- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Protocol for Using CRISPR-Cas9 to Generate a Monocyte Cell Line Harboring a Single-Nucleotide Polymorphism
Published: Vol 16, Iss 8, Apr 20, 2026 DOI: 10.21769/BioProtoc.5660 Views: 711
Reviewed by: Navnita DuttaAnonymous reviewer(s)
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
We established a step-by-step approach for generating a single-nucleotide mutation in the promoter region of an immune regulatory gene in human monocyte THP-1 cells by employing a plasmid-based CRISPR-Cas9 system delivered via transfection with a homology-directed repair template DNA (HDR). Key steps include designing a single-guide RNA (sgRNA), cloning it into a CRISPR plasmid encoding the Cas9 protein, transfection of the plasmid constructs along with single-stranded oligonucleotide repair template (ssODNs) into THP-1 cells, followed by selection and validation. This approach provides a precise and relevant model to investigate the role of single polymorphisms in the regulation of inflammatory gene expression in human monocytes. In addition to the rs1024611 single-nucleotide polymorphism (SNP), this CRISPR/Cas9-based strategy is broadly applicable to functional studies of noncoding and coding variants in innate immune genes.
Key features
• Precise genome editing: CRISPR-Cas9-mediated editing of the rs1024611 SNP in the endogenous CCL2 promoter in THP-1 cells using a single-stranded donor oligonucleotide.
• Generate THP-1 monocytic cell lines: Differing only at the rs1024611 locus, allowing precise comparison of allele-specific effects on the same genetic background.
• The protocol includes strategies for single-cell cloning, PCR-based genotyping, and Sanger sequencing to confirm precise genome editing.
• While this study focuses on the CCL2-2518 SNP, it provides a framework for future investigations of other noncoding variants or regulatory SNPs in immune-related genes.
Keywords: CRISPR-Cas9Graphical overview
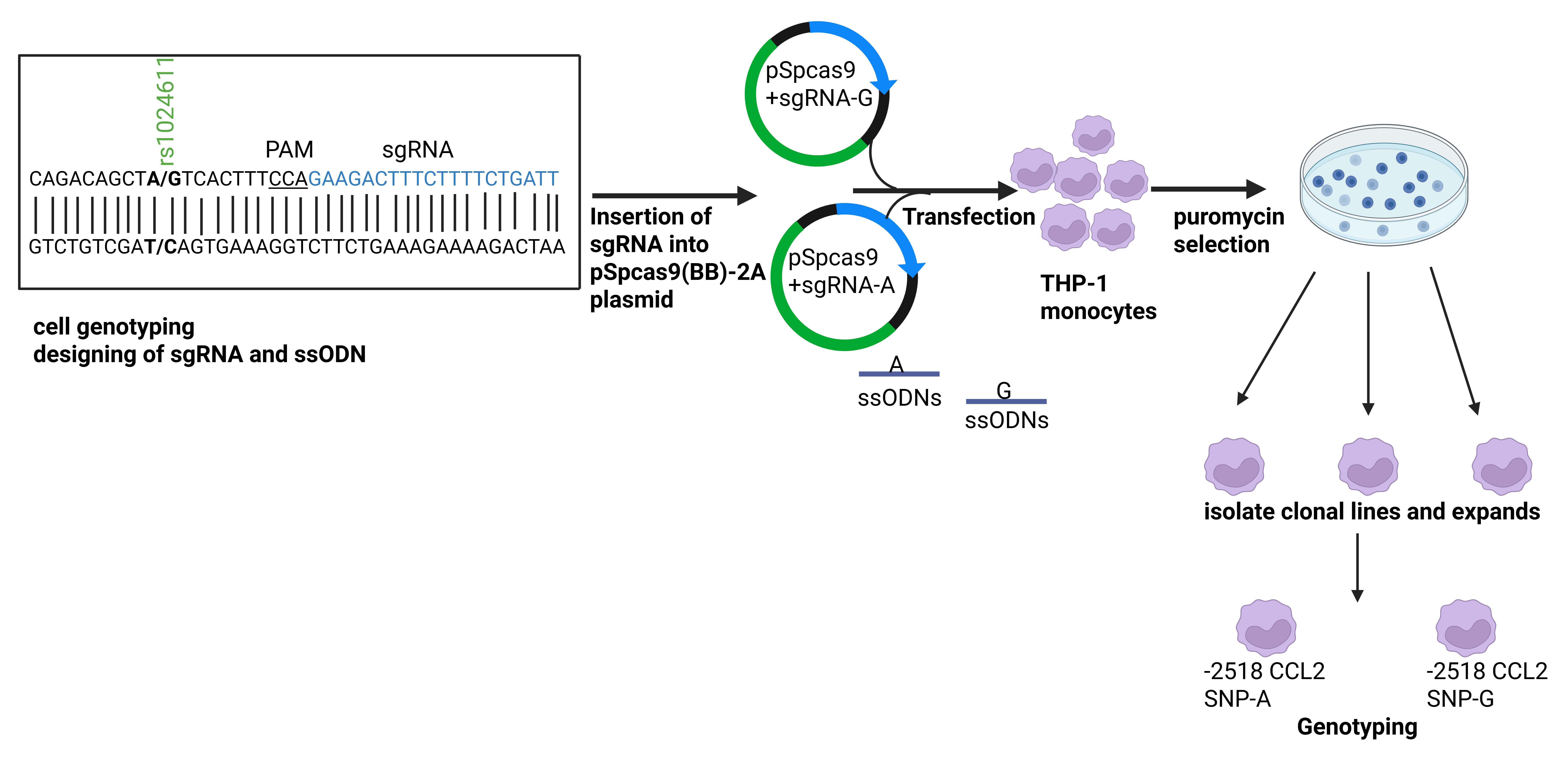
Overview of CRISPR-Cas9-mediated editing in the rs1024611 single-nucleotide polymorphism (SNP) of the CCL2 promoter in THP-1 cells
Background
The human THP-1 cell line, derived from a monocytic leukemia, represents a well-established model for studying monocyte and macrophage biology. THP-1 cells express innate immune receptors and can be differentiated into macrophage-like cells, making them particularly suitable for investigating gene regulation and inflammatory signaling pathways [1–3]. Establishing an isogenic THP-1 model carrying the desired mutation in the gene promoter [in this study, the isogenic THP-1 cells carrying either allele of the CCL2-2518A/G or rs1024611 single-nucleotide polymorphism (SNP) would provide a controlled system for directly comparing allele-specific transcriptional responses and downstream functional effects]. The rs1024611 SNP in the CCL2 promoter influences CCL2 chemokine expression and has been linked to susceptibility to inflammatory and infectious diseases [4]. We generated THP-1 cell lines differing only at this locus using CRISPR-Cas9 and a single-stranded donor oligonucleotide, enabling precise comparisons in an identical genetic background. CRISPR-Cas9 genome editing relies on a guide RNA (gRNA) to target the Cas9 endonuclease to a specific DNA sequence, where Cas9 creates a double-strand break that is subsequently repaired by either non-homologous end joining (NHEJ), resulting in insertions or deletions, or homology-directed repair (HDR) with a donor template, allowing for precise editing in the genes or regulatory regions. It has become a widely used tool for functional genomics studies [5–7].
We established a CRISPR-Cas9 technology to modify THP-1 cells through targeted gene editing, providing a powerful approach to better characterize immune-related mechanisms, including pathogen–host interactions. This protocol is for efficient CRISPR/Cas9-based single-nucleotide editing in THP-1 cells using the transfection method to deliver a designed pSPCas9:sgRNA plasmid construct into the cell nucleus. CRISPR/Cas9-mediated editing at the genetic level was validated by Sanger sequencing or using restriction fragment length polymorphism (RFLP). Advantages of using the plasmid-based CRISPR-Cas9 method for genome editing over other traditional methods are its higher efficiency, cost-effectiveness, and plasmid stability compared to the viral delivery or electroporation-based ribonucleoprotein (RNP) systems [6,8]. Using this protocol, up to 20% single-nucleotide substitution efficiency in the targeted locus was achieved. Our protocol provides an efficient and easy method to create single-cell clones with the desired genotype. Previous studies have reported HDR-mediated single-nucleotide substitution efficiencies ranging from 1% to 10% in human cell lines without enrichment, with improved efficiencies following antibiotic or fluorescence-based selection [6,7]. In THP-1, precise knock-in efficiencies are often lower due to reduced transfection efficiency and limited HDR activity. Using puromycin selection to enrich transfected cells, our plasmid-based CRISPR-Cas9 approach achieved up to 20% single-nucleotide substitution efficiency at the targeted locus, representing a comparatively high editing rate for this challenging cell type.
Materials and reagents
Biological materials
1. THP-1 cell line (ATCC)
2. Bacterial strain (XL10-Gold ultracompetent cells) (VWR)
Reagents
1. AnzaTM T4 DNA ligase master mix (Invitrogen, catalog number: IVGN2102)
2. BbsI (New England BioLabs, catalog number: R0539S)
3. T4 polynucleotide kinase (PNK) (Thermoscientific, catalog number: EK0031)
4. rCutSmart buffer, 10× (New England BioLabs, catalog number: B6004S)
5. T4 DNA ligase reaction buffer, 5× (Invitrogen, catalog number: 15224-017)
6. Alkaline phosphatase (rSAP) (New England BioLabs, catalog number: M0371S)
7. RPMI-1640 (Sigma-Aldrich, catalog number: R8758)
8. Fetal bovine serum (FBS) (Sigma-Aldrich, catalog number: 0926)
9. Penicillin-streptomycin (10,000 U/mL) (Sigma-Aldrich, catalog number: P0781)
10. Trypsin-EDTA solution (Sigma-Aldrich, catalog number: T3924)
11. Opti-MEMTM I reduced serum medium, no phenol red (Gibco, catalog number: 31985-070)
12. Lipofectamine 3000 (Invitrogen, catalog number: L3000-015)
13. Puromycin (Sigma-Aldrich, catalog number: P8833)
14. Taq DNA Polymerase kit (Invitrogen, catalog number: 18038-042)
15. Agarose (Sigma-Aldrich, catalog number: A9539)
16. Dulbecco's phosphate-buffered saline (DPBS) (Sigma-Aldrich, catalog number: D8537)
17. Gene Ruler 1 kb Plus DNA ladder (Thermoscientific, catalog number: SM1333)
18. Ampicillin (Sigma-Aldrich, catalog number: A9518)
19. Gene Elute Gel Extraction and PCR Purification kit (Sigma-Aldrich, catalog number: NA1111-1KT/NA1020-KT)
20. Gene Elute Plasmid Miniprep kit (Sigma-Aldrich, catalog number: PLN350-1KT)
21. Gene Elute Mammalian Genomic DNA Miniprep kit Gene Elutes (Sigma-Aldrich, catalog number: G1N70-1KT)
22. LB medium (Sigma-Aldrich, catalog number: L3022)
23. Agar, bacteriological (Amresco, catalog number: J637)
24. PvuII-HF (New England BioLabs, catalog number: R3151S)
25. 2-Beta-ME or 2-ME (Sigma-Aldrich, catalog number: M6250)
26. S.O.C. medium (Invitrogen, catalog number: 15544-034)
27. Trizma-base (Sigma-Aldrich, catalog number: T6066)
28. Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) (Sigma-Aldrich, catalog number: E5134)
29. Glacial acetic acid (Sigma-Aldrich, catalog number: A6283)
30. L-Glutamine (Sigma-Aldrich, catalog number: G7513)
Solutions
1. 50× TAE buffer (see Recipes)
2. LB medium (see Recipes)
3. LB broth medium (see Recipes)
4. Complete RPMI 1640 (see Recipes)
5. Puromycin solution (1 mg/mL) (see Recipes)
Recipes
1. 50× TAE buffer
Weigh 242 g of Trizma-base (molecular weight 121.14 g/mol) and add to a 1 L Duran bottle. Dissolve it in 700 mL of MilliQ water. Add 100 mL of 0.5 M EDTA and carefully add 57.1 mL of glacial acetic acid. Finally, top up the solution to 1 L with MilliQ water.
2. LB agar medium
Add 2 g of agar powder and 2 g of LB medium to 100 mL of distilled water. Autoclave it before use.
3. LB broth medium
Add 2 g of LB broth powder to 100 mL of distilled water and autoclave it before use.
4. Complete RPMI 1640
To RPMI-1640, add 10% FBS (heat-inactivated), 1% penicillin-streptomycin antibiotics, and 1% glutamine.
5. Puromycin solution (1 mg/mL)
Dissolve 10 mg of puromycin in 1 mL of water and then dilute 1:10 to make a 1 mg/mL concentration.
Laboratory supplies
1. 96-well cell culture plate (Cellstar, catalog number: 655180)
2. 24-well cell culture plate (Cellstar catalog number: 665180)
3. 15 mL centrifuge tube (VWR, catalog number: 525-1069)
4. 50 mL centrifuge tube (VWR, catalog number: 525-0610)
5. 1.7 mL centrifuge tube (VWR, catalog number: 87003-294)
6. 10 μL tip (VWR, catalog number: 76323-388)
7. 200 μL tip (VWR, catalog number: 76323-390)
8. 1,250 μL tip (VWR, catalog number: 76323-404)
9. 10 mL serological pipette (Corning, catalog number: 75816-100)
10. 10 cm cell culture dish (Corning, catalog number: 430167)
11. 14 mL polystyrene round-bottom tube (Falcon, catalog number: 352057)
Plasmid and DNA sequences (Table 1)
Table 1. Plasmids, primers, sgRNAs, and ssODNs used for CRISPR-Cas9 editing of rs1024611
| Description | Source |
|---|---|
| pSpCas9 (BB)-2A-Puro (PX459) | Addgene |
| Sequencing primer: primer: U6-Fwd primer: GAGGGCCTATTTCCCATGATTCC | [8] |
| sgRNA1: sgRNA-A-top: CACCGCTTGACAGAGCAGAAGTGGG | IDT |
| sgRNA1: sgRNA-A-bottom: AAACCCCACTTCTGCTCTGTCAAGC | IDT |
| sgRNA2: sgRNA-G-top: CACCGAATCAGAAAAGAAAGTCTTC | IDT |
| sgRNA2: sgRNA-G-bottom: AAACGAAGACTTTCTTTTCTGATTC | IDT |
| Genotyping primer: rs1024611-Forward: GCTCCGGGCCCAGTATCT | IDT |
| Genotyping primer: rs1024611-Reverse: GAGTGTTGGAAGCATGTCTCTACTT | IDT |
| ssODN: rs1024611-A-F: GAGGGCATCTTTTCTTGACAGAGCAGAAGTGGGAGACAGACAGCTATCACTTTTCAGAAGACTTTCTTTTCTGATTCATACCCTTCACCTT | IDT |
| ssODN: rs1024611-A-R: AAGGTGAAGGGTATGAATCAGAAAAGAAAGTCTTCTGAAAAGTGATAGCTGTCTGTCTCCCACTTCTGCTCTGTCAAGAAAAGATGCCCTC | IDT |
| ssODN: rs1024611-G-F: GAGGGCATCTTTTCTTGACAGAGCAGAAGTGGGAGACAGACAGCTGTCACTTTTCAGAAGACTTTCTTTTCTGATTCATACCCTTCACCTT | IDT |
| ssODN: rs1024611-G-R: AAGGTGAAGGGTATGAATCAGAAAAGAAAGTCTTCTGAAAAGTGACAGCTGTCTGTCTCCCACTTCTGCTCTGTCAAGAAAAGATGCCCTC | IDT |
Equipment
1. Pipettes (Eppendorf)
2. PCR thermocycler (Bio-Rad, model: T100)
3. DNA electrophoresis apparatus (Bio-Rad, model: Wide-mini subcell GT)
4. Gel image analysis system (Invitrogen, model: iBright1500)
5. Nanodrop spectrophotometer (Thermo Scientific, model: Nanodrop One)
6. Centrifuge (Eppendorf, model: 5415 R)
7. 37 °C CO2 incubator (Thermo Scientific, model: 3578)
8. Shaker (New Brunswick Scientific I2400 incubator shaker)
9. Water bath (Fisher Scientific, model: ISOTEMP 215)
Software and datasets
1. Online sgRNA designing tools (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design) (free tool)
Procedure
A. Genotyping THP-1 cells
236 bp encompassing the CCL2-2518 polymorphic region was Sanger-sequenced using GENEWIZ Pvt Ltd. Sequencing results indicate that the polymorphic region is heterozygous with A/G nucleotide in parental THP-1 cells. To generate the THP-1 cells having only one nucleotide at the -2518 position of the CCL2 gene, we performed the CRISPR-based genomic editing in the THP-1 cells using the following method.
B. Cloning of sgRNA sequences into the pSPcas9 (BB)-2A-puro vector
Note: We designed and ordered sgRNAs from a qualified company (e.g., Integrated DNA Technology, IDT), listed in Table 1.
1. Preparation of the sgRNA oligos for vector insertion: Design sgRNAs that have a cut site near the SNP using online sgRNA designing tools (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design).
2. Order 100 μM top and bottom sgRNA from IDT and reconstitute with IDT buffer in 0.2 mL tubes according to Table 2.
Table 2. Components and concentrations for sgRNA annealing and phosphorylation reaction
| Reagent | Final concentration | Amount |
|---|---|---|
| sgRNA TOP (100 μM) | 10 μM | 1 μL |
| sgRNA BOTTOM (100 μM) | 10 μM | 1 μL |
| T4 DNA ligase reaction buffer (5×) | 1× | 2 μL |
| T4 PNK | 1 U/μL | 1 μL |
| ddH2O | n/a | 5 μL |
| Total | n/a | 10 μL |
3. Put the tubes into the thermocycler for oligo annealing: 37 °C for 30 min, 95 °C for 5 min, ramp down to 25 °C at 0.1 °C/s.
4. Dilute the annealed oligos 200× using ddH2O.
5. Digest plasmid pSpCas9 (BB)-2A-Puro. Prepare the mixture in the 1.5 mL tubes according to Table 3 and incubate the tubes at 37 °C for 12–16 h.
Table 3. Reagents and volumes for BbsI-mediated plasmid digestion
| Reagent | Final concentration | Amount |
|---|---|---|
| Vector | 0.04 μg/μL | 2 μg |
| CutSmart buffer (10×) | 1× | 5 μL |
| ddH2O | Up to 50 μL | |
| BbsI | 2 μL | |
| Total | 50 μL |
6. Transfer 42 μL of the mixture to a new tube and perform dephosphorylation using rSAP in 1× CutSmart buffer, following the manufacturer’s protocol. Incubate the tube at 37 °C for 15 min and at 50 °C for 15 min. Run the digested product on 1% agarose gel, purify the plasmid using the GeneElute Gel Extraction kit, and dilute the digested plasmid to 25 ng/μL for the next step.
7. Ligate the sgRNA and vector according to Table 4 and incubate at room temperature for 1 h.
Table 4. Amount of sgRNA, vector, and reagents for the ligation mixture
| Reagent | Final concentration | Amount |
|---|---|---|
| ddH2O | 4.5 μL | |
| Diluted sgRNA | 2 μL | |
| Vector 25 ng/μL | 2.5 ng/μL | 1 μL |
| Anza ligation master mix (4×) | 1× | 2.5 μL |
Critical: 1 μL of ddH2O instead of sgRNA in the ligation system serves as a negative control.
C. Transformation
1. Mix 4 μL of 2-Beta-ME to each aliquot of XL10-Gold ultracompetent cells and keep on ice for 10 min.
2. Add 20–40 μL of competent cells + 5 μL of ligation mix into an ice-cold 14 mL Falcon tube and incubate on ice for 30 min.
3. Subject cells to heat-shock at 42 °C for 30 s, followed by incubation on ice for 2 min. Add 250–450 μL of prewarmed S.O.C. medium and allow to grow at 37 °C under shaking conditions at 250 rpm for 1 h.
4. Plate 50–100 μL of ligation mixture (Table 4) on an Amp-LB-agar plate and place the plate in the incubator for 14–16 h at 37 °C. Use a 100 μg/mL concentration of ampicillin to make the LB-agar plates.
5. Pick a single clone and place it in the tube containing 3 mL of LB medium with 100 μg/mL ampicillin. Then, put the tube in the shaker at 37 °C for 12–16 h.
Notes:
1. More clones from one plate can be picked up at the same time to ensure positive clones.
2. Plasmids were purified and verified using the U6-Fwd primer for sequencing.
D. Cell culture and transfection
Note: THP-1 cells were cultured in complete RPMI-1640 medium at 37 °C with 5% CO2. One 10 cm plate of 70%–80% confluent cells was prepared for this experiment. At the same time, the SNP-centered repair templates should be designed and ordered (ssODNs) from IDT.
D1. Prepare cells for transfection
1. Remove the old medium and rinse the cells once with DPBS. Add 10 mL of ice-cold DPBS to a 10 cm plate, and detach superficially attached cells using gentle pipetting or scraping.
2. Pipette the cells gently, transfer the cell suspension into 15 mL tubes, and spin at 500× g for 5 min.
3. Remove supernatant. Homogenize cells in 5 mL of fresh media and transfer 0.5 mL of the mixture to each well in a 24-well plate. Note that one extra well should be added as control cells. Put the 24-well plate in the cell culture incubator at 37 °C with 5% CO2.
4. Sixteen hours later, replace media with 0.5 mL of fresh media.
D2. Plasmid transfection
1. Dilute Lipofectamine 3000 reagent and plasmid DNAs with Opti-MEM, according to Table 5.
Table 5. Volume of plasmids, ssODN, and reagents for the transfection mixture
| Mixture | Reagent | Experimental group | Control group |
|---|---|---|---|
| #1 | Opti-MEM | 25 μL | 25 μL |
| Lipofectamine 3000 | 1.5 μL | 1.5 μL | |
| #2 | Opti-MEM | 25 μL | 25 μL |
| pSPcas9(BB)-sgRNA | 350 ng | - | |
| ssODN (10 μM) | 2 μL | 2 μL | |
| P3000 | 1 μL | 1 μL |
2. Mix the solution from mixture #2 with mixture #1 and incubate at room temperature for 10–15 min. Add 50 μL to each well and mix gently by rocking.
3. Sixteen hours later, replace the transfection medium with 0.5 mL of fresh warm complete RPMI medium. At 48 h post-transfection, apply puromycin selection at a final concentration of 3 μg/mL.
E. Isolating clonal lines
Note: After 3–5 days, when the control cells all died in the medium with 3 μg/mL puromycin, the experiments are ready for isolating single-cell clones. For seeding single cells on a 10 cm plate, remove the spent culture medium, rinse the cells twice with DPBS, and then dissociate cells using gentle scraping or pipetting and transfer to a 10 cm Petri dish, followed by incubation at 37 °C in a CO2 incubator.
1. Pick single-cell clones for genotyping, as shown in Figure 1. When the clones are visible to the naked eye, they can be picked up from the 10 cm plate using the following steps:
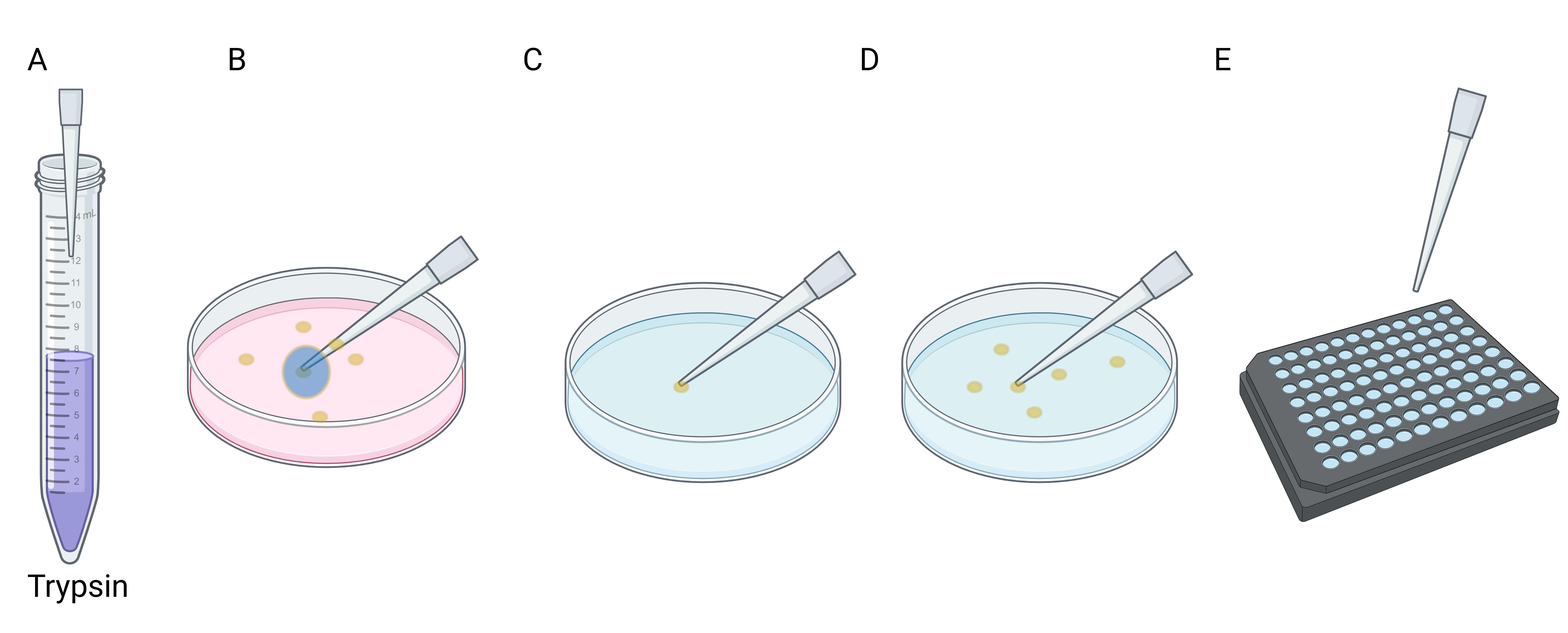
Figure 1. Workflow of picking a single clone for genotyping. (A) Aspirate 2.5 μL of trypsin in the pipette tip. (B and C) Scrape the clone from the plate using the pipette tips (B), then slowly release the pipette to carefully aspirate the clone into the tip (C). (D) Transfer the single clone into one well of a 96-well plate with 20 μL of trypsin and incubate for 5 min at 37 °C. (E) Inactivate trypsin and separate the cells by pipetting; transfer 10 μL of cell suspension from each well into a 96-well PCR plate.
a. Add 20 μL of trypsin-EDTA into each well of the 96-well plate.
b. Take out the 10 cm plate from the incubator, discard the old medium completely, and add 4 mL of DPBS into the plate. Be sure that the DPBS will not spill out while rotating the plate.
c. Use 10 μL pipette tips to pick clones of moderate size. When picking clones from a 10 cm plate, we recommend aspirating 2.5 μL of trypsin first in the pipette tip and then pipetting it out on the clone and holding the push-button.
d. Scrape the clone from the plate with the pipette tip. Then, carefully aspirate the cell into the tip for transfer.
e. Transfer these single clones quickly into the wells of a 96-well plate.
f. Pick 10–20 single clones from each plate with different genotypes (GG or AA repair template) and incubate at 37 °C for 5 min. Then, add 30 μL of medium supplemented with 10% (v/v) FBS to each well of the 96-well plate to inactivate trypsin. g. Pipette the cells to separate the adherent cells. Transfer 10 μL of cell suspension from each well into an 8-tube PCR strip (for genotyping). Then, add 100 μL of fresh medium to each well. Place the 96-well plate back in the cell incubator and, 24 h later, change the old medium of the 96-well plate with fresh medium to remove the trypsin.
F. Genotyping cell clones
1. After collecting all samples in an 8-tube PCR strip, perform DNA extraction using Gene Elute Mammalian Genomic DNA miniprep kit solution and column, followed by PCR for genotype.
Note: We run the following PCR program: an initial denaturation at 94 °C for 3 min; 35 cycles of denaturation at 94 °C for 30 s, annealing at 58 °C for 30 s, and extension at 72 °C for 30 s; and a final extension at 72 °C for 10 min, followed by a hold at 4 °C. We use the PCR mix in Table 6 to amplify a specific PCR product of 236 bp harboring the CCL2 promoter polymorphism at the -2518 position (rs1024611).
Table 6. PCR reaction mixture for amplification of the CCL2 promoter (rs1024611 genotyping in THP-1 clones)
| Reagent | Final concentration | Amount |
|---|---|---|
| 10× PCR buffer-MgCl2 | 1× | 5 μL |
| dNTP (10 mM) | 0.2 nM | 1 μL |
| MgCl2 (50 mM) | 1 mM | 1 μL |
| Primer P1 (5 μM) | 0.5 μM | 5 μL |
| Primer P2 (5 μM) | 0.5 μM | 5 μL |
| Genomic DNA (from clones) | 10–50 ng | |
| DNA Taq polymerase | 0.05 U/μL | 1 μL |
| ddH2O | Up to 50 μL | |
| Total | 50 μL |
2. Purify the PCR product using the Gene Elute DNA Extraction and PCR Purification kit. Take the purified DNA from each clone as a template for genotyping PCR.
3. Genotype the clones as soon as possible, before they reach confluence in the 96-well plate. The clones in the 96-well plate should be checked every day.
G. Expansion of cell clones
1. After the genotyping step, culture-expand the cells with the successfully converted SNP genotypes: When cells reach 50%–70% confluence in the 96-well plate, remove the old medium, rinse the cells twice with DPBS, add 100 μL of fresh complete RPMI medium, and dissociate cells by pipetting. Transfer the entire cell suspension into wells prefilled with 400 μL of fresh medium in a 24-well plate.
Data analysis
To assess genome editing efficiency, 10 single-cell clones were isolated from the transfected THP-1 population and expanded. Genomic DNA was extracted from each clone, and the target locus was PCR-amplified and sequenced. Two out of the 10 clones carried the desired CCL2-2518 SNP edit, corresponding to an editing efficiency of approximately 20%. This clonal analysis provides a direct and precise measure of successful genome editing in THP-1 cells.
Validation of protocol
We have confirmed the single-nucleotide mutation using either Sanger sequencing (Figure 2A–C) or using restriction fragment length polymorphism (PvuII restriction enzyme used to digest the 321 bp of amplified DNA from parental THP-1 carrying G/A, mutant THP-1 cells carrying G/G or A/A allele in the CCL2 promoter at -2518 position, also referred to as rs1024611 SNP) (Figure 2D). After PvuII restriction digestion of THP-1 cells, parental G/A cells show three DNA bands of 321, 182, and 139 bp. G/G mutant cells display two bands of 182 and 139 bp, as the G nucleotide creates the PvuII recognition site (CAGCTG). A/A mutant cells show a single intact band of 321 bp, indicating loss of the restriction site. Using this method, the mutated efficiency at rs1024611 in THP-1 cells is approximately –20%. Thus, the efficiency of this method can vary at different SNP sites and cell lines. This is probably due to the differences in local genomic sequence features, chromatin structure, and cellular properties.
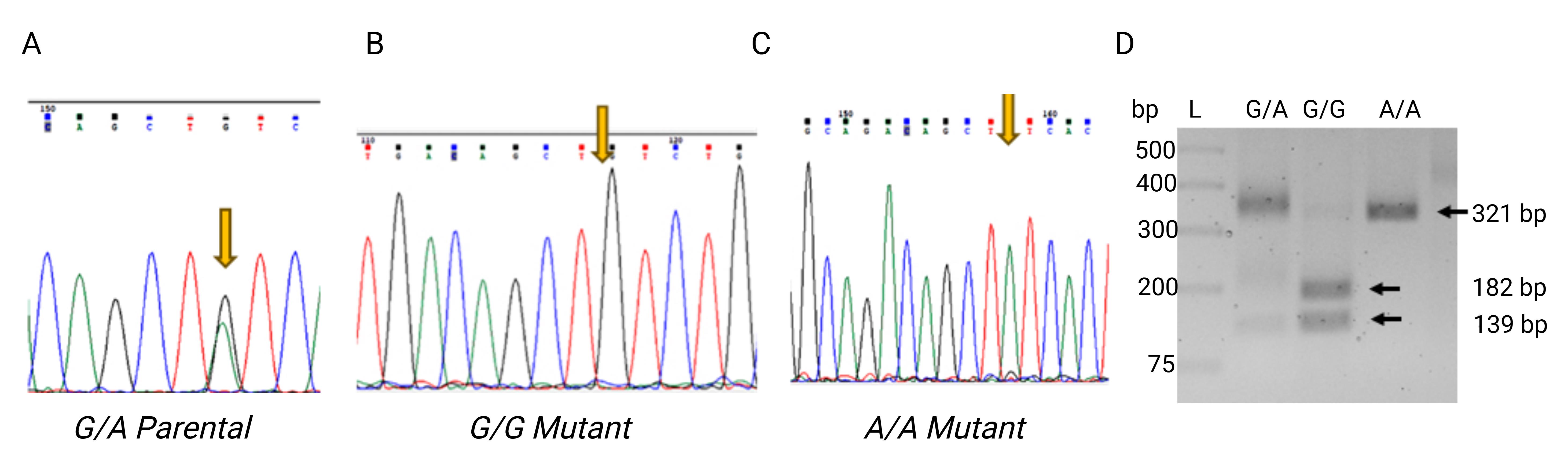
Figure 2. Genotype at rs1024611 in THP-1 cells was successfully converted from G/A to G/G and A/A. (A–C) DNA sequencing chromatogram peak analysis, indicating that the genotype at rs1024611 in THP-1 cells was successfully converted from G/A (A) to G/G (B) and A/A (C). (D) PCR-amplified 321 bp fragments spanning the SNP site were digested with PvuII to distinguish parental and edited alleles. Parental cells (G/A heterozygous) show three bands of 321, 182, and 139 bp, reflecting the presence of both alleles. G/G mutant clones display two bands of 182 and 139 bp, indicating homozygosity for the G allele. A/A mutant clones show a single 321 bp band, consistent with homozygosity for the A allele. This restriction digest confirms the genotypes of the edited THP-1 clones. The GeneRuler 1 kb Plus DNA Ladder was used as a molecular size marker. The chromatograms shown in Figure 2 were generated and analyzed using Chromas version 2.6.6 software or Geneious R8 version.
General notes and troubleshooting
General notes
1. Cells should be initially seeded at a lower density (40%–50%); they must reach 70%–80% confluence at the time of transfection.
2. Change the position after selecting a single clone, or remember to rotate the plate frequently to prevent the plate from drying out.
3. The cells on the 96-well plate usually grow very slowly. Remember to change the medium every three days until the cells are confluent for passaging. Earlier transfer of the cells to a bigger plate may cause cell death when the density of the cells is too low.
Troubleshooting
Problem 1: Genotyping THP-1 cells.
Possible cause: Primers have low specificity for genotyping.
Solution: The SNP genotype of the target cells should be determined before performing CRISPR/Cas9-mediated SNP editing. Accurate genotyping is essential for the proper design of sgRNAs and ssODN repair templates. If sequencing results do not show a single, unambiguous peak, the genotyping primers should be redesigned and revalidated. Careful design of allele-specific primers is critical to ensure reliable and accurate final genotyping results.
Problem 2: Inserting sgRNAs into vector pSpcas9 (BB)-2A-Puro using BbsI restriction digestion.
Possible cause: Ligation efficiency was low.
Solution: Digest vector pSpCas9 (BB)-2A-Puro with BbsI by incubating at 37 °C for 12–16 h to ensure complete digestion. A shorter digestion time results in incomplete digestion of vector DNA, which can significantly reduce the ligation efficiency of sgRNAs into the vector.
Problem 3: THP-1 cell culture and transfection.
Possible cause: Low transfection efficiency.
Solution: THP-1 cells were transfected using Lipofectamine 3000 following the manufacturer’s protocol. Optimization experiments were performed using this reagent; alternative transfection reagents were not comparatively evaluated in this study.
Problem 4: Pick a single clone for genotyping.
Possible cause: The single clone died after being transferred to the 96-well plate.
Solution: Once clones are visible to the naked eye, individual clones can be picked from the 10 cm plate. Select the moderate-sized clones using a pipette with a 10 μL tip. Very small clones fail to proliferate after transferring clones from a 10 cm plate to a 96-well plate and are likely to undergo apoptosis. In addition, large clones are unlikely to be single clones, likely being a mixture of two or more single clones.
Problem 5: ≥90% cell death is not observed in non-transfected controls within 3–5 days.
Possible cause: The puromycin concentration may be insufficient.
Solution: In such cases, a puromycin kill curve should be performed to determine the minimal effective concentration required to eliminate non-transfected cells within 72 h. Antibiotic potency and cell density at the start of selection should also be verified.
Acknowledgments
This work was supported in part by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Numbers R21AI151936, R21AI171734, and R01AI168543 to J.L. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
pSpCas9(BB)-2A-Puro (PX459) V2.0 was a gift from Feng Zhang (Addgene plasmid #62988; http://n2t.net/addgene:62988; RRID: Addgene_62988). The graphical abstract was created using BioRender. This protocol is adapted from [9].
Competing interests
The authors declare that they have no competing interests.
Ethical considerations
No human or animal subjects were involved in this protocol.
References
- Tsuchiya, S., Yamabe, M., Yamaguchi, Y., Kobayashi, Y., Konno, T. and Tada, K. (1980). Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int J Cancer. 26(2): 171–176. https://doi.org/10.1002/ijc.2910260208
- Chanput, W., Mes, J. J. and Wichers, H. J. (2014). THP-1 cell line: An in vitro cell model for immune modulation approach. Int Immunopharmacol. 23(1): 37–45. https://doi.org/10.1016/j.intimp.2014.08.002
- Bosshart, H. and Heinzelmann, M. (2016). THP-1 cells as a model for human monocytes. Ann Transl Med. 4(21): 438–438. https://doi.org/10.21037/atm.2016.08.53
- Pham, M. H., Bonello, G. B., Castiblanco, J., Le, T., Sigala, J., He, W. and Mummidi, S. (2012). The rs1024611 regulatory region polymorphism is associated with CCL2 allelic expression imbalance. PLoS One. 7(11): e49498. https://doi.org/10.1371/journal.pone.0049498
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. and Charpentier, E. (2012). A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science. 337(6096): 816–821. https://doi.org/10.1126/science.1225829
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A., et al. (2013). Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 339(6121): 819–823. https://doi.org/10.1126/science.1231143
- Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., Norville, J. E. and Church, G. M. (2013). RNA-Guided Human Genome Engineering via Cas9. Science. 339(6121): 823–826. https://doi.org/10.1126/science.1232033
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A. and Zhang, F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 8(11): 2281–2308. https://doi.org/10.1038/nprot.2013.143
- Gao, P., Dong, X., Wang, Y. and Wei, G. H. (2021). Optimized CRISPR/Cas9-mediated single nucleotide mutation in adherent cancer cell lines. STAR Protoc. 2(2): 100419. https://doi.org/10.1016/j.xpro.2021.100419
Article Information
Publication history
Received: Jan 23, 2026
Accepted: Mar 11, 2026
Available online: Mar 23, 2026
Published: Apr 20, 2026
Copyright
© 2026 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Mahatha, A. C., Ramos-Espinosa, O., De Lima, D. S., Liu, E. Y., Vaidyan, S. and Liu, J. (2026). Protocol for Using CRISPR-Cas9 to Generate a Monocyte Cell Line Harboring a Single-Nucleotide Polymorphism. Bio-protocol 16(8): e5660. DOI: 10.21769/BioProtoc.5660.
Category
Immunology > Host defense > Human
Molecular Biology > DNA > Chromosome engineering
Cell Biology > Cell engineering > CRISPR-cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.