- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Simple and Easy Method for RNA Extraction from the Cyanobacterium Synechocystis sp. PCC 6803
Published: Vol 16, Iss 7, Apr 5, 2026 DOI: 10.21769/BioProtoc.5654 Views: 40
Reviewed by: Dennis J NürnbergSamujjal BhattacharjeeSrujana Samhita Yadavalli
Original research article
The authors used this protocol in:

2025
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Cyanobacteria have been widely used as model organisms in photobiochemical research and have recently been exploited as hosts in numerous pilot studies to produce valuable biochemicals via genetic and metabolic modifications. Analyzing cellular RNA is a suitable method for studying genetic changes in cells. Several methods have previously been reported for cyanobacterial RNA extraction. However, the majority of these methods rely heavily on phenol and chloroform, which are hazardous. Additionally, these methods are time-consuming and difficult to perform. Using Synechocystis sp. PCC 6803 as a model, this study developed a novel method for extracting total ribonucleic acid (RNA) using standard centrifugation techniques and laboratory chemicals such as citric acid, ethylenediaminetetraacetic acid, sodium dodecyl sulfate, sodium chloride, and tri-sodium citrate dihydrate to extract RNA from cyanobacterial cells. This method does not necessitate the use of hazardous chemicals, especially phenol and chloroform. Furthermore, it is cost-effective since it does not require expensive chemicals. The results of the quantification, purity, and integrity checks show the effectiveness of this method for extracting good-quality RNA. Furthermore, RT-qPCR results demonstrate that the quality of the extracted RNA is suitable for downstream applications.
Key features
• Simple and efficient RNA extraction method.
• Requires less than an hour to extract total RNA.
• Provides high-quality RNA suitable for downstream applications.
• This method might be used to extract RNA from other cyanobacteria and algae.
Keywords: CyanobacteriaGraphical overview
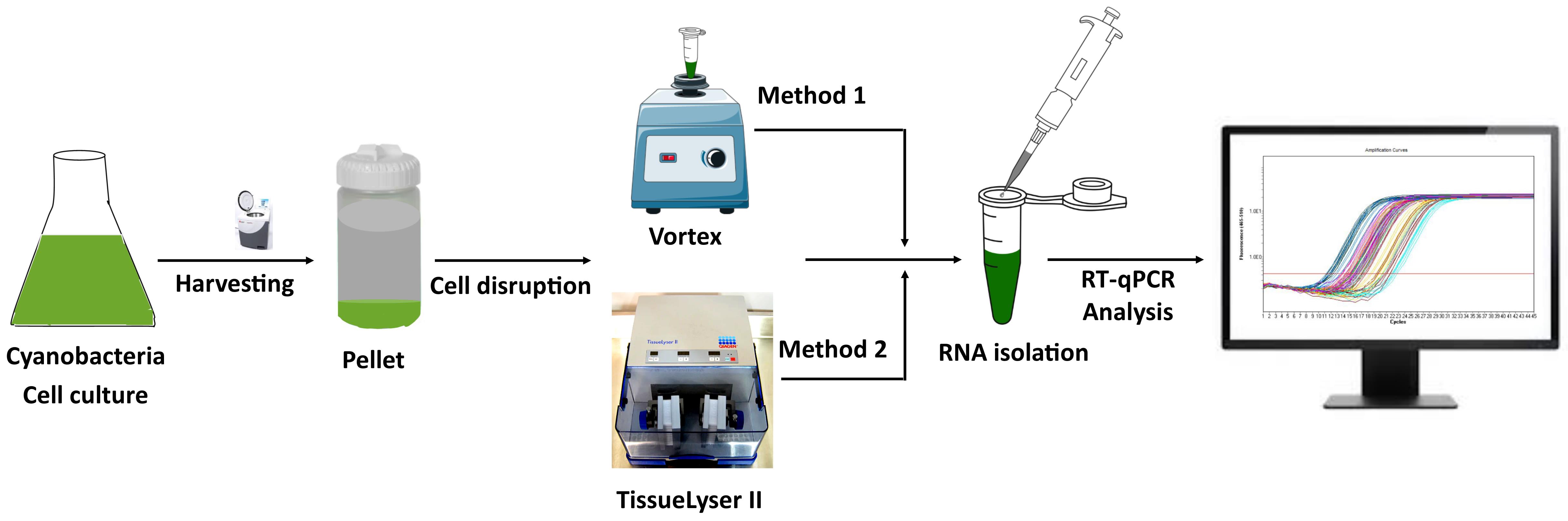
Overview of total RNA extraction from cyanobacteria and its downstream application
Background
Cyanobacteria are oxygenic photosynthetic organisms that have been used in various studies to investigate photosynthesis and biochemical production within the cell [1–4]. They have superior natural characteristics, including photoautotrophic growth [5], uptake of exogenous DNA in conjunction with homologous recombination (in some cyanobacteria) [6,7], and minimal growth requirements compared to other photosynthetic organisms [8], making them ideal candidates for biotechnological applications. Synechocystis sp. PCC 6803 is a unicellular freshwater cyanobacterium widely used as a model organism in photosynthesis research [9]. It has recently emerged as an efficient host for the production of a variety of valuable chemicals, including biofuels [3], biopharmaceutical proteins [4], and bioplastics [2]. The production of these chemicals within the cell requires effective genetic and metabolic changes. Modifying and introducing foreign genes into the genome to synthesize desired products necessitates their effective expression. RNA analysis is an efficient and reliable method for studying gene expression levels [10]. Several techniques have been developed to analyze RNA and evaluate gene expression, including quantitative polymerase chain reaction (qPCR), RNA-seq, RT-qPCR, complementary DNA (cDNA) libraries, and northern blotting [11–13]. Therefore, the quality and quantity of RNA are particularly critical for the many possible downstream applications.
Extracting high-quality RNA from cyanobacteria is always challenging. Cyanobacteria have a complex, gram-negative-type cell wall [14], which poses challenges to the currently available RNA extraction methods. Furthermore, cyanobacteria contain a variety of secondary metabolites, including polysaccharides and phenolic compounds, which hinder the effectiveness of RNA extraction methods in producing high-quality RNA [15]. Several methods, including chemical, physical, and hot and cold treatments, have been successfully used to extract RNA from cyanobacteria; however, they produce low yields [15–18]. Extracting RNA with chemicals such as organic solvents and phenol has been shown to work [15,19]; however, these chemicals constitute possible health hazards [20–22]. Physical and mechanical methods, on the other hand, produce higher yields than chemical ones, but require more time and processes to break the cells [18]. Commercial kits have been used to circumvent the multiple challenges of extracting large amounts of high-quality RNA in a short period of time. Although these commercial kits have a standard procedure for extracting high-quality RNA, they can be prohibitively expensive. Furthermore, the majority of commercially available RNA purification kits are not intended for organisms that contain polysaccharides and phenolic compounds, such as plants, algae, and cyanobacteria, because these compounds irreversibly bind to nucleic acids and co-precipitate them [23–27].
This study developed a novel method for extracting total RNA from Synechocystis sp. PCC 6803, using simple laboratory chemicals and no hazardous reagents. This new method is a combination of physical and chemical procedures. It is simple, quick, inexpensive, and produces a high yield. We used two different approaches to break the cells in order to avoid the use of expensive equipment and compared the effectiveness and yields of the two approaches. The cyanobacterium Synechocystis sp. PCC 6803 was used as a model organism for this study, but the protocol might also be applied to other cyanobacteria and algae. The method using a TissueLyser to break the cells showed better RNA yield than the method using a vortex mixer.
Materials and reagents
Biological materials
1. Cyanobacterium Synechocystis sp. PCC 6803
2. PCR primers (Table 1)
Table 1. List of primers used in RT-qPCR
| Protein name | Gene name | Primer name | Sequence (5-3) | Amplicon size (bp) |
|---|---|---|---|---|
| Photosystem II protein D1 | psbA2 | psbA2_qPCR-Fwd | TGTAATCGGCATCTGGTTCACT | 141 |
| psbA2_qPCR-Rev | ATGTTGGCTCGGTTCAATACA | |||
| Photosystem II protein D1 | psbA3 | psbA3_qPCR-Fwd | TCATCAAGGTACCGAACCAAC | 203 |
| psbA3_qPCR-Rev | TCTCTGAGCTTGAGGCCAA | |||
| Photosystem II CP47 chlorophyll-binding protein | psbB | psbB_qPCR-Fwd | TGCTGCTGGTATTGTCGGTAT | 124 |
| psbB_qPCR-Rev | AGCGGCAATACTACTGGACAA | |||
| Photosystem II CP43 chlorophyll-binding protein | psbC | psbC_qPCR-Fwd | TGGGTTGTGGTGCTCTGTTA | 126 |
| psbC_qPCR-Rev | AGATAATCGCCGGGTTGAG | |||
| Photosystem II D2 protein | psbDI | psbDI_qPCR-Fwd | TGCGGTGTTTGTCAGTGTCT | 196 |
| psbDI_qPCR-Rev | ACCGTGGATAGCACAGAGGA | |||
| Photosystem II D2 protein | psbDII | psbDII_qPCR-Fwd | AGACGGTGAAGATTCCAACAC | 184 |
| psbDII_qPCR-Rev | ACCCACAGAACTCATCCACAA | |||
| Cytochrome b559 α subunit | psbE | psbE_qPCR-Fwd | TGTCACCAGCATTCGCTACT | 208 |
| psbE_qPCR-Rev | TTATTGATTAAACTCTTGAATTTCC | |||
| Cytochrome b559 β subunit | psbF | psbF_qPCR-Fwd | GTTGGTAATTAACAATGGCAACC | 150 |
| psbF_qPCR-Rev | CCTAGCGTTGAATAAATTGCATC | |||
| Photosystem II protein J | psbJ | psbJ_qPCR-Fwd | CAATTTAGGAGGCATGGTATGTTC | 144 |
| psbJ_qPCR-Rev | CCTCGATTACATGGAAGAACCTAA | |||
| Photosystem I chlorophyll-binding reaction center protein A | psaA | psaA_qPCR-Fwd | CACCAGATCCACGTCTCCAT | 116 |
| psaA_qPCR-Rev | TAGAGTTCCGCCATCTTGCT | |||
| Photosystem I chlorophyll-binding reaction center protein B | psaB | psaB_qPCR-Fwd | TTCTAAGTTGATGCCGGACAA | 120 |
| psaB_qPCR-Rev | AAGCAGAGATGTCGCAGGTA | |||
| Bacterial RNase P | rnpB | rnpB_qPCR-Fwd | AGAGCGCACCAGCAGTATC | 118 |
| rnpB_qPCR-Rev | ATTCCTCAAGCGGTTCCA |
Reagents
1. EDTA (Sigma-Aldrich, catalog number: E9884)
2. Anhydrous citric acid (Sigma-Aldrich, catalog number: 1.93026)
3. Sodium chloride (NaCl) (Merck, catalog number: 1.06404)
4. Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: 436143)
5. Tri-sodium citrate dehydrate (Sigma-Aldrich, catalog number: 1.06448)
6. RNaseOUT (InvitrogenTM, catalog number: 10777019)
7. DNase I (InvitrogenTM, catalog number: 18080044)
8. Superscript III (InvitrogenTM, catalog number: 18068015)
9. qPCRBIO SyGreen Blue Mix Separate Rox (PCRBIOSYSTEMS, catalog number: PB20.17)
10. Liquid nitrogen
11. Diethyl pyrocarbonate (DEPC)-treated MilliQ water (Thermo Fisher Scientific, catalog number: AM9922)
12. Agarose (Sigma-Aldrich, catalog number: A2576)
13. Tris-borate-EDTA (TBE), 10× buffer (Thermo Scientific Chemicals, catalog number: J62788.K2)
14. Deoxyribonucleotide triphosphate (dNTP) mix (Thermo Scientific, catalog number: FERR0192)
15. 96-well plates (Bio-Rad, catalog number: HSP9601)
16. Dithiothreitol (DTT) (Thermo Scientific Chemicals, catalog number: AC327190010)
17. Phusion DNA polymerase (NEB, catalog number: M0530)
18. 1 kb Plus DNA ladder (Thermo Fisher Scientific, catalog number: 10787018)
19. RNA loading dye (NEB, catalog number: B0363S)
20. DNA loading dye (NEB, catalog number: B7024S)
21. BG-11 media (Gibco, catalog number: A1379901)
22. Isopropanol (Thermo Scientific Chemicals, catalog number: 327272500)
Solutions
1. Lysis buffer (see recipes)
2. Precipitation buffer (see recipes)
Recipes
1. Lysis buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| EDTA | 0.01 M | 0.372 g |
| Anhydrous citric acid | 0.132 M | 2.536 g |
| Tri-sodium citrate dihydrate | 0.068 M | 2.82 g |
| SDS, pH 5.0 | 0.069 M | 1.99 g |
| DEPC-treated MilliQ water | n/a | to 100 mL |
2. Precipitation buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| NaCl | 4 M | 23.37 g |
| Anhydrous citric acid | 0.033 M | 0.634 g |
| Tri-sodium citrate dihydrate | 0.017 M | 0.50 g |
| DEPC-treated MilliQ water | n/a | to 100 mL |
Note: Lysis and precipitation buffer can be stored at room temperature up to one year. Filtration/autoclaving is not needed.
Laboratory supplies
1. Nuclease-free microfuge tubes (Eppendorf, catalog number: 022364111)
2. Nuclease-free pipette tips (Eppendorf, catalog number: 0030078748)
Equipment
1. Pipettes (GILSON, catalog numbers: F167500, F167300)
2. Vortex (USA Scientific Inc, catalog number: 74045600)
3. Refrigerated tabletop micro centrifuge (Eppendorf centrifuge 5415 R, with rotor, catalog number: 022621431)
4. Mini centrifuge (Thermo ScientificTM, catalog number: 75004061)
5. Thermocycler (Eppendorf Mastercycler® nexus X2, catalog number: 6337000043)
6. TissueLyser II (QIAGEN, catalog number: 85300)
8. Agarose gel electrophoresis system (Analytik Jena, model: Biometra Horizon 58)
9. LightCycler® 480 system (Roche, catalog number: 05 015 278 001)
10. Gel Doc XR+ System (Bio-Rad, catalog number: 170–8195)
11. NanoDrop (Thermo Fisher Scientific, catalog number: ND-ONE-W)
12. Steel beads (5 mm) (QIAGEN, catalog number: 69989)
13. UV-Visible spectrophotometer (JASCO, model: V-550)
Procedure
A. Sample collection
1. Grow 150 mL of liquid culture of cyanobacteria in BG-11 medium until it reaches an OD730 of 0.8–1.0.
2. Harvest the cells by spinning them in a Beckman centrifuge at 5,000× g for 10 min at room temperature.
3. Discard the supernatant and resuspend the pellet in 5 mL of BG-11 medium.
4. Transfer 1 mL of sample into a 1.7 mL microfuge tube.
B. RNA extraction
B1. Method 1
1. Vigorously vortex the sample for 5 min at maximum speed.
2. Add 300–400 μL of lysis buffer to the cell suspension and gently mix by pipetting up and down 5 times using a 1,000 μL pipette.
3. Add 150 μL of precipitation buffer to the cell lysate and mix by inversion (10 times).
4. Incubate cell lysate on ice for 5–7 min.
5. After incubation, centrifuge the sample at 15,000× g for 3 min on a small benchtop centrifuge to pellet the unbroken cells and membranes.
6. Transfer the supernatant to a new microfuge tube without disturbing the pellet, then centrifuge at 16,000× g for 4 min to remove any remnants.
7. After centrifugation, transfer the supernatant to another clean microfuge tube and mix with 100% isopropanol in a 1:1 ratio.
8. Incubate the sample for 7 min on ice, followed by 10 min at room temperature.
9. Following incubation, centrifuge the sample mixture at 15,000× g for 5 min using a small benchtop centrifuge.
10. Discard supernatant and wash the pellet with 400 μL of cold 70% ethanol. Centrifuge again at 15,000× g for 5 min.
11. Remove the ethanol carefully without disturbing the RNA pellet.
12. Air dry the pellets by inverting the tubes on a clean paper towel with the lid open for 10–15 min.
13. Gently resuspend the dried pellet in 40 μL of DEPC-treated Milli-Q water and centrifuge at 16,000× g for 5 min to remove impurities and remnants. Impurities and remnants go to the pellet.
14. Transfer the RNA-containing supernatant to a clean microfuge tube and store it at -80 °C for later analysis.
B2. Method 2
1. Snap-freeze resuspended cells in liquid nitrogen and break them using one stainless steel bead per tube in a TissueLyser II (bead bashing for 1 min at 30 rps). Transfer the cell lysates to a new microfuge tube and proceed with step B1.2 and subsequent steps from method 1. See Figure 1 for the graphical presentation of RNA extraction methods.
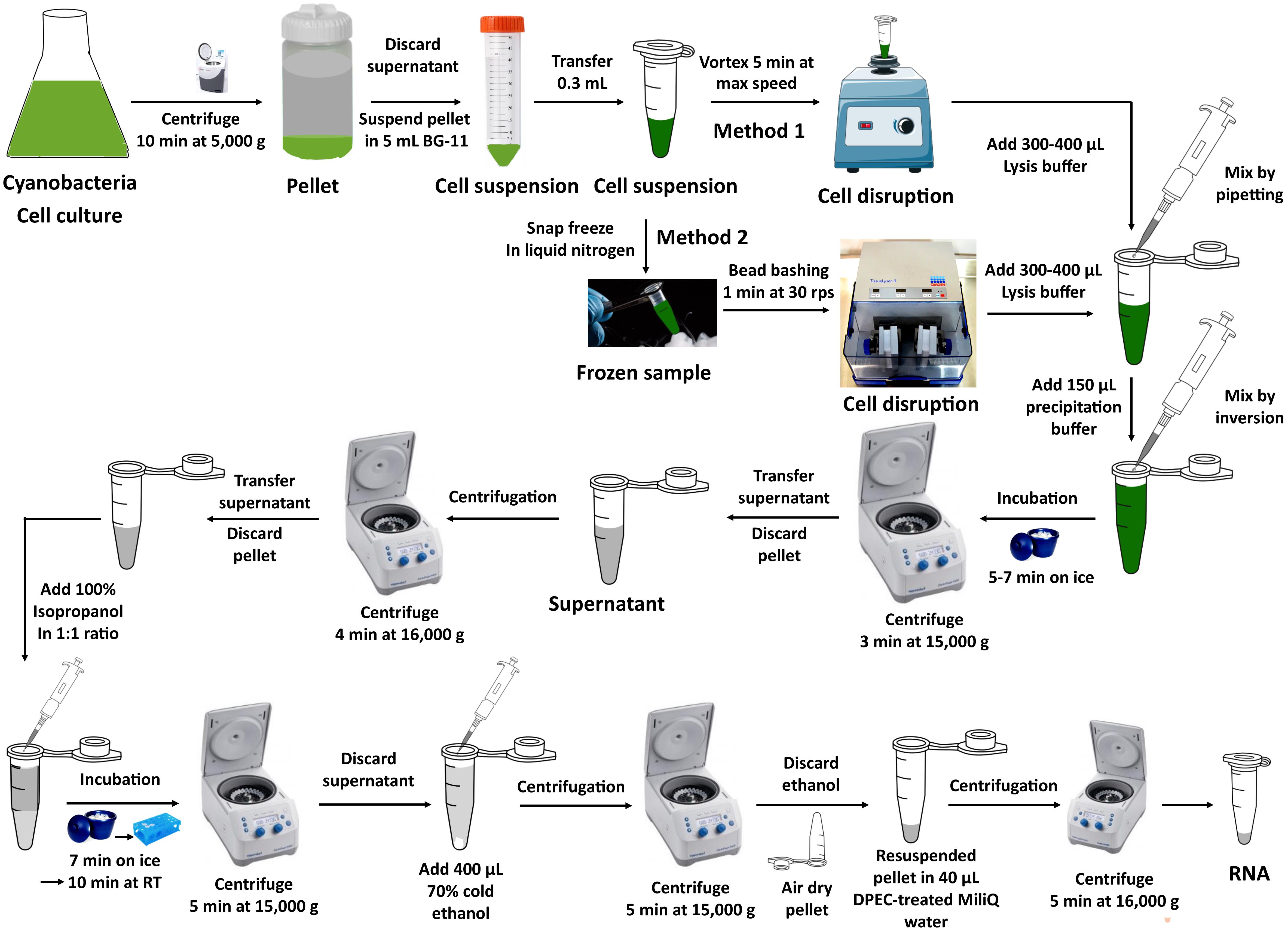
Figure 1. Overview of RNA extraction method
C. Quantification and purity check
Note: Data corresponding to this section are presented in Table 2.
1. Assess the purity and quantity of extracted RNA samples with a NanoDrop. Take 1 μL of isolated RNA and measure its concentration and purity.
2. The absorbance values at 260 and 280 nm (A260/280) indicate the purity and concentration of the isolated RNA.
3. The A260/280 and A260/230 ratios should be in the range of 1.9–2.1 and 2.0–2.2, respectively. The purity check results for the extracted RNA samples show the A260/280 and A260/230 ratios in the above range, indicating that the extracted RNA is of good quality and devoid of contaminants.
Table 2. Concentration and purity of RNA samples
| Method 1 | Method 2 | |||||
|---|---|---|---|---|---|---|
| Sample number | A260/280 nm | A260/230 nm | Concentration (μg/μL) | A260/280 nm | A260/230 nm | Concentration (μg/μL) |
| 1 | 2.05 | 2.10 | 0.125 | 1.99 | 2.19 | 1.53 |
| 2 | 2.00 | 2.08 | 0.109 | 2.04 | 2.16 | 1.98 |
| 3 | 1.99 | 2.01 | 0.150 | 2.06 | 2.0 | 1.89 |
| 4 | 1.95 | 1.89 | 0.138 | 2.03 | 2.08 | 1.25 |
| 5 | 1.98 | 2.15 | 0.132 | 1.97 | 1.98 | 1.77 |
| 6 | 1.99 | 2.13 | 0.145 | 1.95 | 1.90 | 1.99 |
| 7 | 1.96 | 1.95 | 0.149 | 1.99 | 2.13 | 1.36 |
D. Integrity check
Note: Data corresponding to this section are presented in Figure 2.
1. Use agarose gel electrophoresis to determine the integrity of RNA.
2. Load 1 μL of extracted RNA, 2 μL of loading dye, and a molecular marker onto a 1% agarose gel. Run for 50 min at 150 V using TBE (1×) buffer.
3. Use a Gel Doc XR+ System to capture and analyze the gel image. A good RNA sample should display obvious, distinct bands on the gel that correspond to 23S, 16S, and 5S RNA. If the RNA sample shows bands or smears below the 5S RNA band, this indicates that the RNA has been degraded or contaminated.
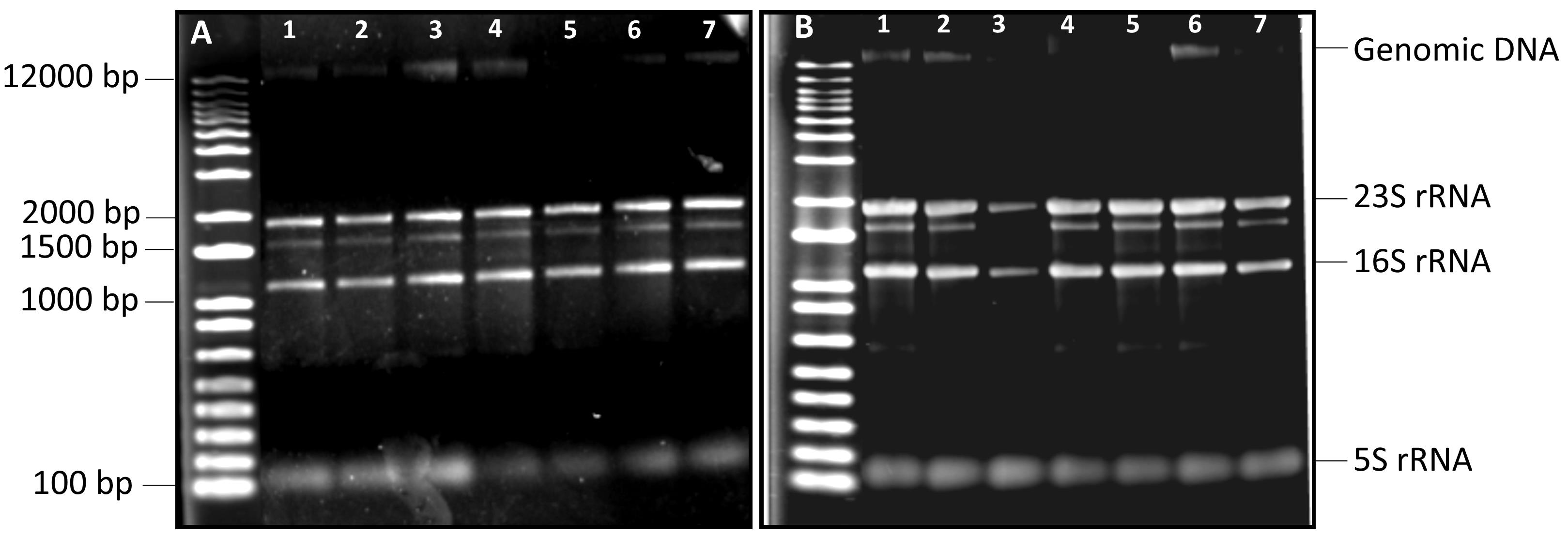
Figure 2. Electrophoretic mobility of extracted RNA on a 1% agarose gel. (A) Total RNA extracted from samples using Method 1 (section B1). (B) Total RNA extracted from samples using Method 2 (section B2). Samples 1–7 represent independent biological repeats. A 1 kb Plus DNA ladder was used in each gel.
E. DNase I treatment
Note: Data corresponding to this section are presented in Figure 3.
1. In a thermocycler, incubate 1 μg of extracted RNA with 1 μL of DNase I for 30 min at 37 °C.
2. To assess genomic DNA degradation, run 1 μL of DNase I-treated RNA on a 1% agarose gel alongside 1 μL of the untreated sample.
3. To further verify DNA degradation, run a PCR using samples with and without DNase I treatment as template. Add the following to each tube: 1.25 μL of forward and reverse primers (10 μM), 0.5 μL of dNTP mix (10 mM), 5 μL of 5× GC buffer, 0.75 μL of MgCl2 (50 mM), 0.25 μL of Phusion DNA polymerase, 1 μL of template, and H2O up to 25 μL. PCR conditions are as follows:
a. Initial denaturation: 98 °C for 30 s
b. Denaturation (35 cycles): 98 °C for 10 s
c. Annealing: (according to the primers) for 30 s
d. Extension: 72 °C for 30 s/kb
e. Final extension: 72 °C for 10 min
4. Run 1 μL of PCR product from DNase I–treated sample on a 1% agarose gel alongside 1 μL of PCR product from DNase I–untreated sample. The treated sample should not provide any PCR products on the gel, whereas an amplified product from the untreated sample is expected.
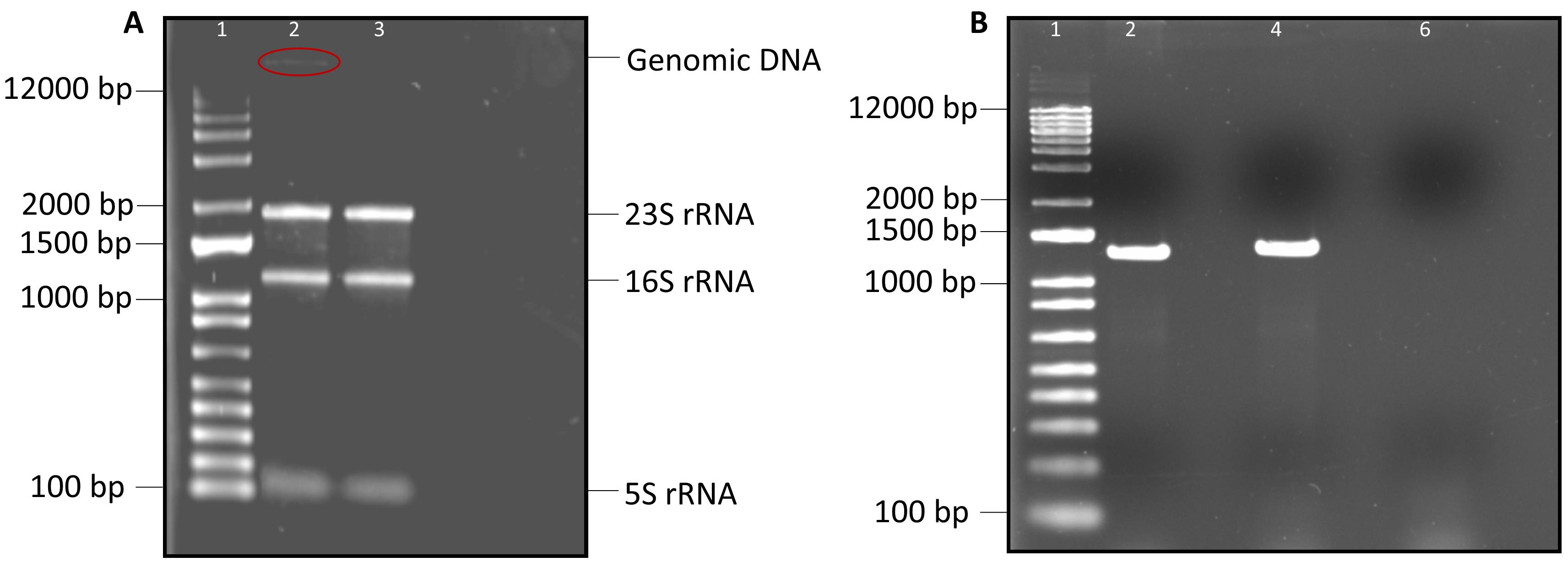
Figure 3. Gel electrophoresis of DNase I–treated RNA sample and the amplified PCR products. (A) Gel represents the electrophoretic mobility of DNase I–treated and untreated RNA samples in lanes 3 and 2, respectively. (B) Amplified PCR products from DNase I–untreated samples are presented in lanes 2 and 4; treated sample is in lane 6. The psbDII gene was amplified using fwd: AGTGTGTTTAGCGGCCCAAG, and rev: GTGGTGCAAACAGCGCAACA primers. Lane 1 represents the molecular marker (1 kb plus DNA ladder).
F. Complementary DNA (cDNA) synthesis
Note: Data corresponding to this section are presented in Figure 4.
1. Transfer approximately 500 ng of DNase I–treated RNA to a clean, RNase-free microfuge tube, leaving the remaining 500 ng of RNA in the original tube. Keep the tubes on ice. Label one tube as “RT” and the other as “NO RT”.
2. Add the following to each tube: 0.65 μL of random primers (250 ng/μL) (6:2:1:1 mix of 7, 8, 9, and 10-mers), 1 μL of dNTP mix (40 mM), 4 μL of 5× first-strand synthesis buffer, 2 μL of 0.1 M DTT (ensure all DTT is resuspended before use), and 0.75 μL of RNaseOUT.
3. Mix them by inversion followed by a quick spin.
4. Incubate the mixture at 25 °C for 5 min prior to the addition of Superscript III reverse transcriptase.
5. Add 1 μL of Superscript III to the “RT” tube and 1 μL of water to the “NO RT” tube. Mix them with gentle pipetting and inversion only, followed by a quick spin.
6. Incubate in a thermocycler for 60 min at 50 °C (55 °C if using a gene-specific primer).
7. After incubation, heat-inactivate the sample for 15 min at 70 °C.
8. Keep the cDNA at -20 °C for later use.
9. Check the cDNA synthesis on a 1% agarose gel. The cDNA sample should show multiple bands on the agarose gel, confirming the presence of cDNA.
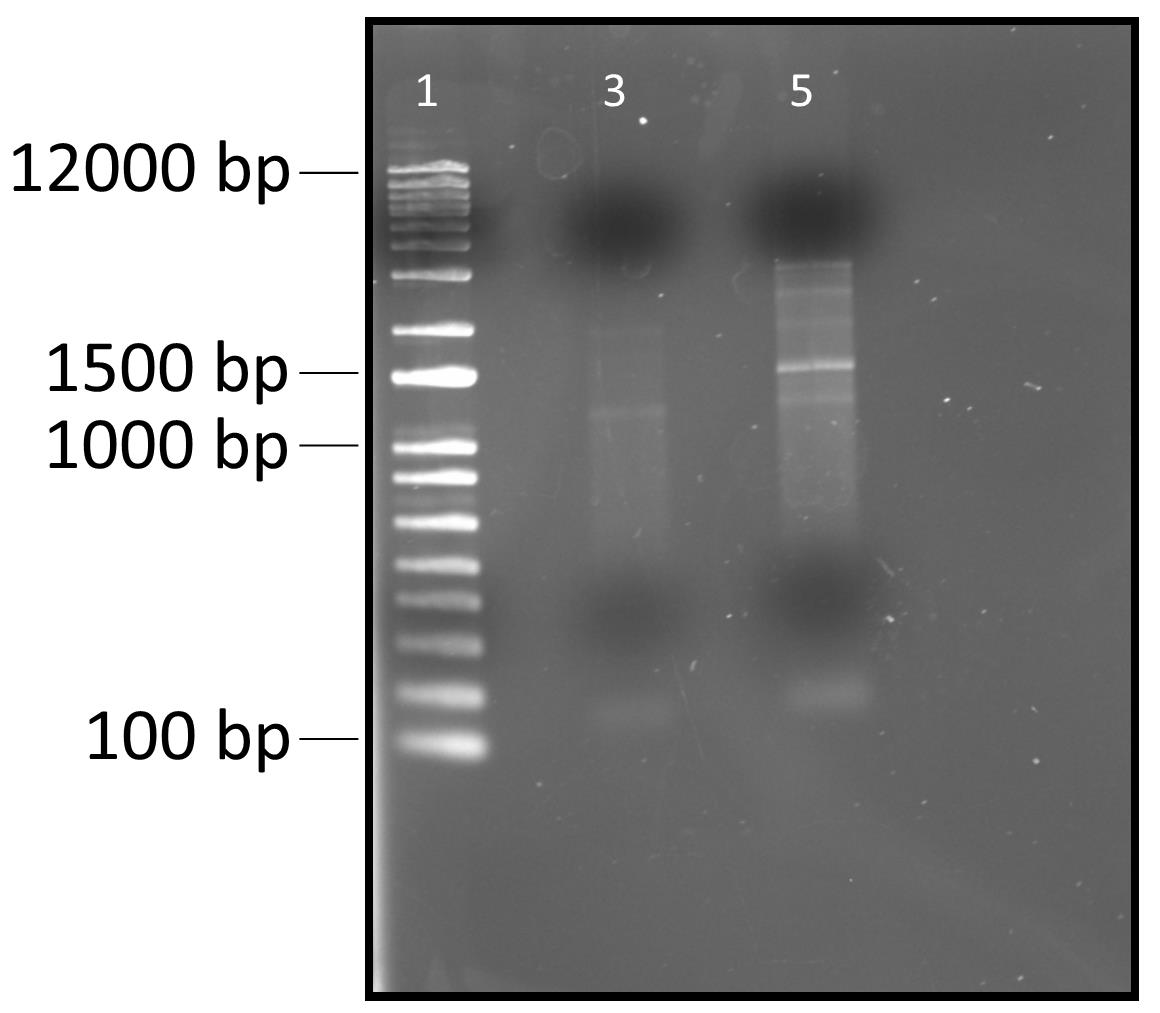
Figure 4. Electrophoretic mobility of RT-PCR products (cDNA) on 1% agarose gel. Lane 1 represents 1 Kb plus DNA ladder. Lane 3 represents PCR product without Superscript III reverse transcriptase. Lane 5 represents PCR products with Superscript III reverse transcriptase.
G. Quantitative reverse transcription polymerase chain reaction (RT-qPCR)
Note: Data corresponding to this section are presented in Figures 5 and 6 and Table 3.
1. Dilute cDNA 1:4 in DEPC-treated MilliQ water.
2. Transfer 4 μL of diluted cDNA to a 96-well plate. Add 1 μL of gene-specific 10 μM primer mix (forward and reverse primers) and 5 μL of 2× qPCRBIO SyGreen Blue Mix to the well containing cDNA.
3. PCR conditions:
a. Pre-incubation: 95 °C for 5 min
b. Amplification (45 cycles): 95 °C for 10 s
58 °C for 10 s
72 °C for 08 s
c. Melting curve: 95 °C for 5 s
65 °C for 1 min
97 °C [acquisition mode: continuous; acquisition (per °C): 5]
d. Cooling: 40 °C for 30 s
4. RT-qPCR results for the non-template control and no-reverse transcriptase control should not show any amplification curves, whereas the sample containing template and enzyme should exhibit one amplification curve and one melting peak for a particular gene of interest. Expect an RT-qPCR efficiency of 90%–100% for high-quality RNA and cDNA samples.
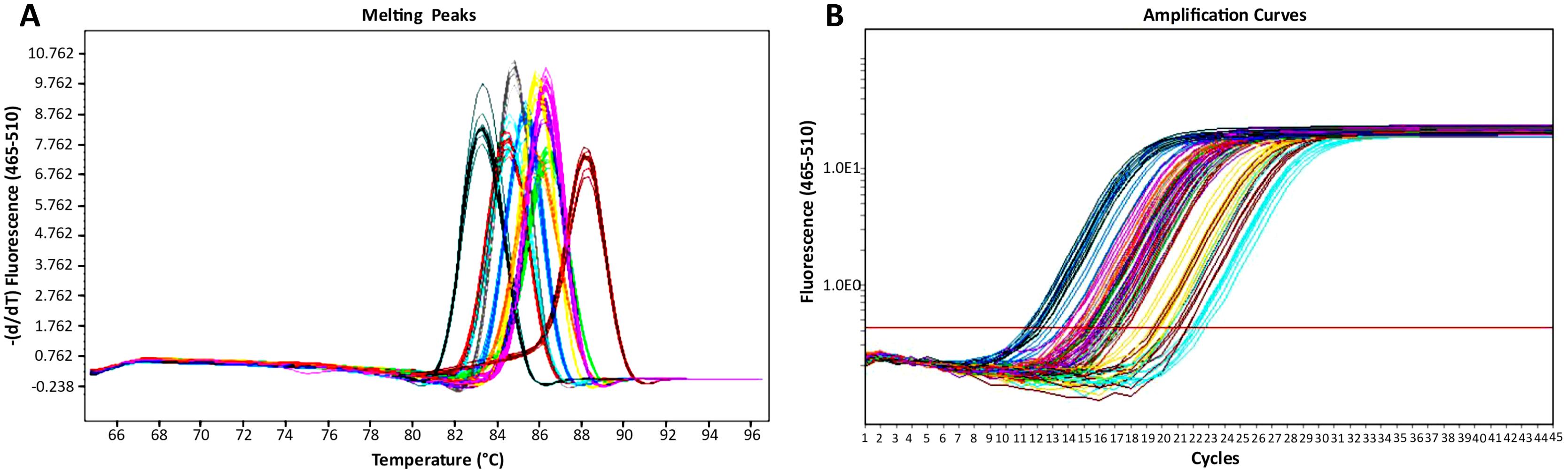
Figure 5. qPCR analysis results. (A) Melting peaks. (B) Amplification curves. Data are shown for psbA2 (dark blue), psbA3 (purple), psbB (yellow green) psbC (orange), psbDI (red), psbDII (magenta), psbE (yellow), psbF (maroon), psbJ (cyan), psaA (light blue), psaB (pink), and rnpB (green).
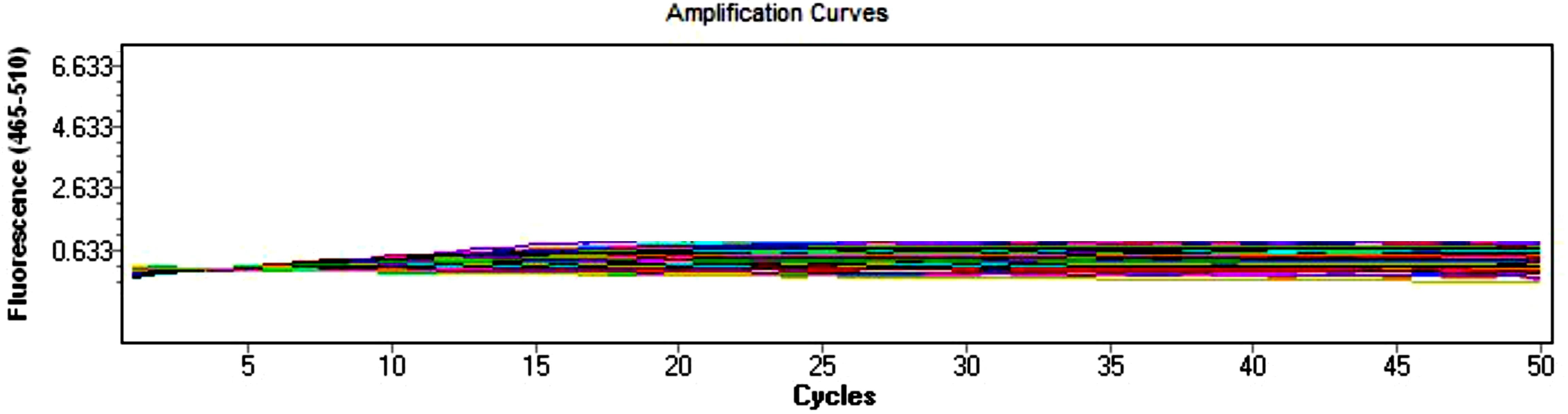
Figure 6. qPCR analysis results of non-template control and no-reverse transcriptase control. The non-template samples are blue, green, olive, cyan, brown, light blue, maroon, gray, purple, lime, and salmon. The no-reverse transcriptase samples are red, pink, yellow, magenta, orange, light green, violet, indigo, navy blue, mustard, and pale green.
Table 3. qPCR analysis results
| Protein name | Gene name | Amplicon Tm (°C) | Ct values | Efficiency (%) |
|---|---|---|---|---|
| Photosystem II protein D1 | psbA2 | 85.31 | 15.96 | 100 |
| Photosystem II protein D1 | psbA3 | 83.68 | 18.46 | 96 |
| Photosystem II CP47 chlorophyll- binding protein | psbB | 84.48 | 17.51 | 90 |
| Photosystem II CP43 chlorophyll- binding protein | psbC | 86.35 | 17.05 | 99 |
| Photosystem II D2 protein | psbDI | 86.28 | 15.92 | 89 |
| Photosystem II D2 protein | psbDII | 84.83 | 18.71 | 100 |
| Cytochrome b559 α subunit | psbE | 85.92 | 20.81 | 99 |
| Cytochrome b559 β subunit | psbF | 88.15 | 21.65 | 100 |
| Photosystem II protein J | psbJ | 84.63 | 22.25 | 98 |
| Photosystem I chlorophyll-binding reaction center protein A | psaA | 83.35 | 12.96 | 100 |
| Photosystem I chlorophyll-binding reaction center protein B | psaB | 85.81 | 15.09 | 100 |
| Bacterial RNase P | rnpB | 86.22 | 18.49 | 100 |
Data analysis
Raw data collected from Nanodrop and RT-qPCR are presented in tables. Gel images were created using Microsoft Publisher imaging software. The RT-qPCR data of the targeted genes can be calculated (relative expression or fold change) using the 2-ΔΔCT or comparative CT method [35–36].
Validation of protocol
This protocol has been used and validated in the following research article:
Majhi and Eaton-Rye [1]. PsbE:Phe10 of cytochrome b559 modifies inactivation and recovery of Photosystem II in response to high-light stress. Physiologia Plantarum (Figure S16 and S17).
In this study, this protocol was used to measure the expression of various photosynthetic genes. Cyanobacterial cells were treated with different light conditions, and the extracted RNA samples were used to assess the expression of genes for photosystem proteins in response to varied light conditions. At least three biological replicates were performed to verify the robustness of this protocol. The results presented in [1] demonstrate the effectiveness of this protocol.
General notes and troubleshooting
General notes
1. RNA is very sensitive to temperature. As such, keep the RNA sample on ice to prevent degradation.
2. Breaking cells using the TissueLyser improves yield by 15–20 times compared to vortexing the cells. Extracted RNA using both methods had a small quantity of remnant genomic DNA.
3. DNase treatment is necessary for downstream applications.
4. The whole extraction process generally takes less than 1 h.
Troubleshooting
Problem 1: Low yield.
Possible cause: Cells were not broken completely.
Solution: Vortex or bead-beat a little longer.
Problem 2: Low or high A260/280 nm values.
Possible causes: Impurities or protein contamination.
Solution: Centrifuge the dissolved RNA pellet again and carefully take out the supernatant without disturbing the pellet.
Acknowledgments
Conceptualization, B.K.M.; Investigation, B.K.M.; Writing—Original Draft, B.K.M.; Writing—Review & Editing, B.K.M., J.E.-R.; Funding acquisition, J.E.-R.; Supervision, J.E.-R.
Funding sources: University of Otago Ph.D. scholarship. University of Otago, Biochemistry Department internal funds. Original research paper in which this protocol was described: [1].
Competing interests
The authors declare no conflicts of interest.
References
- Majhi, B. K. and Eaton‐Rye, J. J. (2025). PsbE:Phe10 of Cytochrome b 559 Modifies Inactivation and Recovery of Photosystem II in Response to High‐Light Stress. Physiol Plant. 177(5): e70585. https://doi.org/10.1111/ppl.70585
- Majhi, B. K. (2025). Carbon capture, storage, and utilization in cyanobacteria. Carbon Resour Convers. 100388. https://doi.org/10.1016/j.crcon.2025.100388
- Majhi, B. K. (2025). Cyanobacteria: Photosynthetic cell factories for biofuel production. J Bioresour Bioprod. 10(2): 128–144. https://doi.org/10.1016/j.jobab.2024.10.001
- Majhi, B. K. and Melis, A. (2024). Recombinant protein synthesis and isolation of human interferon alpha-2 in cyanobacteria. Bioresour Technol. 400: 130664. https://doi.org/10.1016/j.biortech.2024.130664
- Muñoz-Marín, M. d. C., López-Lozano, A., Moreno-Cabezuelo, J. Ã., Díez, J. and García-Fernández, J. M. (2024). Mixotrophy in cyanobacteria. Curr Opin Microbiol. 78: 102432. https://doi.org/10.1016/j.mib.2024.102432
- Barten, R. and Lill, H. (1995). DNA-uptake in the naturally competent cyanobacterium,Synechocystis sp.PCC 6803. FEMS Microbiol Lett. 129(1): 83–87. https://doi.org/10.1111/j.1574-6968.1995.tb07561.x
- Kufryk, G. I., Sachet, M., Schmetterer, G. and Vermaas, W. F. (2002). Transformation of the cyanobacterium Synechocystissp. PCC 6803 as a tool for genetic mapping: optimization of efficiency. FEMS Microbiol Lett. 206(2): 215–219. https://doi.org/10.1111/j.1574-6968.2002.tb11012.x
- Majhi, B. K. (2025). Cyanobacteria: A Biological Chassis for Industrial Applications. Cyanobacteria - Exploring Their Role in Energy, Environment and Industry [Working Title]. e1012978. https://doi.org/10.5772/intechopen.1012978
- Ikeuchi, M. and Tabata, S. (2001). Synechocystis sp. PCC 6803 — a useful tool in the study of the genetics of cyanobacteria. Photosynth Res. 70(1): 73–83. https://doi.org/10.1023/a:1013887908680
- Corchete, L. A., Rojas, E. A., Alonso-López, D., De Las Rivas, J., Gutiérrez, N. C. and Burguillo, F. J. (2020). Systematic comparison and assessment of RNA-seq procedures for gene expression quantitative analysis. Sci Rep. 10(1): e1038/s41598–020–76881–x. https://doi.org/10.1038/s41598-020-76881-x
- Stanton, L. (2001). Methods to Profile Gene Expression. Trends Cardiovasc Med. 11(2): 49–54. https://doi.org/10.1016/s1050-1738(01)00085-8
- Singh, K. P., Miaskowski, C., Dhruva, A. A., Flowers, E. and Kober, K. M. (2018). Mechanisms and Measurement of Changes in Gene Expression. Biol Res Nurs. 20(4): 369–382. https://doi.org/10.1177/1099800418772161
- Zhang, M., Liu, Y. H., Chang, C. S., Zhi, H., Wang, S., Xu, W., Smith, C. W. and Zhang, H. B. (2019). Quantification of gene expression while taking into account RNA alternative splicing. Genomics. 111(6): 1517–1528. https://doi.org/10.1016/j.ygeno.2018.10.009
- Mohr, K. I., Brinkmann, N. and Friedl, T. (2011). Cyanobacteria. Reitner, J. and Thiel, V. (Eds.) Encyclopedia of Geobiology. Springer Netherlands, 306–311. https://doi.org/10.1007/978-1-4020-9212-1_221
- Pinto, F. L., Thapper, A., Sontheim, W. and Lindblad, P. (2009). Analysis of current and alternative phenol based RNA extraction methodologies for cyanobacteria. BMC Mol Biol. 10(1): e1186/1471–2199–10–79. https://doi.org/10.1186/1471-2199-10-79
- Kim, B., Oh, H., Lee, Y., Choi, G., Ahn, C., Yoon, B. and Kim, H. (2006). SIMPLE METHOD FOR RNA PREPARATION FROM CYANOBACTERIA1. J Phycol. 42(5): 1137–1141. https://doi.org/10.1111/j.1529-8817.2006.00263.x
- Singh, S. P., Sinha, R. P., Daiker, V. and Häder, D. P. (2009). Quantitative and qualitative extraction of RNA from a filamentous cyanobacterium Anabaena variabilis PCC 7937. J Appl Phycol. 22(1): 113–116. https://doi.org/10.1007/s10811-009-9428-7
- Kim Tiam, S., Comte, K., Dalle, C., Duval, C., Pancrace, C., Gugger, M., Marie, B., Yéprémian, C. and Bernard, C. (2019). Development of a new extraction method based on high-intensity ultra-sonication to study RNA regulation of the filamentous cyanobacteria Planktothrix. PLoS One. 14(9): e0222029. https://doi.org/10.1371/journal.pone.0222029
- Cárdenas Espinosa, M. J., Schmidgall, T., Wagner, G., Kappelmeyer, U., Schreiber, S., Heipieper, H. J. and Eberlein, C. (2021). An optimized method for RNA extraction from the polyurethane oligomer degrading strain Pseudomonas capeferrum TDA1 growing on aromatic substrates such as phenol and 2,4-diaminotoluene. PLoS One. 16(11): e0260002. https://doi.org/10.1371/journal.pone.0260002
- Babich, H. and Davis, D. (1981). Phenol: A review of environmental and health risks. Regul Toxicol Pharm. 1(1): 90–109. https://doi.org/10.1016/0273-2300(81)90071-4
- Joshi, D. R. and Adhikari, N. (2019). An Overview on Common Organic Solvents and Their Toxicity. J Pharm Res Int. : 1–18. https://doi.org/10.9734/jpri/2019/v28i330203
- Rahman, M. M., Rahaman, M. S., Islam, M. R., Rahman, F., Mithi, F. M., Alqahtani, T., Almikhlafi, M. A., Alghamdi, S. Q., Alruwaili, A. S., Hossain, M. S., et al. (2021). Role of Phenolic Compounds in Human Disease: Current Knowledge and Future Prospects. Molecules. 27(1): 233. https://doi.org/10.3390/molecules27010233
- Dos Reis Falcão, V., Pedroso Tonon, A., Cabral Oliveira, M. and Colepicolo, P. (2007). RNA Isolation method for polysaccharide rich algae: agar producing Gracilaria tenuistipitata (Rhodophyta). J Appl Phycol. 20(1): 9–12. https://doi.org/10.1007/s10811-007-9174-7
- Wang, X., Tian, W. and Li, Y. (2007). Development of an Efficient Protocol of RNA Isolation from Recalcitrant Tree Tissues. Mol Biotechnol. 38(1): 57–64. https://doi.org/10.1007/s12033-007-0073-6
- Sim, M. C., Ho, C. L. and Phang, S. M. (2013). A simple and effective method for RNA isolation and cDNA library construction from the brown seaweed Sargassum polycystum (Fucales, Phaeophyceae). J Appl Phycol. 25(5): 1277–1285. https://doi.org/10.1007/s10811-013-9980-z
- Greco, M., Sáez, C. A., Brown, M. T. and Bitonti, M. B. (2014). A Simple and Effective Method for High Quality Co-Extraction of Genomic DNA and Total RNA from Low Biomass Ectocarpus siliculosus, the Model Brown Alga. PLoS One. 9(5): e96470. https://doi.org/10.1371/journal.pone.0096470
- Jensen, T., Saleh, L., Bents, D., Krohn, S., Wu, Y. C., Mucke, M., Boje, A. S., Veltel, S., Hennig, S., Piker, L., et al. (2023). Optimised protocols for RNA extraction from a broad taxonomic range of algae. J Appl Phycol. 35(4): 1743–1753. https://doi.org/10.1007/s10811-023-02980-7
- Pathak, R. R. and Lochab, S. (2010). A method for rapid isolation of total RNA of high purity and yield from Arthrospira platensis. Can J Microbiol. 56(7): 578–584. https://doi.org/10.1139/w10-045
- Yu, L., Wu, X., Yu, Y., Shi, L. and Zhang, M. (2019). Recruitment of cyanobacteria by reverse transcription quantitative real-time PCR based on expression of Microcystis gene. PeerJ. 7: e7188. https://doi.org/10.7717/peerj.7188
- Rump, L. V., Asamoah, B. and Gonzalez-Escalona, N. (2010). Comparison of commercial RNA extraction kits for preparation of DNA-free total RNA from Salmonella cells. BMC Res Notes. 3(1): e1186/1756–0500–3–211. https://doi.org/10.1186/1756-0500-3-211
- Joseph, L. J. (2016). Setting Up a Laboratory. Genetic Diagnosis of Endocrine Disorders. 409–426. https://doi.org/10.1016/b978-0-12-800892-8.00029-4
- Schroeder, A., Mueller, O., Stocker, S., Salowsky, R., Leiber, M., Gassmann, M., Lightfoot, S., Menzel, W., Granzow, M., Ragg, T., et al. (2006). The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol. 7(1): e1186/1471–2199–7–3. https://doi.org/10.1186/1471-2199-7-3
- Ma, H., Bell, K. N. and Loker, R. N. (2021). qPCR and qRT-PCR analysis: Regulatory points to consider when conducting biodistribution and vector shedding studies. Mol Ther Methods clin dev. 20: 152–168. https://doi.org/10.1016/j.omtm.2020.11.007
- Harshitha, R. and Arunraj, D. R. (2021). Real‐time quantitative PCR: A tool for absolute and relative quantification. Biochem Mol Biol Educ. 49(5): 800–812. https://doi.org/10.1002/bmb.21552
- Livak, K. J. and Schmittgen, T. D. (2001). Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods. 25(4): 402–408. https://doi.org/10.1006/meth.2001.1262
- Schmittgen, T. D. and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 3(6): 1101–1108. https://doi.org/10.1038/nprot.2008.73
Article Information
Publication history
Received: Dec 19, 2025
Accepted: Mar 2, 2026
Available online: Mar 13, 2026
Published: Apr 5, 2026
Copyright
© 2026 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Majhi, B. K. and Eaton-Rye, J. J. (2026). A Simple and Easy Method for RNA Extraction from the Cyanobacterium Synechocystis sp. PCC 6803. Bio-protocol 16(7): e5654. DOI: 10.21769/BioProtoc.5654.
Category
Microbiology > Microbial biochemistry > RNA
Molecular Biology > RNA > RNA extraction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.