Here, we provide a detailed protocol of a pulse-chase method using the SNAP-tag labeling system which has utilized quantitative histone variant detection with single cell resolution. Using this protocol, distinct histone kinetics, dissociation of pre-existing histones and association of newly synthesized histones, can be detected simultaneously. We describe two detection approaches, fluorescence microscopy and flow cytometry, as well as the detail of imaging analysis using FIJI/ImageJ software which is freely available (https://fiji.sc/).
Advantages and Limitations
The pulse-chase method using a chemical protein labeling system is an easy, non-hazardous and sensitive approach compared with a conventional pulse-chase approach using radioactive molecules (see in Alternative methods section). Most of the required reagents and fluorophores are commercially available. Global turnover of histones can be addressed using imaging and flow cytometric applications and notably, timing-specific localization at specific chromatin architectures also can be characterized with imaging approaches. Using this protocol, both pre-existing and newly incorporated histone variants can be detected simultaneously in the same cells with single cell resolution. Unlike radioisotope or metabolic labeling approaches, which detect endogenous histones, this labeling approach relies on the genetically encoded tags (e.g., SNAP-, CLIP-, Halo-tag) that can be linked with specific substrates. Therefore, a plasmid construct that expresses the target histones with SNAP-tag and its use in a stably expressing cell line are required. In addition, the localization and other biological functions of desired tagging histones must be examined to determine whether it remains functioning as an endogenous histone. Optimization of the construct (e.g., N-terminus or C-terminus tagging, changing the choice of promoter) and levels of expression might be necessary for obtaining accurate observations. Another limitation of this approach is that it is not applicable for detection of post-translational modifications of histones.
Alternative methods
Isotope labelling with proteomic detection
SILAC (Stable Isotope Labelling with Amino acids in Culture) followed by mass spectrometry is a powerful approach to investigate global turnover of endogenous histone variants and post-translational modifications (Yuan et al., 2014). In this approach, newly synthesized histones are labeled with radioactive heavy isotope and chase the turnover of labeled his tones compared with pre-existing histones containing light amino acids. Following mass spectrometry analysis determines the pre-existing and deposition of post-translational modifications or variants. This approach is suitable to detect global histone turnover, but is not able to detect histone marks at specific chromatin architecture or genomic loci.
Metabolic labeling with genome-wide approaches
Non-radioactive metabolic labeling of nascent proteins can be an alternative approach to label global newly synthesized histones. The approach, “Covalent Attachment of Tags to Capture Histones and Identify Turnover”, also called ‘CATCH-IT’, enables genome-wide investigation to characterize active histone replacement (Deal et al., 2010). This approach is based on the labeling scheme of nascent peptide by incorporation of methionine homolog, azidohomoalanine (Aha), which is generally used for the detection of active translation in cells. In this approach, the nucleosomes containing Aha-labeled newly synthesized histones are bioconjugated with biotin by a cycloaddition reaction (as known as “click” chemistry), and pulled down with streptavidin beads. Isolated DNA from pull-down was applied on a tiling microarray to determine the genomic loci that exhibit active histone replacement in Drosophila S2 cells. The characterized genomic sites that have active histone turnover correspond with the site of incorporation of histone H3 variant, H3.3, which is detected at transcriptionally active loci (Henikoff et al., 2009). This approach enables the detection of genomic loci with active turnover of histones.
Chemical labeling approaches such as the SNAP-tagging system can also be the alternative option to investigate genome-wide histone variant incorporation (Sato et al., 2019). In this approach, newly incorporated SNAP-tagged histones are linked with SNAP-biotin after the treatment of SNAP-Cell® Block (bromothenylpteridine, BTP), a non-fluorescent substrate to mask the reactivity of pre-existing histones. Then, biotin-linked, newly incorporated histones can be pulled down with streptavidin beads. The purified DNA fraction from pull-down samples can be sequenced with massive parallel sequencing. This approach might be useful to detect timing and genomic loci dependent incorporation of histone variants but unable to detect post-translational modifications.
Materials and Reagents
- 50-ml Falcon tubes
- 15-ml Falcon tubes
- 1.5-ml tubes
- Tissue culture plates (12 or 24-well; Falcon, catalog number: 353043 or 353047 )
- Tissue culture dishes (35 x 10 mm or 60 x 15 mm, and 100 x 20 mm, Falcon, catalog numbers: 353001 or 353002 and 353003 )
- Coverslips (18 or 12 mm No.1.5)
- Microscope slides (e.g., Fisher Scientific, catalog number: 12-544-2 )
- 5-ml FACS tubes (Fisher Scientific, catalog number: 149595 )
- Adherent cells of interest (e.g., mouse embryonic fibroblasts [MEFs], HEK293 cells)
- SNAP-Cell Oregon Green (New England BioLabs, catalog number: S9104S )
- SNAP-Cell Block (New England BioLabs, catalog number: S9106S )
- SNAP-Cell TMR-Star (New England BioLabs, catalog number: S9105S )
- Thymidine (Sigma, catalog number: T1895 )
- RO-3306 (Santa Cruz Biotechnology, catalog number: sc-358700 )
- Nocodazole (Sigma, catalog number: M1404 )
- Prolong Diamond Antifade Mountant (Thermo Fisher Scientific, catalog number: P36970 )
- Prolong Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific, catalog number: P36962 )
- Drug for selection of stably expressing cells [e.g., 600-1,200 µg/ml G418 (geneticin), Zeocin (Thermo Fisher Scientific, catalog number: R25001 )]
- Click-iTTM EdU Cell Proliferation Kit for Imaging, Alexa FluorTM 647 dye (Thermo Fisher Scientific, catalog number: C10340 )
- pSNAPf Vector (New England BioLabs, catalog number: N9183S )
- pCCL-CellCycle (Sato et al., 2019)
- Suitable culture media and supplements [e.g., DMEM (Sigma, catalog number: D6429 ) supplemented with 10% FBS (Atlanta Biologicals, Inc., catalog number: S11150H ) and 100 U/ml Penicillin-Streptomycin (Sigma, catalog number: 15140148 )]
- EDTA, 0.5 M, pH 8.0, Molecular Biology Grade (Sigma, catalog number: 324506 )
- HEPES solution, 1 M, pH 7.0-7.6, sterile-filtered (Sigma, catalog number: H0887 )
- Coverslips coating solution, e.g., 0.01% Poly-L-lysine solution (Sigma, catalog number: P4707 ), collagen coating solution (according to the manufacturer's instruction, Sigma, catalog number: SAFC-125-50 )
- Sterile 1x PBS pH 7.4, no calcium, no magnesium for cell culture (e.g., Corning, catalog number: 21-031-CV )
- 10× DPBS, (e.g., Roche, catalog number: 11666789001 )
- 32% (wt/vol) paraformaldehyde (PFA) (Electron Microscopy Sciences, catalog number: 15714 )
!CAUTION: Paraformaldehyde is a hazardous solution and a cross-linking agent. Wear protective gloves and handle it under a fume hood. - Triton X-100, 0.1% (vol/vol) (Thermo Fisher Scientific, catalog number: BD 151500 )
- Nuclease-free water (Thermo Fisher Scientific, catalog number: 10977-015 )
- Immersion oil 1.518, for the microscope/objective
- Bovine serum albumin (BSA) (Sigma, catalog number: A7030 )
- Cloning of SNAP-histone expression vector and its stably expressing cell line (see Recipes)
- Fixation Buffer (see Recipes)
- Permeabilization buffer (see Recipes)
- Blocking buffer (see Recipes)
- Sorting Buffer (see Recipes)
Equipment
- Vortex mixer
- Tabletop centrifuge
- Pointed tip tweezer
- Water bath
- Biological hood/biosafety cabinet (for cell culture work)
- Cell culture incubator suitable for cell culture of your choice (e.g., 37 °C, 5% CO2)
- Wide-field fluorescent microscope (e.g., Olympus, model: BX-61 , equipped with four filter sets for DAPI (Semrock, model: DAPI-5060C-Zero ), Cy3 (Chroma, model: 41007 ), FITC (Semrock, model: FITC-5050A-Zero), and Cy5 (Semrock, model: Cy5-4040C-Zero ), an EXFO X-Cite Series 120 PC metal halide light source, Photometrics Cool SNAP HQ CCD camera, and molecular Devices Metamorph acquisition software or equivalent microscope set-ups or equivalent microscope system
- Flow cytometer (BD Biosciences, model: LSR II )
Software
- Metamorph software for image acquisition (https://www.moleculardevices.com/systems/metamorph-research-imaging/metamorph-microscopy-automation-and-image-analysis-software)
- ImageJ/FIJI (Schindelin et al., 2012) (avalable at https://fiji.sc/)
- FlowJo (https://www.flowjo.com/solutions/flowjo/downloads)
Procedure
The protocol contains five main processes: (i) Generation of the SNAP-tagged histone expression vector and stable expressing cell lines, (ii) Optimization of cell cycle synchronization, (iii) Detection of global and local histone incorporation using imaging, (iv) Detection of global histone incorporation using flow cytometry, (v) Analysis. Although the first process (i) explains general procedures for cloning and establishing stably expressing cell lines, we emphasize the tips on how to design the SNAP-tagged histone expression vector and isolation of stably expressing cells. This protocol mainly describes the procedures for the fluorescent labeling approaches to investigate histone turnover using microscope (iii) and flow cytometry (iv) (Figure 1).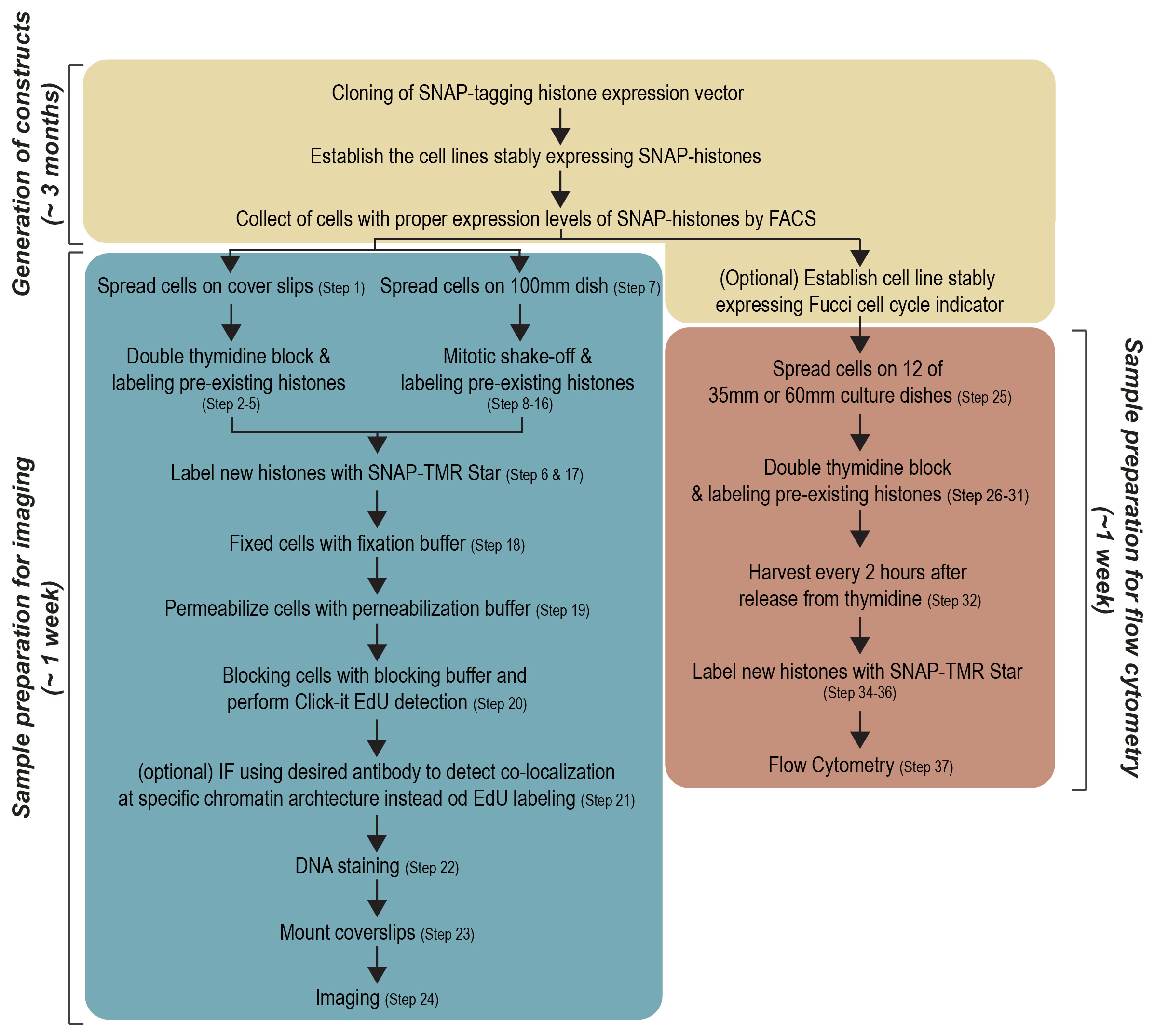
Figure 1. Workflow of protocol. Three sections of pipelines, Generation of constructs, Sample preparation for imaging and Sample preparation for flow cytometry, are shown as boxes.
(i) Generation of the SNAP-tagged histone expression vector and stable expressing cell lines
Protein tagging with a small epitope is valuable for detection of the protein of interest in various biochemical approaches. However, tagging a small peptide sometimes interferes with the biological function of the target protein and localization of the protein. In general, it is desirable to test whether the N-terminal or C-terminal tagging of protein alters its functions. The localization of the SNAP-tagged histone variant can be confirmed using immunofluorescence by comparing with the endogenous histone variant whether it is localized at the expected chromatin sites. Additionally, it may be necessary to test if the SNAP-tagged construct retains its specific function in chromatin.
To determine the precise turnover of histone variants, the generation of stably expressing cell lines is highly recommended, since expression level is expected to be altered after mitosis when a transiently transfected SNAP-tagged histone is used. Inserting the SNAP-tag sequence into the endogenous histone locus using CRISPR/Cas9 technology is ideal but not necessary. The SNAP-expressing vector is commercially available from NEB, which contains a neomycin selection gene. In the process of establishing stably expressing cell lines, titration of the drug concentration in your cells is required for the isolation of positive cells. If your cell line already obtains neomycin resistance, such as HEK 293T cell which is immortalized with the large T antigen with neomycin resistance, the neomycin selection marker needs to be replaced.
The expression level of the SNAP-tagged histone may also influence the timing of incorporation of histones. Since overexpression of histone variants may cause undesirable non-specific incorporation into chromatin, isolating a cell population with a moderate to lower expression of SNAP-tagging histones by Fluorescence-activated cell sorting (FACS) before the pulse-chase experiment is recommended. We also recommend testing the level of SNAP-histone variants compared with endogenous histone variants in purified chromatin fractions by western blotting after establishment of the cell line.
(ii) Optimization of cell cycle synchronization
Cell cycle synchronization is a common method to arrest the whole cell population into a particular cell cycle phase. Most cell cycle synchronization methods rely on the use of a drug which blocks a specific function required for cell cycle progression. While various cell cycle synchronization methods were established in the past (Table 1), the efficiency of synchronization with drug concentration might depend on the cell type used. Any cell synchronization methods can be used in this protocol after the optimization of drug treatment and staining scheme of pre-existing and newly incorporated histones. Here we introduce the protocol in HEK293T cells using a double thymidine block for synchronization at G1/S phase and mitotic shake-off for synchronization at G2/M phase (Figure 2). Both are widely utilized as general cell cycle synchronization methods. Successful cell synchronization and release should be confirmed by flow cytometry, western blotting or immunofluorescence (IF) with cell cycle indicators or markers such as co-expressing the Fucci cell cycle reporter (Sakaue-Sawano et al., 2008), DNA staining with Hoechst 33342, anti-phosphorylated Histone-3 at Serine-28 (M phase marker) or any other cell cycle markers.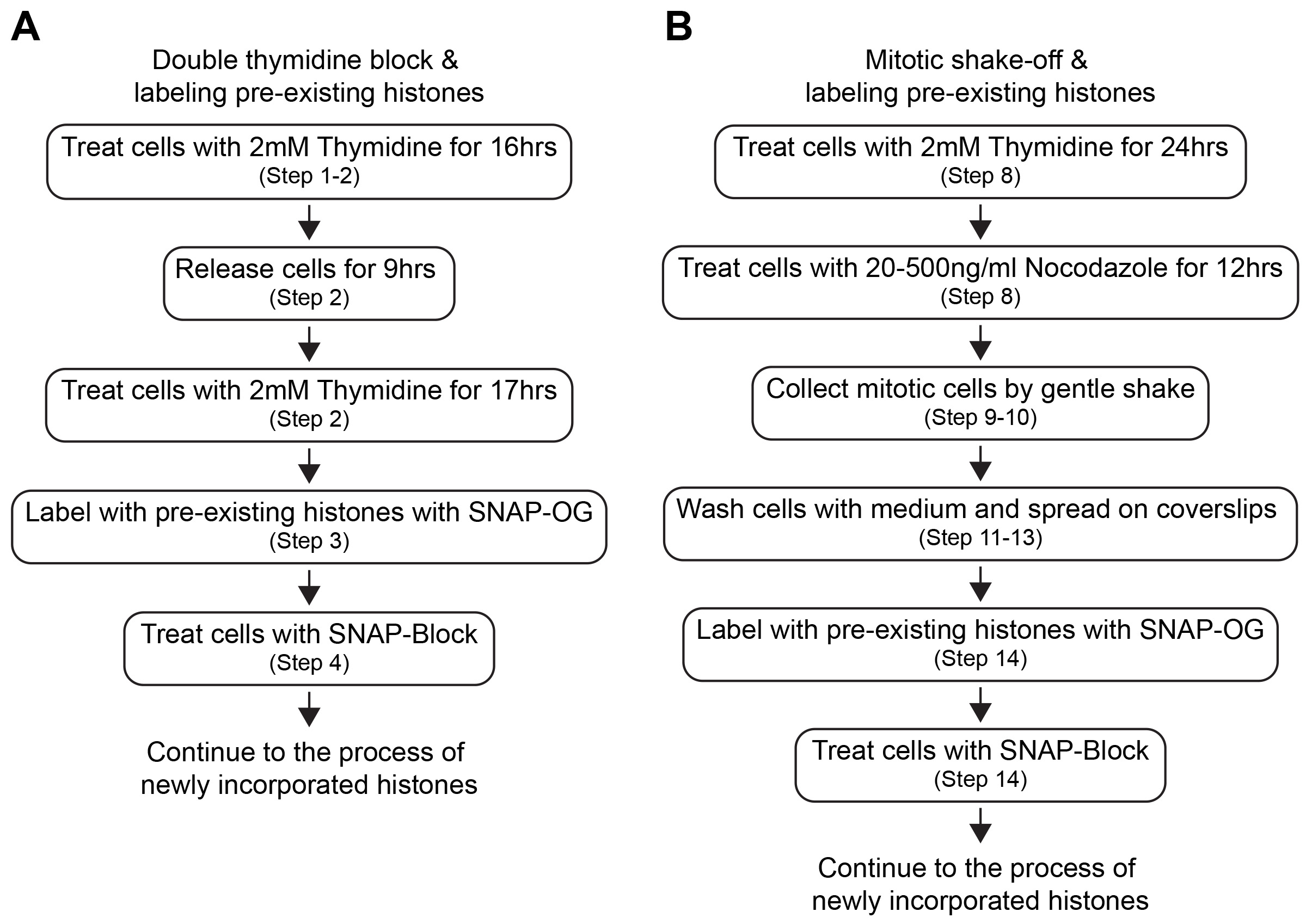
Figure 2. Workflow of cell synchronization and labeling of histones. Each step of cell synchronization methods (A. double thymidine block and B. mitotic shake-off) and timing of labeling of pre-existing histones are shown.
Table 1. Common drug list for cell synchronization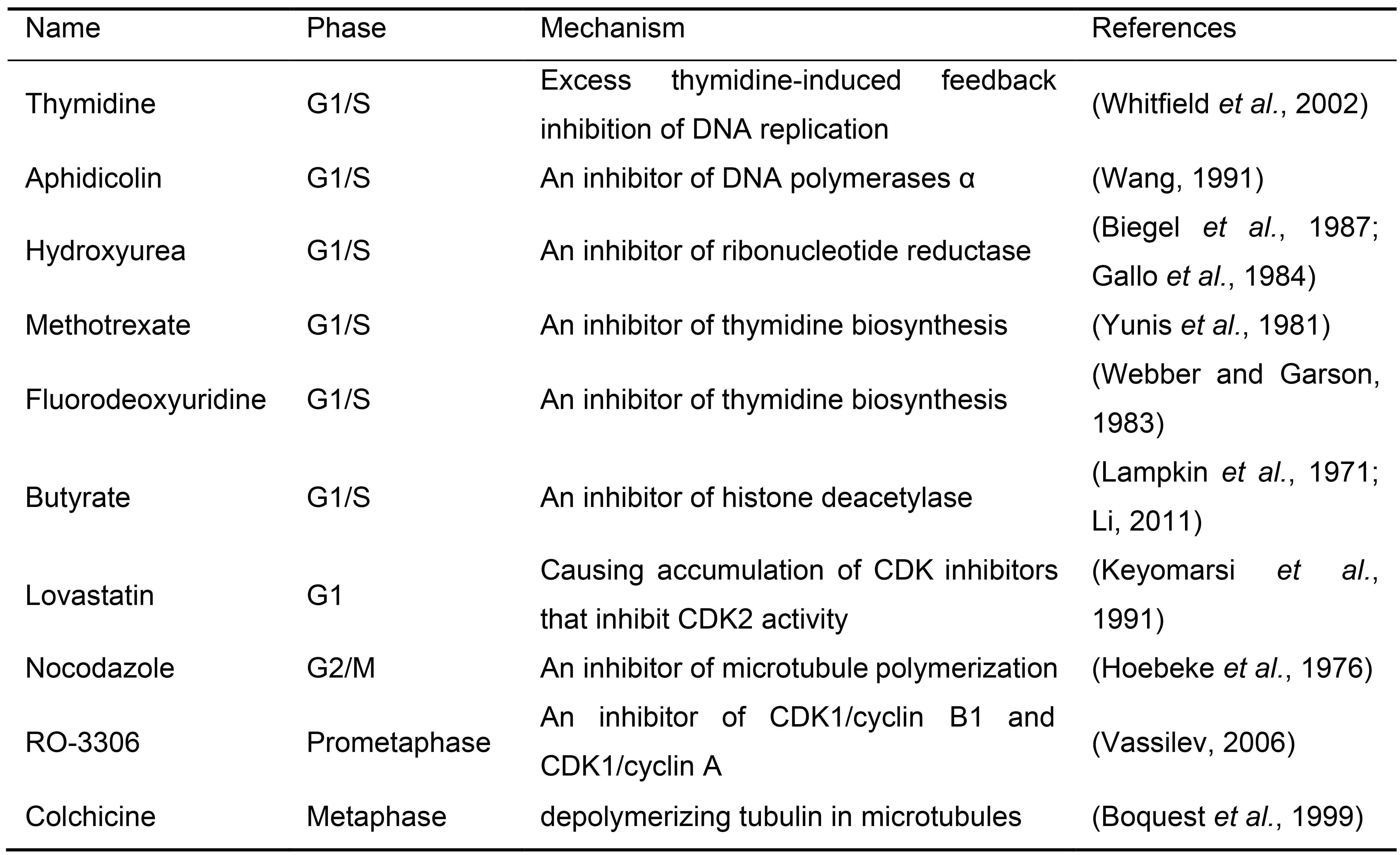
(iii) Detection of global and local histone incorporation using imaging
A key procedure for imaging detection is to grow cells on coverslips. Specific procedures for coating of coverslips might be required for different cell types. Careful handling is important to keep the adherent cells attached to the coverslips during the entire procedure. Localization of the histone variant with specific chromatin structure can be addressed by colocalization analysis with a marker of desired chromatin structure using IF after the fixation of cells. In this case, carefully consider the choice of fluorophores and the filter setting of your microscope to avoid the incompatible bleed-through detection of fluorophores.
- Labeling of histone incorporation during S-G2 phase ● Timing 5 days
- Place coverslips in 12-well or 24-well plate and perform coating coverslips (e.g., 0.01% poly-L lysine, 0.01% collagen) at room temperature for 1 h. After washing 3 times with double distilled water, spread cells on coated coverslips and grow cells at least 24 h.
- Cell synchronization at the G1/S can be performed using double thymidine block methods (Jackman and O'Connor, 2001). Incubation time and drug concentrations must be optimized for each cell type. Synchronize the cells at the G1/S transition with a treatment of 2 mM thymidine for 16 h, release from thymidine for 9 h and treat again with 2 mM thymidine again for 17 h.
? TROUBLESHOOTING (Table 2)
Table 2. Troubleshooting Table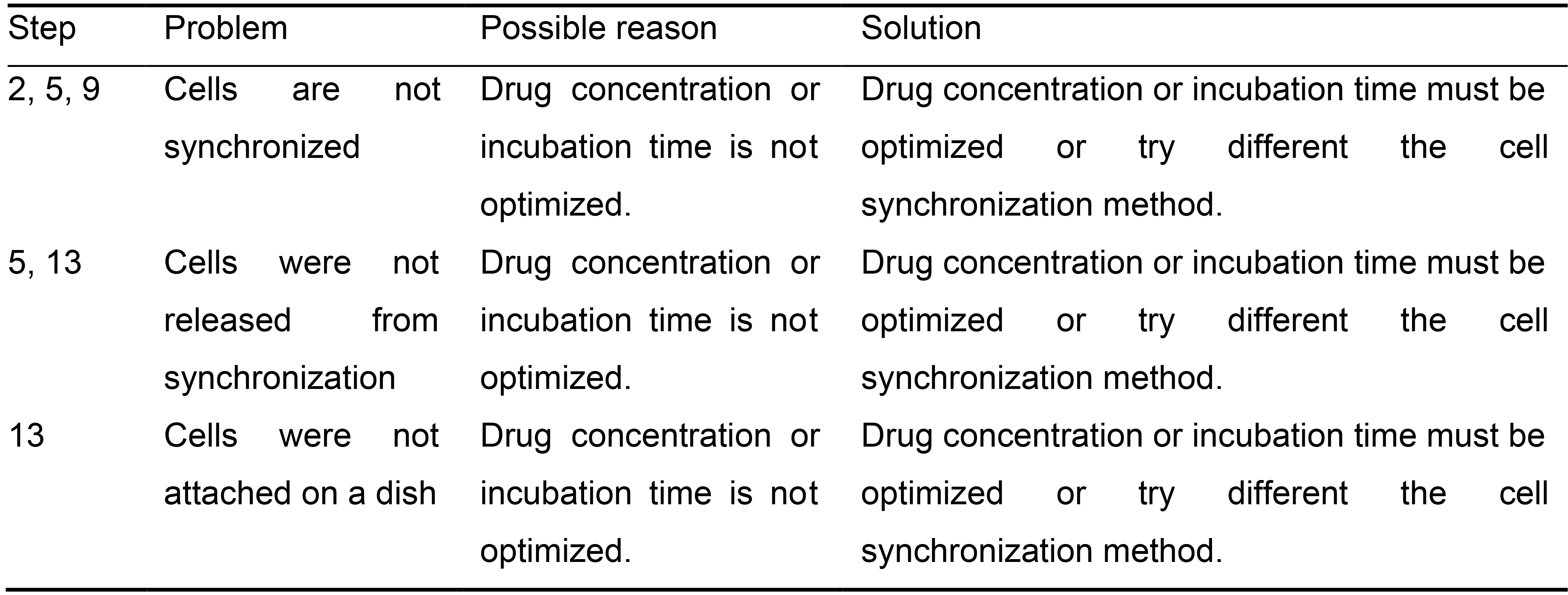
- Label pre-existing SNAP-tagged histones with SNAP-Cell Oregon Green (1 µM) in culture medium for 30 min at 37 °C in 5% CO2.
- Block unlabeled pre-existing SNAP-tagged histones with non-fluorescent SNAP-substrates, SNAP-cell Block (10 µM) for 30 min in culture medium at 37 °C in 5% CO2.
CRITICAL STEP: This step is important to avoid insufficient initial labeling of pre-existing histones that will result in inaccurate detection of newly incorporated histones. - Add culture medium containing 10 μM of EdU and 10 μM RO-3306 and incubate for 12 h at 37 °C in 5% CO2.
? TROUBLESHOOTING (Table 2) - Label newly incorporated SNAP histones using SNAP-Cell TMR (1 µM) in culture medium with RO-3306 for 30 min at 37 °C in 5% CO2.
- Labeling of histone incorporation during G1 phase ● Timing 5 days
To collect mitotic cells, a mitotic shake off can be performed.- Spread cells in 100 mm dish.
- Synchronize the cells at S/G1 border with 2 mM thymidine for 24 h, then treat with 20-500 nM nocodazole for 12 h. Incubation time and drug concentrations must be optimized for each cell type.
- Check the cells forming rounding shape and weak attachment using light microscopy.
CRITICAL STEP: If cells are not forming rounded shape, synchronizing at M phase is not successful.
? TROUBLESHOOTING (Table 2) - Gently shake the dish to collect mitotic cells into 1.5 ml Eppendorf tube or 15 ml Falcon tubes.
- Centrifuge the cells at 500 x g for 4 min and wash cells with a culture medium.
- Wash cells twice with culture medium and spread cells on pre-coated coverslips as described in Step 1.
- One to two hours later, check the cells if they attached to the coverslips.
CRITICAL STEP: The cells must be attached to the cover glass. If not, releasing from nocodazole treatment is not successful.
? TROUBLESHOOTING (Table 2) - Label pre-existing SNAP-tagged histones with SNAP-Cell Oregon Green (1 µM) in culture medium for 30 min at 37 °C in 5% CO2.
- Block unlabeled pre-existing SNAP-tagged histones with non-fluorescent SNAP-substrates, SNAP-cell Block (10 µM) for 30 min in culture medium at 37 °C in 5% CO2.
CRITICAL STEP: This step is important to avoid insufficient initial labeling of pre-existing histones that will result in inaccurate detection of newly incorporated histones. - Add 10 µM of EdU and 2 mM thymidine in culture medium for 12-18 h at 37 °C in 5% CO2.
- Label newly incorporated SNAP histones using SNAP-Cell TMR (1 µM) in culture medium with 2 mM thymidine for 30 min at 37 °C in 5% CO2.
- Cell fixation, Permeabilization and Click-it EdU labeling ● Timing 2 days
- Fix cells with fixation buffer for 15 min at room temperature.
PAUSE POINT: The fixed cells can be stored in PBS at 4 °C for up to 1 month.
! CAUTION Paraformaldehyde is a hazardous solution and a cross-linking agent. Wear protective gloves and handle it under a fume hood. - Permeabilize the cells with permeabilization buffer for 15 min at room temperature.
- Block cells using 1x PBS containing 3% BSA for 1 h and perform click-it EdU labeling as described in the manufacturer's instruction.
- (Optional) Immunofluorescence (IF) can be performed using the desired antibody if co-localization with histones needs to be addressed. However, cautious optimization is required for the microscope filter setup and wavelength range to avoid bleed-through from other fluorophores that co-labeled in the same cells.
- DNA staining with DAPI or Hoechst 33342.
- Mount coverslips using anti-Prolong Diamond Antifade Mountant.
- Fix cells with fixation buffer for 15 min at room temperature.
- Detection using microscope ● Timing 1-2 days
- Images can be acquired with an Olympus BX61 widefield, epifluorescent microscope using a 60x 1.4 PlanApo objective or equivalent. Filter sets for DAPI (Semrock), Cy3 (Chroma), FITC (Semrock), and Cy5 (Semrock), with an EXFO X-Cite Series 120 PC metal halide light source, Photometrics Cool SNAP HQ CCD camera, Olympus Type-F immersion oil (nd 1.516) and Molecular Devices Metamorph acquisition software or equivalent microscope set-ups are required. Images can be taken with optically sectioned using a 0.5 μm Z step, spanning a 6~10.0 μm Z depth in total depending on the thickness of each cell type. Exposure times of 10 to 200 ms are typically used to acquire each plane in the Cy3, Cy5, FITC and DAPI channels.
(iv) Detection of global incorporation using flow cytometry
We also introduce the protocol addressing the global histone turnover using flow cytometry detection. Although this approach can determine the global histone kinetics, it is unable to detect the histone kinetics at specific genomic loci or chromatin architectures.
- Labeling of histone incorporation
- Spread cells in 12 of 35 mm or 60 mm dishes.
- Synchronize the cells at the G1/S transition by double thymidine block. Briefly, treated cells with 2 mM thymidine for 16 h, wash 3 times with 1x PBS, and release from thymidine by replacing culture medium for 9 h and treat with 2 mM thymidine again for 17 h.
- Label pre-existing SNAP-tagged histones with SNAP-Cell Oregon Green (1 µM) in culture medium for 30 min at 37 °C in 5% CO2.
- Treat cells with SNAP-cell Block (10 µM) for 30 min in culture medium at 37 °C in 5% CO2 to mask insufficient labeling of pre-existing SNAP-tagged histones with non-fluorescent SNAP-substrates.
CRITICAL STEP: This step is important to avoid insufficient initial labeling of pre-existing histones that will result in inaccurate detection of newly incorporated histones. - Release 6 dishes from cell synchronization for detection of 12- to 22-hour time points. Keep synchronizing other 6 dishes for detection of 2- to 12-hour time point.
- Incubate cells for 12 h.
- Release the other 6 dishes from cell synchronization for detection of 2- to 12-hour time point.
- Harvest cells every 2 h and store cells at -80 °C.
PAUSE POINT: The frozen medium and protocol are depending on cell types. - Thaw cells and wash one time with a pre-warmed medium.
- Label newly incorporated SNAP histones by adding culture medium with SNAP-JF646 (1 µM) to cell pellets. Pipet cells and incubate for 30 min at 37 °C water bath.
- Centrifuge the cells at 500 x g for 4 min at room temperature and wash twice with pre-warmed culture medium.
- (Optional) Commercially available Dyes detecting cell cycle (e.g., Hoechst 33342) are also optional instead of the Fucci cell cycle indicator.
- Detection of expression levels of Oregon green (Pre-existing histones), JF646 (newly incorporated histones), mCherry (S-G2 phase cell cycle indicator) and TagBFP (G1 phase cell cycle indicator) using Flow cytometry.
BOX 1. Manual imaging Analysis workflow of global pre-existing and newly incorporated histones (see also Figure 3)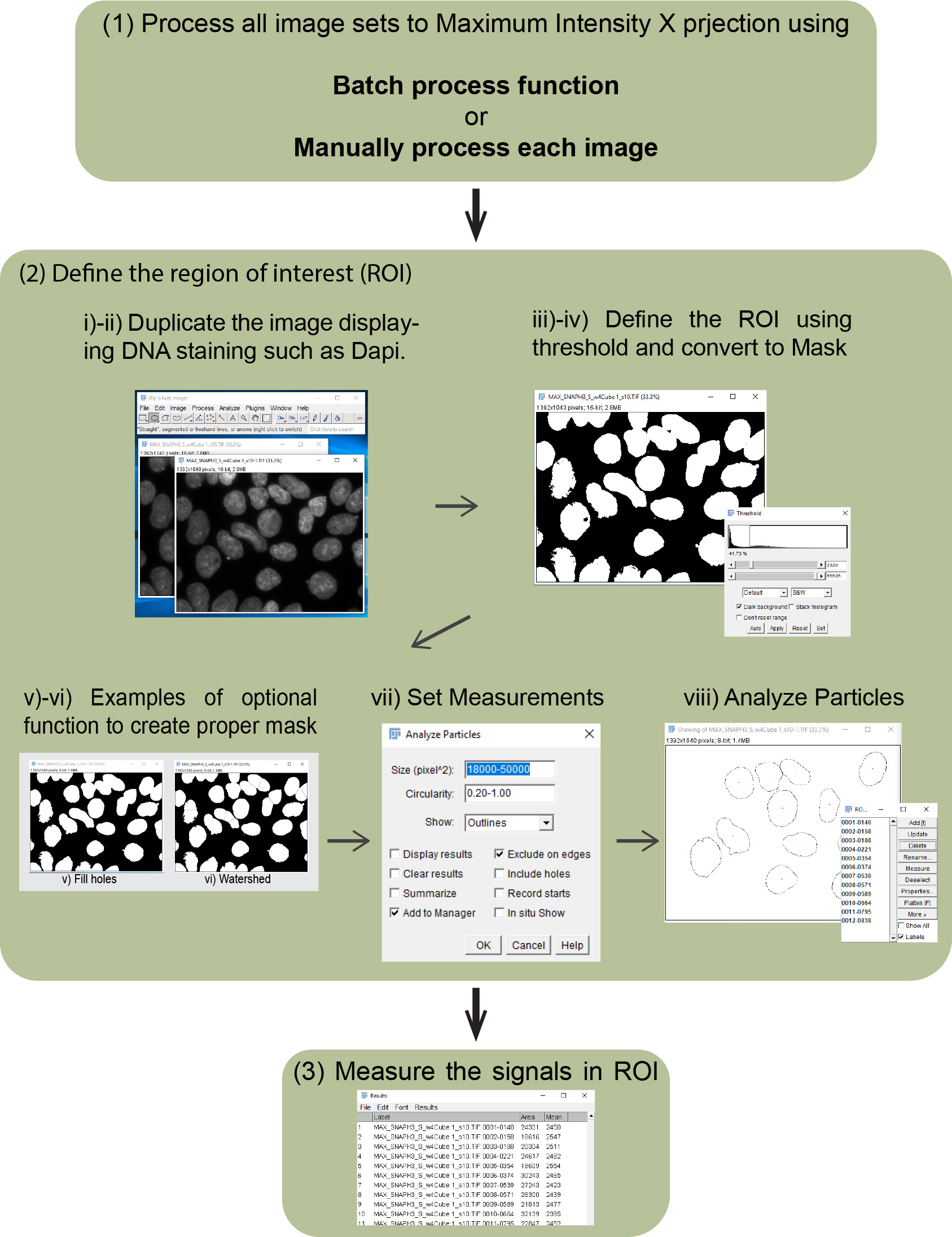
Figure 3. Imaging analysis workflow. Outlines of imaging analysis to detect intensity of pre-existing, newly incorporated histones, nuclear staining (e.g., DAPI and Hoechst 33342) and EdU (S phase marker) are shown.- Process all image sets to Maximum Intensity Z-projection using the batch process function as follows:
- Process/Batch/Macro.
- Choose the Input folder in "Input…".
- Choose the output folder in "Output…".
- (Optional) Put specific name in “File name contains:” if specific files in selected folder need to be processed.
- Put macro code “run("Z Project...", "projection=[Max Intensity]");” in the script box.
- Click “Process”.
- Open four images by dragging the files into the main interface of FIJI or following:
File/Open/, then choose your image. - Image/Stacks/Z Project/, then choose projection type “Max Intensity”.
- Define the region of interest (ROI) to measure the intensities of pre-existing and newly incorporated histones, and EdU incorporation by making a mask using nuclear staining such as DNA staining with DAPI.
Duplicate the image displaying DNA staining such as DAPI.- Select duplicated DAPI channel image.
- Image/Duplicate…
- Image/Adjust/Threshold; then choose suitable threshold method (Default can be chosen as a start), check “Dark background” and Apply.
- Process/Binary/Convert to Mask.
- (Optional) Process/Binary/Fill holes.
- (Optional) Process/Binary/Watershed (https://imagej.net/Nuclei_Watershed_Separation).
- Analyze/Set Measurements, check “Area”, “Mean gray value” and “Display label”, then OK.
- Analyze/Analyze Particles, add proper number to accomplish the selection of proper area into “Size (pixel^2)” and “Circularity”, check “Add to Manager”, then OK.
- Measure DAPI signal (DNA) in the masked area using Region of interest (ROI).
- Select channel image.
- Click “Measure” in ROI Manager.
- Measure Oregon green signal (Pre-existing histones) using ROI.
- Select Oregon green channel image.
- Click “Measure” in ROI Manager.
- Measure TMR-STAR signal (newly incorporated histones) signal using ROI.
- Select TMR-STAR channel image.
- Click “Measure” in ROI Manager.
- Measure EdU signal (Nascent DNA) using ROI.
- Select EdU channel image.
- Click “Measure” in ROI Manager.
- Save results and analyze.
BOX 2. Analysis workflow of local pre-existing and newly incorporated histones using color profiler or RBG profiler (see also Figure 5A)- Install the plugin in Fiji/ImageJ software.
- Open all image sets that will be compared by dragging the files into the main interface of FIJI or following:
File/Open/, then choose your image. - Merge these images into a single image.
Image/Color/Merge Channels. - Change the image type to RGB color.
Image/Type/RGB color. - Make a line to make a profile.
- Choose a line tool.
- Make a line.
- Plugins/RGB Profiler.
Imaging analysis (~1 week)
Steps 1-6, cell synchronization using double thymidine block and labeling pre-existing histones for imaging analysis: 4 days.
Steps 7-17, cell synchronization using mitotic shake off, and labeling pre-existing and newly incorporated histones for imaging analysis: 4 days.
Steps 18- 19, fixation and permeabilization of the cells: 1 h.
Step 20, labeling of EdU using click-it kit: 1 h.
Step 21, (optional) IF using the desired antibody to determine the histone deposition at specific chromatin architecture or factors: 3 h.
Steps 22- 23, DNA staining and mount coverslips: Overnight.
Step 24, microscopy imaging: 1 day.
Flow cytometry analysis (~1 week)
Step 25, Spread cells and cell synchronization using double thymidine block and labeling pre-existing histones: 4 days.
Step 32, Release from cell synchronization and harvest cells: 2 days.
Steps 34-37, labeling of newly incorporated cells and Flow cytometry analysis: 2 h.
Data analysis
We introduce a basic analytical pipeline for imaging approach using FIJI/ImageJ which is a free open source image processing package. An automated analysis using ImageJ macro code would be time-saving for large data sets if you are familiar with programming. Additionally, many other imaging processing programs (e.g., Matlab) can be also a great option to process enormous data sets.
Data analysis for global histone turnover using flow cytometry detection can be easily accomplished using available flow cytometers such as LSR II (BD Bioscience). The software such as FlowJo (BD Bioscience) can assess the FCS data sets and permit visualizing complex cytometric data sets. In this protocol, we can use a mean or median of the total intensity of fluorophore in the entire cell population with single-cell resolution to investigate the timing of incorporation and deposition of histones. Displaying histograms of fluorescent intensities from labeling with pre-existing and newly incorporated histones as well as cell cycle markers in this protocol, can help to visualize the timing of global histone kinetics including the dissociation of pre-existing histones and incorporation of newly synthesized histones during the cell cycle.
Anticipated results
In this protocol, we summarized our previously published results for the cell cycle-specific dynamics of histone H3.1 and macroH2A1.2 in HEK293T cells to show that the SNAP-labeling pulse-chase system works reliably and to illustrate how to analyze the imaging data sets (Sato et al., 2019).
Although we illustrate three parts in this protocol (Figure 1) including generation of constructs, sample preparation for imaging and sample preparation for flow cytometry, we mainly described the detailed protocol for the latter two. We also illustrate how to analyze the imaging data sets using ImageJ/FIJI (Figure 3 and Box 1). Here we present an example of the SNAP pulse-chase detection during the cell cycle using an imaging approach detecting SNAP-histone H3.1 (Figure 4). We demonstrated two types of cell synchronization, double thymidine block and mitotic shake off, to investigate histone incorporation and dissociation during the S-G2 or G1 phase. The pre-existing and newly incorporated SNAP-H3.1 stably expressed in HEK 293T were labeled as shown in Figures 4A-4B. EdU click-it detection was also performed to confirm successful cell synchronization. The global histone incorporation rates in individual cells were determined by the mean of pixel intensity of newly incorporated histones (TMR-Star) normalized by the mean of pixel intensity of pre-existing histones (Oregon Green) in the nucleus. Using this approach, we detected H3.1 incorporation during S-G2 phase as it is known as DNA replication-dependent deposition (Figures 4C-4D).
We also illustrate the detection of colocalization in imaging analysis using RGB Profiler (Figure 5A and Box 2). Here we present the colocalization of newly incorporated macroH2A histone variant at inactivate X chromosomes (Xi) in HEK 293T cells (Figures 5A-5B). The pre-existing and newly incorporated SNAP-macroH2A1.2 were labeled as shown in Figure 4A and incorporation of SNAP-macroH2A1.2 in Xi during the cell cycle was determined by the colocalization with pre-existing SNAP-macroH2A1.2. As shown in Figure 5B, macroH2A1.2 is incorporated into the Xi during the G1 phase.
Global histone incorporation and dissociation also can be detected by flow cytometry. We demonstrated the SNAP-macroH2A1.2 pulse-chase approach and detected the global SNAP-macroH2A dynamics by flow cytometry as shown in Figures 5C-5D. Here we synchronized cells at G1/S border and labeled pre-existing SNAP-macroH2A1.2 with Oregon green, then detected newly incorporated SNAP-macroH2A1.2 every two h after releasing from cell synchronization. Although we determined the cell cycle transition using the Fucci cell cycle sensor (Figure 5C [two right panels] and 5D [top]), other cell cycle indicator such as Hoechst 33342 DNA staining also could be the option (Figure 6). A histogram shows the fluorescent intensities of cell populations. The transitions of signal intensity of pre-existing SNAP-macroH2A1.2 (Oregon green, Figure 5C [left] and 5D [middle]) show that the signal intensity decreased when cells entered mitosis but little pre-existing SNAP-macroH2A dissociate from chromatin through the entire cell cycle. The transitions of newly incorporated SNAP-macroH2A1.2 (JF646, Figure 5C [second left], and 5D [bottom]).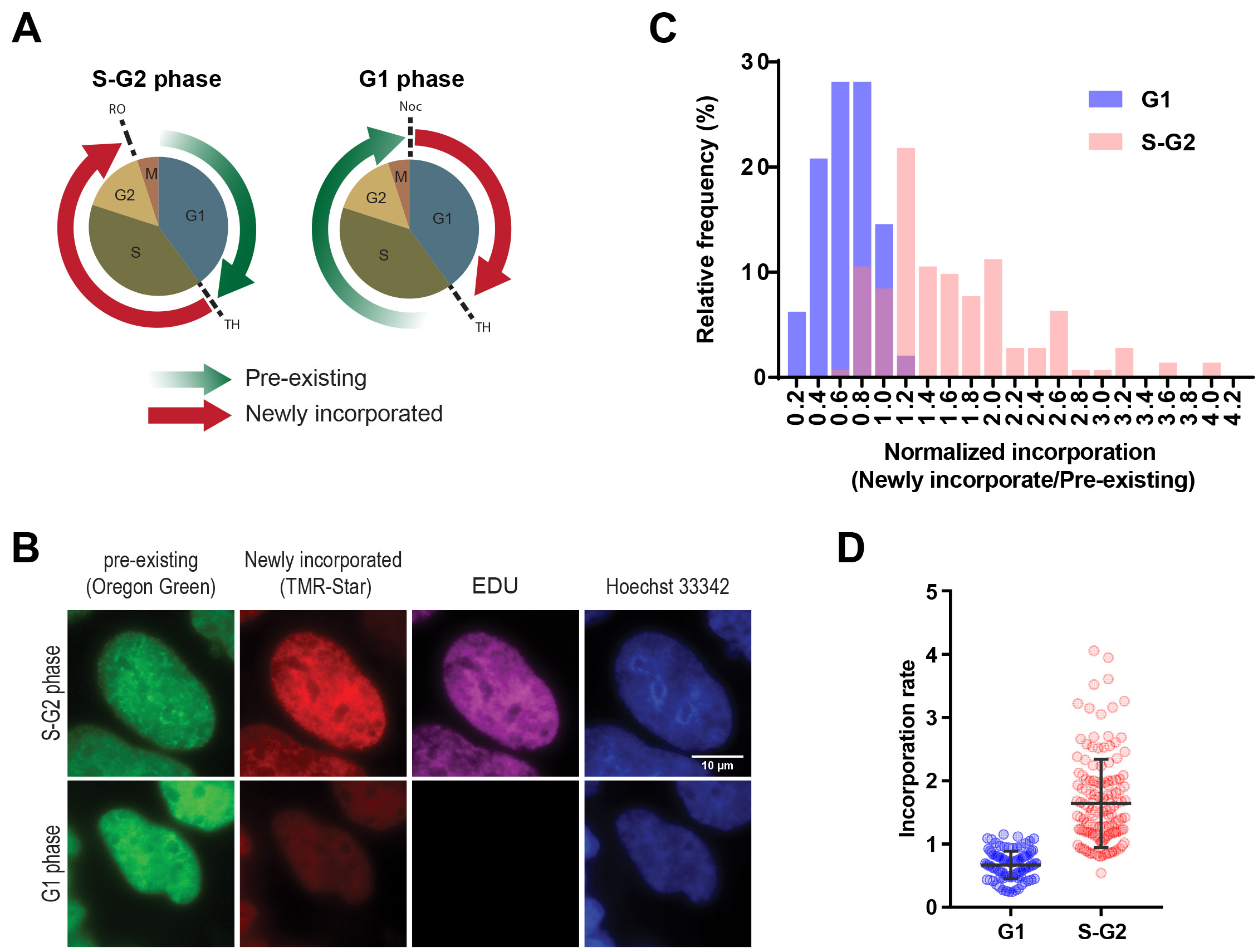
Figure 4. Detection of global pre-existing and newly incorporated histone H3.1 during specific cell cycle. A. The illustration of labelling histones at S/G2 and G1 phases for the analysis in B-D. (Left panel) To label newly incorporated histones in S/G2 phase, HEK 293T cells stably expressing SNAP-tagged H3 were synchronized at the G1/S phase border by double thymidine block as shown as TH. Pre-existing histones were labeled with SNAP-Oregon Green (green arrow), subsequently blocked using the non-fluorescent SNAP-Block reagent to avoid non-sufficient labeling. The cells were released from synchronization to progress to the G2/M transition until they were synchronized at the G2/M border using RO-3306 (a CDK1/cyclin B1 and CDK1/cyclin A inhibitor, shown as RO), then newly-incorporated SNAP-tagged histones were labeled with SNAP-TMR Star (red arrow). (Right panel) To detect newly incorporated histones in the G1 phase, mitotic cells were collected by shake-off following nocodazole treatment for 12 h and spread onto coverslips (shown as Noc). After 2 h, pre-existing SNAP-H3 were labelled with Oregon Green and treated with the blocking reagent. Cells were released from synchronization and progressed to the G1/S transition using double thymidine block, then newly-incorporated SNAP-tagged histones were labeled with SNAP-TMR Star. After being released from the first synchronization, the cells were also treated with EdU until the second synchronization, allowing cells that had undergone DNA synthesis to be identified. B. Representative images of pre-existing and newly-synthesized SNAP-tagged H3.1 in S/G2 or G1 phase. Scale bar = 10 µm. Image analysis showing a higher incorporation of histone H3 during the S-G2 phase (C-D). C. Frequency distribution of histone incorporation rate in S-G2 and G1 cell cycle phase. Histone incorporation rate was calculated by the mean pixel intensity of red (newly incorporated histones) normalized by mean pixel intensity of green (Pre-existing histones) in the nucleus during the S/G2 and in G1 phases. Successful cell synchronization was confirmed by EdU labeling. Hoechst 33342 labeling was performed to define the area of a nucleus in the following analysis. D. Quantification of histone incorporation rate during S-G2 and G2 phase. Each dot denotes the incorporation rate as described in Figure 4C from single cells. The error bars represent one standard deviation from the number of single cells (n = that indicated on each data set. *P < 0.0001. The P values were determined using two-tailed unpaired t-tests.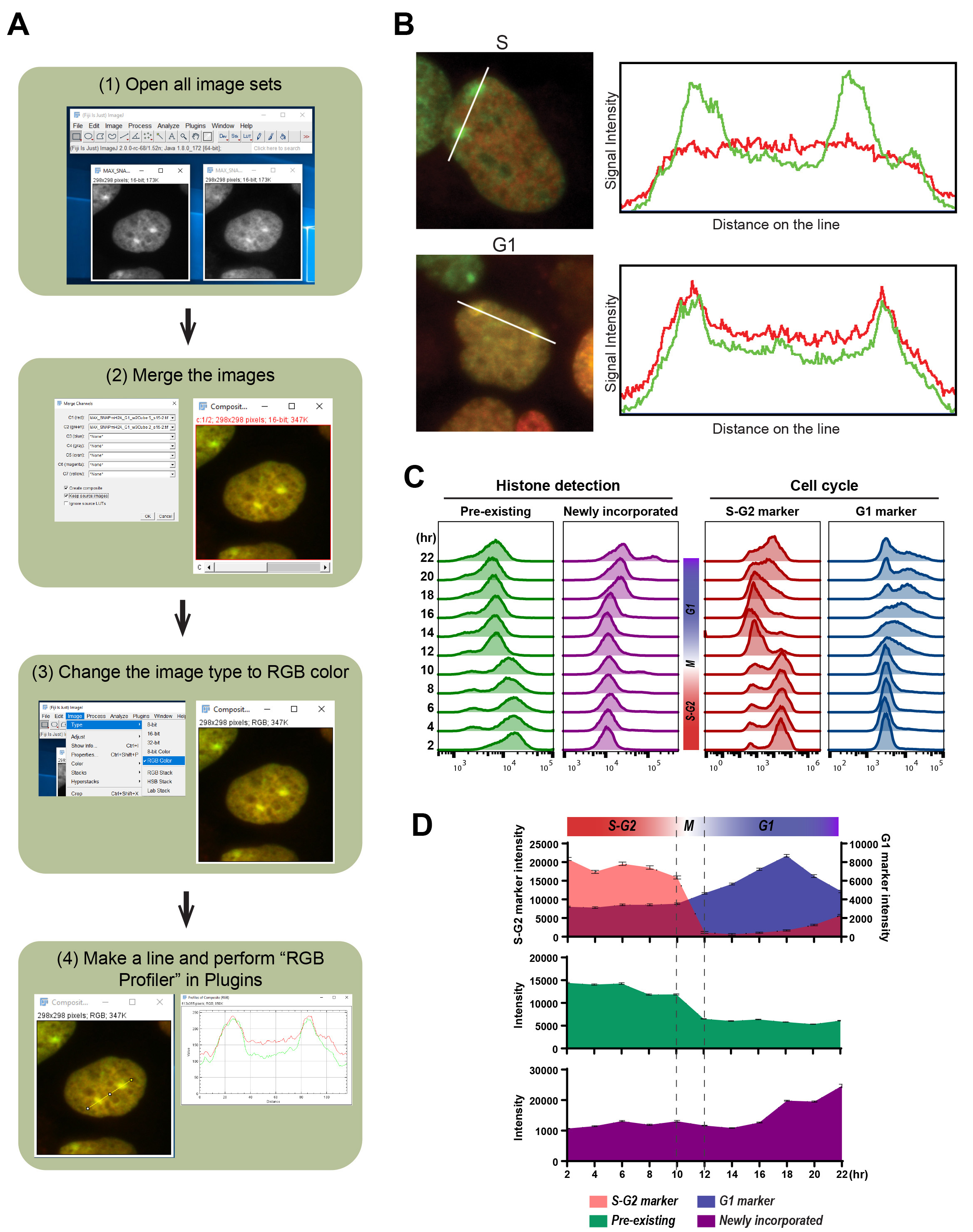
Figure 5. Detection of local and global pre-existing and newly incorporated histone macroH2A1.2 during specific cell cycle. A. The outline of image analysis using RBG Profiler plugin (https://imagej.nih.gov/ij/plugins/rgb-profiler.html, Color Profiler plugin (https://imagej.nih.gov/ij/plugins/color-profiler.html) also provides the same functionality) to address the colocalization of newly incorporated histones with pre-existing histones. Cell cycle-specific histone incorporation at specific chromatin architecture can also be addressed with the detection of chromatin structure using IF. Here we show the example detecting colocalization with pre-existing and newly incorporating histones. B. the example of image analysis using cell profiler. The left images of pre-existing (Oregon Green: green), newly incorporated (TMR: red) SNAP-tagged macroH2A during S-G2 (upper) and G1 (lower) phase were merged into single images and colocalization on the two inactive X chromosomes (Xi) were determined in HEK 293T cells. In the upper and lower right panels, the signal intensities measured along the white lines in the images of pre-existing and newly incorporated are shown. C. Flow cytometric analysis of pre-existing (left: Oregon Green) and newly incorporated macroH2A (second left: JF646), with S-G2 (second right: mCherry-tagged hGeminin) and G1 (right: TagBFP-tagged hCdt1) cell cycle markers derived from Fucci sensor. Histograms indicate the fluorescent intensity at each time point (2-22 hour) after releasing from synchronization at the G1/S border. The cell cycles at each time point were estimated from the Fucci cell cycle sensor. D. the median of fluorescent intensity of pre-existing (middle: Oregon Green) and newly incorporated macroH2A (bottom: JF646), with S-G2 and G1 (top: mCherry-tagged hGeminin and TagBFP-tagged hCdt1) cell cycle markers from Flow cytometric analysis are plotted. Error bars indicate 95% confident intervals.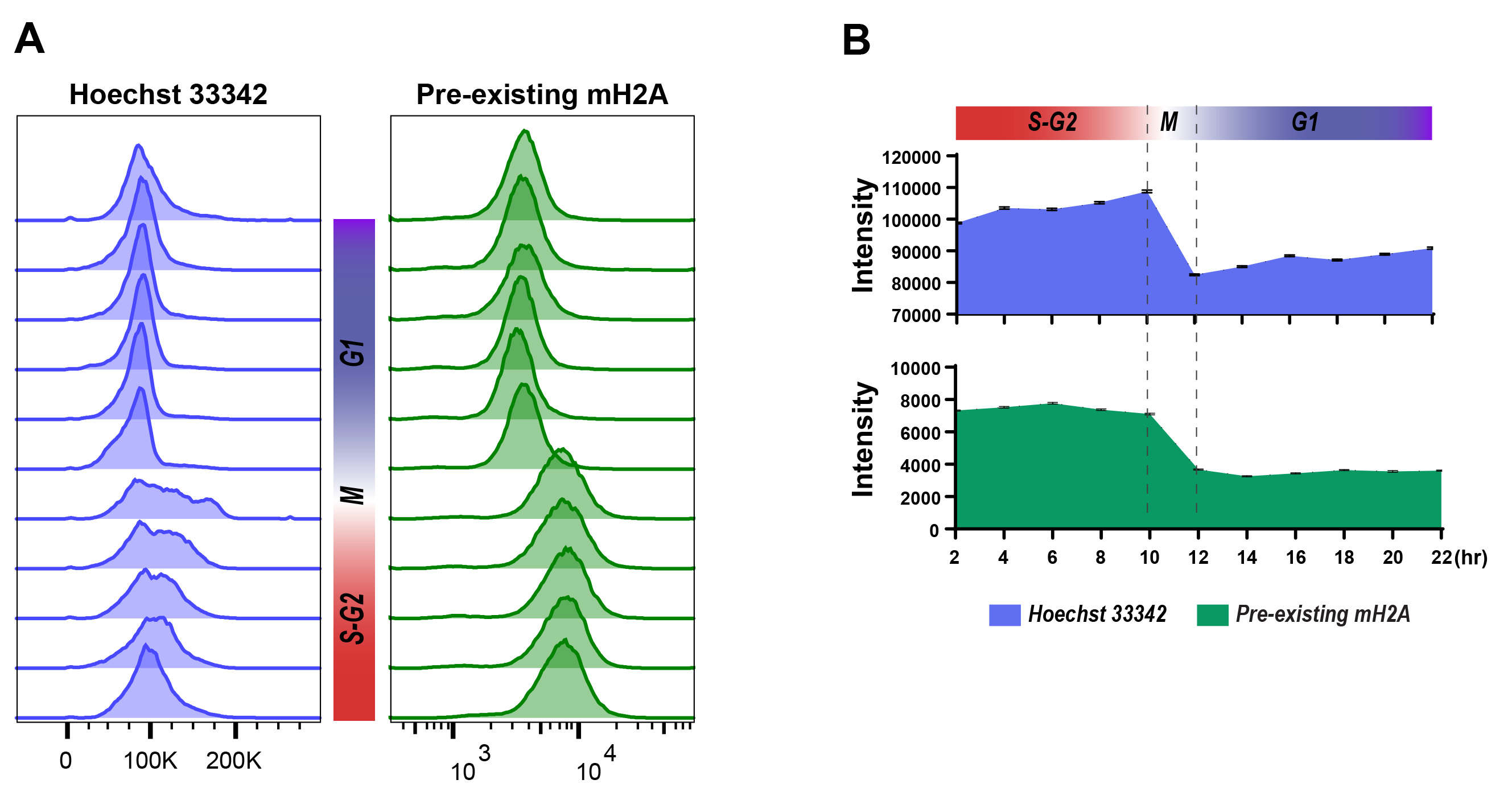
Figure 6. Cell cycle determination using Hoechst 33342 staining and Flow Cytometry detection. A. Flow cytometric analysis of cell cycle (left: Hoechst 33342) and pre-existing SNAP-macroH2A (right: Oregon Green) labeled and released from cell synchronization as described in Steps 25-36. Histograms indicate the fluorescent intensity at each time point (2-22 h) after releasing from synchronization at the G1/S border. The cell cycles at each time point could be determined by the intensity transitions of Hoechst 33342 in the cell population. B. the median of fluorescent intensity of DNA staining (top: Hoechst 33342) and pre-existing SNAP-macorH2A1.2 (middle: Oregon Green) from flow cytometric analysis are plotted. Error bars indicate 95% confident intervals.
Recipes
- Cloning of SNAP-histone expression vector and its stably expressing cell line
A plasmid that encodes desired histone with SNAP-tag needs to be cloned by general cloning methods. pSNAPf Vector (catalog # N9183S from NEB) is useful to establish your construct and stably expressing cell line since it contains neomycin resistance gene for efficient drug selection. - Fixation Buffer
4% (wt/vol) PFA in 1x PBS
Note: This solution can be prepared from 32% (wt/vol) PFA. Protected from light for up to 2 years. - Permeabilization buffer
0.5% Triton in 1x PBS - Blocking buffer
3% BSA in 1x PBS - Sorting Buffer
1 mM EDTA
25 mM HEPES pH 7.0
1% FBS (Heat inactivated) in 1x DPBS
Acknowledgments
This work was supported by NIH grant R01 DA030317 (JMG). We thank members of the Greally and Singer laboratories for discussions, the Einstein FACS and Genomics cores. We also thank L. Lavis for SNAP-JF646. This protocol was adopted from previous work (Sato et al., 2019).
Author contributions statement: H.S. designed and performed the experiments and analyzed the data. H.S., J.M.G. and R.H.S. wrote the manuscript. J.M.G. and R.H.S. supervised the research.
Competing interests
The authors declare no competing financial interests.
References
- Alabert, C., Barth, T. K., Reveron-Gomez, N., Sidoli, S., Schmidt, A., Jensen, O. N., Imhof, A. and Groth, A. (2015). Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev 29(6): 585-590.
- Alabert, C. and Groth, A. (2012). Chromatin replication and epigenome maintenance. Nat Rev Mol Cell Biol 13(3): 153-167.
- Annunziato, A. T. (2012). Assembling chromatin: The long and winding road. Biochimic Biophys Acta 1819(3-4): 196-210.
- Balhorn, R., Jackson, V., Granner, D. and Chalkey, R. (1975). Phosphorylation of the lysine-rich histones throughout the cell cycle. Biochemistry 14(11): 2504-2511.
- Banaszynski, L. A., Allis, C. D. and Lewis, P. W. (2010). Histone variants in metazoan development. Dev Cell 19(5): 662-674.
- Biegel, J. A., Leslie, D. S., Bigner, D. D. and Bigner, S. H. (1987). Hydroxyurea synchronization increases mitotic yield in human glioma cell lines. Acta Neuropathol 73(3): 309-312.
- Boquest, A. C., Day, B. N. and Prather, R. S. (1999). Flow cytometric cell cycle analysis of cultured porcine fetal fibroblast cells. Biol Reprod 60(4): 1013-1019.
- Clapier, C. R. and Cairns, B. R. (2009). The biology of chromatin remodeling complexes. Annu Rev Biochem 78: 273-304.
- Deal, R. B., Henikoff, J. G. and Henikoff, S. (2010). Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 328(5982): 1161-1164.
- Dunleavy, E. M., Roche, D., Tagami, H., Lacoste, N., Ray-Gallet, D., Nakamura, Y., Daigo, Y., Nakatani, Y. and Almouzni-Pettinotti, G. (2009). HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell 137(3): 485-497.
- Gallo, J. H., Ordonez, J. V., Brown, G. E. and Testa, J. R. (1984). Synchronization of human leukemic cells: relevance for high-resolution chromosome banding. Hum Genet 66(2-3): 220-224.
- Geiman, T. M. and Robertson, K. D. (2002). Chromatin remodeling, histone modifications, and DNA methylation-how does it all fit together? J Cell Biochem 87(2): 117-125.
- Henikoff, S., Henikoff, J. G., Sakai, A., Loeb, G. B. and Ahmad, K. (2009). Genome-wide profiling of salt fractions maps physical properties of chromatin. Genome Res 19(3): 460-469.
- Henikoff, S. and Smith, M. M. (2015). Histone variants and epigenetics. Cold Spring Harb Perspect Biol 7(1): a019364.
- Hoebeke, J., Van Nijen, G. and De Brabander, M. (1976). Interaction of oncodazole (R 17934), a new antitumoral drug, with rat brain tubulin. Biochem Biophys Res Commun 69(2): 319-324.
- Jackman, J. and O'Connor, P. M. (2001). Methods for synchronizing cells at specific stages of the cell cycle. Curr Protoc Cell Biol Chapter 8: Unit 8 3.
- Keyomarsi, K., Sandoval, L., Band, V. and Pardee, A. B. (1991). Synchronization of tumor and normal cells from G1 to multiple cell cycles by lovastatin. Cancer Res 51(13): 3602-3609.
- Lampkin, B. C., Nagao, T. and Mauer, A. M. (1971). Synchronization and recruitment in acute leukemia. J Clin Invest 50(10): 2204-2214.
- Li, C. (2011). Specific cell cycle synchronization with butyrate and cell cycle analysis. Methods Mol Biol 761: 125-136.
- Martinez-Garcia, E., Popovic, R., Min, D. J., Sweet, S. M., Thomas, P. M., Zamdborg, L., Heffner, A., Will, C., Lamy, L., Staudt, L. M., Levens, D. L., Kelleher, N. L. and Licht, J. D. (2011). The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 117(1): 211-220.
- Pesavento, J. J., Yang, H., Kelleher, N. L. and Mizzen, C. A. (2008). Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol Cell Biol 28(1): 468-486.
- Petruk, S., Sedkov, Y., Johnston, D. M., Hodgson, J. W., Black, K. L., Kovermann, S. K., Beck, S., Canaani, E., Brock, H. W. and Mazo, A. (2012). TrxG and PcG proteins but not methylated histones remain associated with DNA through replication. Cell 150(5): 922-933.
- Sakaue-Sawano, A., Kurokawa, H., Morimura, T., Hanyu, A., Hama, H., Osawa, H., Kashiwagi, S., Fukami, K., Miyata, T., Miyoshi, H., Imamura, T., Ogawa, M., Masai, H. and Miyawaki, A. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132(3): 487-498.
- Sato, H., Wu, B., Delahaye, F., Singer, R. H. and Greally, J. M. (2019). Retargeting of macroH2A following mitosis to cytogenetic-scale heterochromatic domains. J Cell Biol 218(6): 1810-1823.
- Scharf, A. N., Barth, T. K. and Imhof, A. (2009). Establishment of histone modifications after chromatin assembly. Nucleic Acids Res 37(15): 5032-5040.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Stasevich, T. J., Hayashi-Takanaka, Y., Sato, Y., Maehara, K., Ohkawa, Y., Sakata-Sogawa, K., Tokunaga, M., Nagase, T., Nozaki, N., McNally, J. G. and Kimura, H. (2014). Regulation of RNA polymerase II activation by histone acetylation in single living cells. Nature 516(7530): 272-275.
- Tillo, D., Mukherjee, S. and Vinson, C. (2016). Inheritance of cytosine methylation. J Cell Physiol 231(11): 2346-2352.
- Vardabasso, C., Hasson, D., Ratnakumar, K., Chung, C. Y., Duarte, L. F. and Bernstein, E. (2014). Histone variants: emerging players in cancer biology. Cell Mol Life Sci 71(3): 379-404.
- Vassilev, L. T. (2006). Cell cycle synchronization at the G2/M phase border by reversible inhibition of CDK1. Cell Cycle 5(22): 2555-2556.
- Wang, T. S. (1991). Eukaryotic DNA polymerases. Annu Rev Biochem 60: 513-552.
- Webber, L. M. and Garson, O. M. (1983). Fluorodeoxyuridine synchronization of bone marrow cultures. Cancer Genet Cytogenet 8(2): 123-132.
- Whitfield, M. L., Sherlock, G., Saldanha, A. J., Murray, J. I., Ball, C. A., Alexander, K. E., Matese, J. C., Perou, C. M., Hurt, M. M., Brown, P. O. and Botstein, D. (2002). Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell 13(6): 1977-2000.
- Xu, M., Wang, W., Chen, S. and Zhu, B. (2011). A model for mitotic inheritance of histone lysine methylation. EMBO Rep 13(1): 60-67.
- Yuan, Z. F., Arnaudo, A. M. and Garcia, B. A. (2014). Mass spectrometric analysis of histone proteoforms. Annu Rev Anal Chem (Palo Alto Calif) 7: 113-128.
- Yunis, J. J., Bloomfield, C. D. and Ensrud, K. (1981). All patients with acute nonlymphocytic leukemia may have a chromosomal defect. N Engl J Med 305(3): 135-139.
- Zee, B. M., Britton, L. M., Wolle, D., Haberman, D. M. and Garcia, B. A. (2012). Origins and formation of histone methylation across the human cell cycle. Mol Cell Biol 32(13): 2503-2514.
- Zink, L. M. and Hake, S. B. (2016). Histone variants: nuclear function and disease. Curr Opin Genet Dev 37: 82-89.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Sato, H., Singer, R. H. and Greally, J. M. (2020). Quantitative Kinetic Analyses of Histone Turnover Using Imaging and Flow Cytometry. Bio-protocol 10(17): e3738. DOI: 10.21769/BioProtoc.3738.
- Sato, H., Wu, B., Delahaye, F., Singer, R. H. and Greally, J. M. (2019). Retargeting of macroH2A following mitosis to cytogenetic-scale heterochromatic domains. J Cell Biol 218(6): 1810-1823.
Category
Cell Biology > Cell imaging > Fluorescence
Cell Biology > Cell-based analysis > Flow cytometry
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.