- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Past Issue in 2024
Volume: 14, Issue: 6

Bioinformatics and Computational Biology
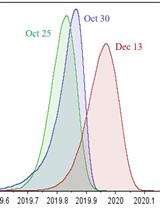
Classification of a Massive Number of Viral Genomes and Estimation of Time of Most Recent Common Ancestor (tMRCA) of SARS-CoV-2 Using Phylodynamic Analsysis
Biological Engineering

From Llama to Nanobody: A Streamlined Workflow for the Generation of Functionalised VHHs
Cell Biology
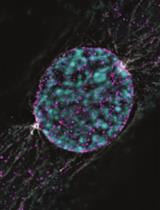
Dissecting the Mechanical Control of Mitotic Entry Using a Cell Confinement Setup
Immunology

Mesenchymal Stromal Cell (MSC) Functional Analysis—Macrophage Activation and Polarization Assays

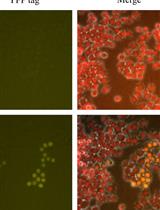
Genetic Knock-Ins of Endogenous Fluorescent Tags in RAW 264.7 Murine Macrophages Using CRISPR/Cas9 Genome Editing
Microbiology
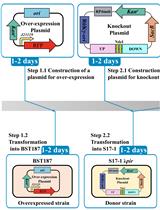
Efficient Genetic Transformation and Suicide Plasmid-mediated Genome Editing System for Non-model Microorganism Erwinia persicina

Preparation and Purification of β-1,3-glucan-Linked Candida glabrata Cell Wall Proteases by Ion-Exchange Chromatography, Gel Filtration, and MDPF-Gelatin-Zymography Assay
Molecular Biology
Proximity Labelling to Quantify Kv7.4 and Dynein Protein Interaction in Freshly Isolated Rat Vascular Smooth Muscle Cells