- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Lipid-Mediated Sequential Recruitment of Proteins Via Dual SLIPT and Dual SLIPTNVOC in Live Cells
(§Technical contact: kristina.bayer@mr.mpg.de) Published: Vol 15, Iss 21, Nov 5, 2025 DOI: 10.21769/BioProtoc.5489 Views: 1812
Reviewed by: Abhilash PadavannilAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2025
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Cellular phenomena such as signal integration and transmission are based on the correct spatiotemporal organization of biomolecules within the cell. Therefore, the targeted manipulation of such processes requires tools that can precisely induce the localizations and interactions of the key players relevant to these processes with high temporal resolution. Chemically induced dimerization (CID) techniques offer a powerful means to manipulate protein function with high temporal resolution and subcellular specificity, enabling direct control over cellular behavior. Here, we present the detailed synthesis and application of dual SLIPT and dual SLIPTNVOC, which expand the SLIPT (self-localizing ligand-induced protein translocation) platform by incorporating a dual-ligand CID system. Dual SLIPT and dual SLIPTNVOC independently sort into the inner leaflet of the plasma membrane via a lipid-like anchoring motif, where they present the two headgroup moieties trimethoprim (TMP) and HaloTag ligand (HTL), which recruit and dimerize any two iK6eDHFR- and HOB-tagged proteins of interest (POIs). Dual-SLIPTNVOC furthermore enables this protein dimerization of POIs at the inner leaflet of the plasma membrane in a pre-determined order and light-controlled manner. In this protocol, we detail the synthetic strategy to access dual SLIPT and dual SLIPTNVOC, while also providing the underlying rationale for key design and synthetic decisions, with the aim of offering a streamlined, accessible, and broadly implementable methodology. In addition to the detailed synthesis, we present representative applications and typical experimental outcomes and recommend strategies for data analysis to support effective use of the system. Notably, dual SLIPT and dual SLIPTNVOC represent the first CID systems to emulate endogenous lipidation-driven membrane targeting, while retaining hallmark advantages of CID technology—the precision over POI identity, recruitment sequence, high spatiotemporal control, and “plug-and-play” flexibility.
Key features
• Expands the original SLIPT technology [1] by enabling plasma membrane (PM) recruitment of any two POIs and their dimerization.
• Dual SLIPTNVOC as the first self-localizing lipid-like probe to induce PM recruitment and dimerization, with a defined recruitment sequence.
• Optimal use case at low probe concentrations: the system mimics physiological lipid-mediated dimerization without globally saturating the plasma membrane with recruited POIs.
• Descriptions of solid phase peptide synthesis and chemical synthesis for facile access to dual SLIPT and dual SLIPTNVOC, experimental results, and their analysis
Keywords: SLIPT technologyGraphical overview
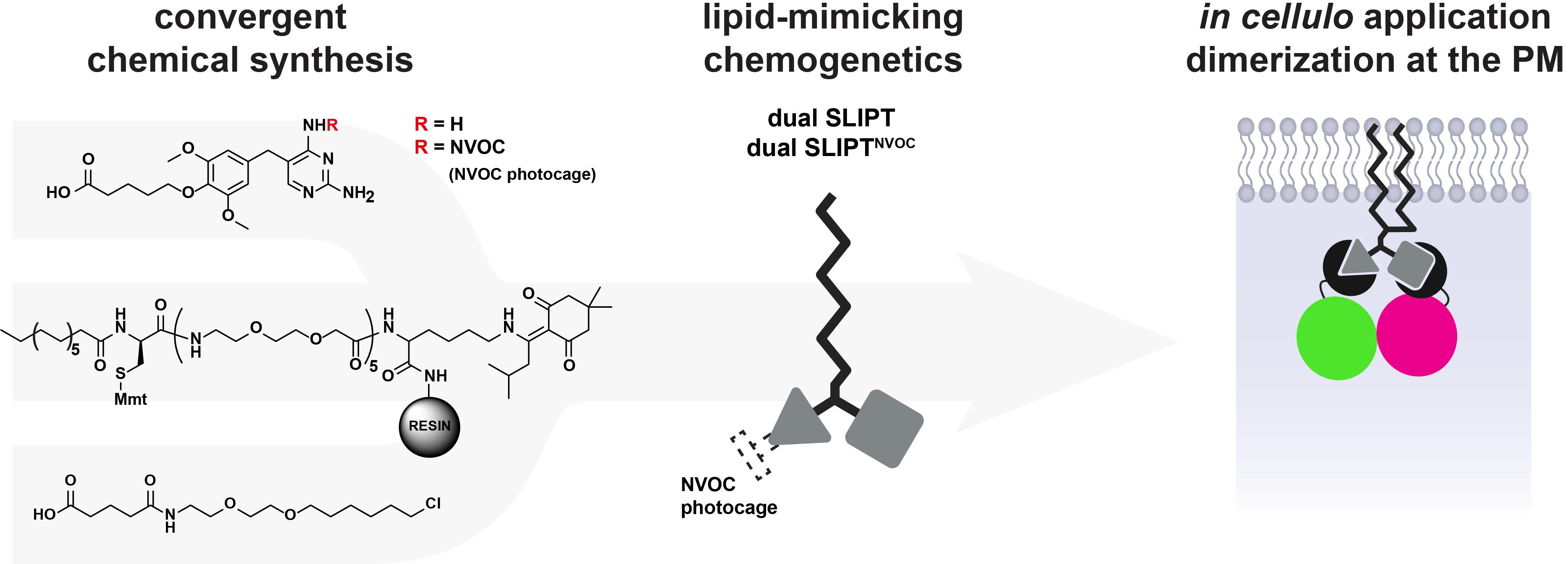
From chemical synthesis to in cellulo dimerization
Background
Spatiotemporal control of the localization and interactions of proteins of interest (POIs) using light as an external trigger is a highly attractive and minimally invasive manipulation strategy. Optogenetic or chemogenetic tools have emerged as a pivotal technique for establishing cause-and-effect and synthetically programming cellular behavior. The protocol presented herein details the synthesis and application of dual SLIPT and dual SLIPTNVOC [2], which expand on the self-localizing ligand-induced protein translocation (SLIPT) technology [1] with a chemically inducible dimerization (CID) approach.
Dual SLIPT and dual SLIPTNVOC comprise a (opto-)chemical tool set that enables recruitment of any two POIs to the inner leaflet of the plasma membrane (PM) (Figure 1). Dual SLIPT independently sorts into the PM via a lipid-like localization motif and recruits the two POIs by presenting two headgroup moieties [Trimethoprim (TMP) and HaloTag ligand (HTL)], which are bound by the two protein-tags iK6eDHFR (eDHFR variant [3]) and Halo-based oligonucleotide binder (HOB [4]). Thus, dual SLIPT makes use of a minimal number of mutually orthogonal protein tags to translocate two POIs in parallel from the cytosol to the entire inner leaflet of the PM and induce dimerization upon addition of the probe to the cell.
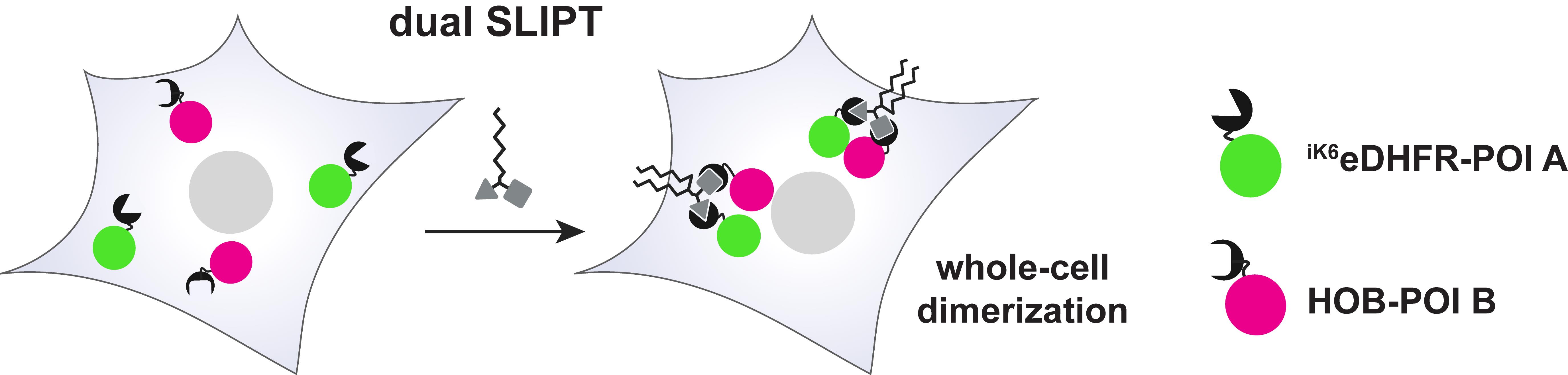
Figure 1. Mode of action of dual SLIPT. Cells expressing iK6eDHFR-POI A and HOB-POI B are treated with dual SLIPT. This causes dual recruitment of both fusion proteins to the inner leaflet of the plasma membrane inside the entire cell (whole-cell dimerization), which induces the dimerization of POI A and POI B.
Dual SLIPTNVOC, on the other hand, presents one constitutively active headgroup (HTL) and one conditionally active (photocaged TMPNVOC) headgroup to the cytosol. Upon addition to the cell, HOB-tagged POIs are immediately recruited to the inner leaflet of the PM, whereas TMPNVOC remains inactive with respect to PM recruitment of iK6eDHFR-tagged POIs (Figure 2). Irradiation with blue light allows for photouncaging the TMP headgroup, rendering it active and causing the co-recruitment of iK6eDHFR-tagged POIs within seconds [2]. Thus, dual SLIPTNVOC offers light-controlled, sequential activation for greater spatiotemporal precision.
Use of dual SLIPT and dual SLIPTNVOC has been demonstrated in identifying sequence-dependent effects of protein localization at the plasma membrane, as occurs during signal integration, propagation, and crosstalk, by controlling lamellipodia via sequential PM recruitment of guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs) [2].
Furthermore, by using dual SLIPTNVOC, the precise control over the light-irradiated area enables spatially confined dimerization to specific subcellular regions, thereby closely emulating endogenous lipid-mediated protein recruitment.
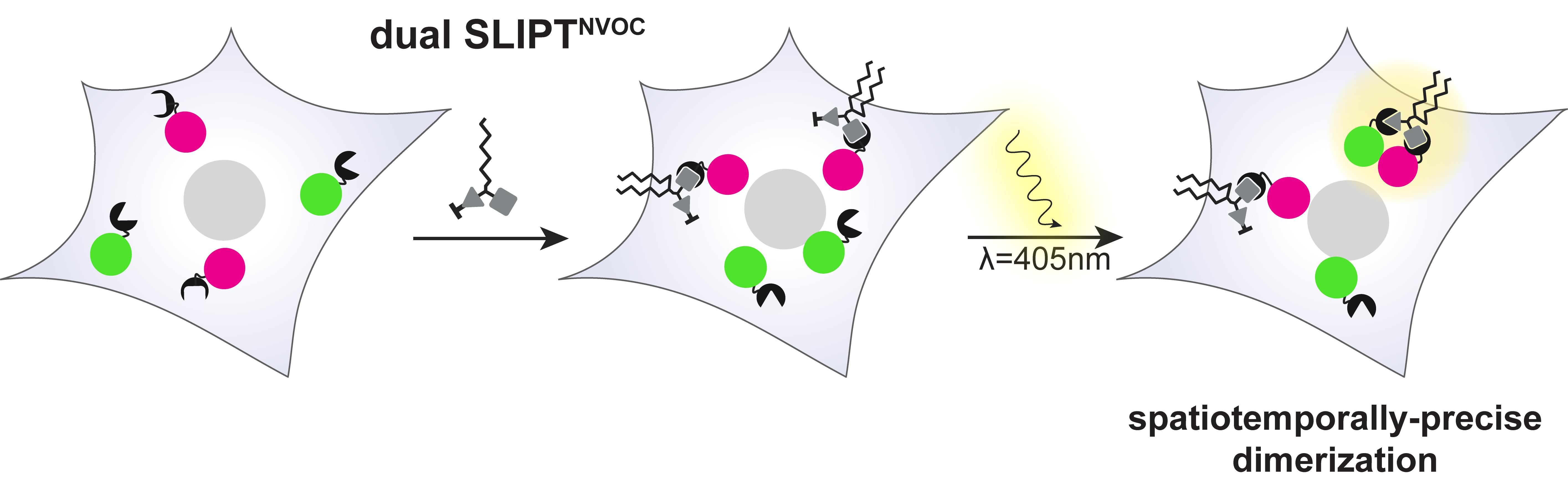
Figure 2. Mode of action of dual SLIPTNVOC. Cells expressing iK6eDHFR-POI A and HOB-POI B are treated with dual SLIPTNVOC. This causes PM recruitment of HOB-POI B inside the entire cell before irradiation of a subcellular region causes availability of the previously photocaged TMP arm and subsequent localized recruitment of iK6eDHFR-POI A. POI A and B are thereupon dimerized with high spatial and temporal precision.
Conventional CID (for reviews, see [5,6]) and optogenetic [7,8] strategies have been used to induce PM recruitment of POIs by prelocalizing one protein at the membrane (localizer) and translocating a second (actuator) upon stimulation (addition of the CID or irradiation with light). While suitable for constitutively membrane-associated POIs, these methods are less effective when both partners are cytosolic until signaling triggers their PM recruitment—a hallmark of many PM-associated signaling pathways, including many members of various actin signaling cascades (e.g., GEFs [9], GAPs [10], and nucleation-promoting factors [11]).
Chemically inducible trimerization (CIT) [12] addresses this limitation by enabling the recruitment of two cytosolic proteins to a membrane-localized third partner. However, similar to other CID and optogenetic systems, CIT requires more protein tags than actuators, increasing steric bulk and diverging from endogenous lipidation-mediated PM recruitment events.
Photocaged lipids (reviewed in [13]), on the contrary, offer the most native approach. They accumulate at the PM, being inactive until light-mediated uncaging, which reveals the active lipid headgroup and causes recruitment of proteins that can bind that specific lipid. While the most physiological approach mentioned here, manipulations with photocaged lipids are less orthogonal due to a lack of exact control over the identity of the recruited proteins. As a result, causality between PM recruitment of a specific POI and observed cellular effect is difficult to establish.
Dual SLIPT and dual SLIPTNVOC offer a compelling combination of the CID approach and the controlled release of endogenous lipids. The self-localizing nature of the lipid-like anchoring motif dispenses with the sterically demanding localizer, while maintaining the precision over POI identity and “plug-and-play” flexibility of traditional CID tools. The additional control over recruitment sequence, combined with the high spatiotemporal control of dual SLIPTNVOC, enables various experimental designs, such as engineering synthetic signaling pathways or the creation of logic circuits at the membrane.
This protocol describes the detailed synthesis, application, and validation of the two final probes, dual SLIPT and dual SLIPTNVOC. We describe how Trimethoprim and HaloTag ligand, both commercially available, are linker-functionalized to append them to a lipid-like anchoring motif. The said anchoring motif can be assembled by facile solid phase peptide synthesis (SPPS). This protocol further describes the use and rationale of various protecting groups that enable synthetic access to the final probes. To complete this protocol, minimal chemical training, as well as standard chemical equipment, is required. Possession of a peptide synthesizer is optional, but advisable.
Materials and reagents
All catalog numbers provided below shall serve as guide; alternative sources may be used as well.
Biological materials
1. HeLa (RRID:CVCL_0030) (DSMZ, catalog number: ACC 57) (see General note 1)
2. Halo-based oligonucleotide binder (Addgene, catalog number: 167268)
3. iK6eDHFR (Addgene, catalog numbers: 172100 and 172101)
Reagents
1. Boc-HaloTag ligand (ABCR, CAS number: 920264-35-3, catalog number: AB632176)
2. Trimethoprim (TCI, CAS number: 738-70-5, catalog number: T2286)
3. Myristic acid (TCI, CAS number: 544-63-8, catalog number: M0476)
4. Fmoc-D-Cysteine (Mmt) (AmBeed, CAS number: 1198791-73-9, catalog number: A311982)
5. Fmoc-8-amino-3,6-dioxaoctanoic (Roth, CAS number: 166108-71-0, catalog number: 7400.1)
6. Fmoc-Lys(ivDde) (Novabiochem, CAS number: 204777-78-6, catalog number: 8520820001)
7. Fmoc-Lys(Dde) (TCI, CAS number: 150629-67-7, catalog number: F1270)
8. Fmoc-4-aminobutyric acid (TCI, CAS number: 116821-47-7, catalog number: F0911)
9. Sieber amide resin (Novabiochem, CAS number: 915706-90-0, catalog number: 8550080001)
10. HBraq (TCI, CAS number: 10035-10-6, catalog number: H1220)
11. Ethyl-bromovalerate (TCI, CAS number: 14660-52-7, catalog number: B1557)
12. ((Nitroveratryl)oxy)chlorocarbamate (Aldrich, CAS number: 42855-00-5, catalog number: 420069-1G)
13. Glutaric anhydride (Aldrich, CAS number: 108-55-4, catalog number: G3806-5G)
14. 60% hydrazine monohydrate solution (TCI, CAS number: 7803-57-8, catalog number: H0172)
15. Diisopropylcarbodiimide (DIC) (TCI, CAS number: 693-13-0, catalog number: D0254)
16. Oxyma Pure (Aldrich, catalog number: 8510860100)
17. Piperidine (Roth, catalog number: A122.2)
18. Hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU) (TCI, catalog number: A1797-5G)
19. Benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) (TCI, catalog number: 11450788)
20. Triisopropylsilane (TIS) (Fisher Scientific, catalog number: 11322157)
21. Dimethylformamide (DMF) (Roth, catalog number: A529.1)
22. DMF (peptide synthesis-grade) (Fisher Scientific, catalog number: 10387221)
23. DMF, anhydrous (Fisher Scientific, catalog number: 16398227)
24. 1-methyl-2-pyrrolidinone (Fisher Scientific, catalog number: 11360358)
25. Imidazole (Fisher Scientific, catalog number: 11369959)
26. Hydroxylamine hydrochloride (TCI, catalog number: H1581-25G)
27. Cesium carbonate (Cs2CO3) (Roth, catalog number: 6873.4)
28. Methanol (MeOH) (Fisher Scientific, catalog number: 30280932)
29. Dichloromethane (DCM) (Fisher Scientific, catalog number: 15538744)
30. DCM, anhydrous (TCI, catalog number: D3478-500ML)
31. N’, N’-Diisopropylethylamine (DIPEA) (Roth, catalog number: 4105.1)
32. Tetrahydrofuran (THF) (Thermo Scientific Alfa Aesar, catalog number: 11378016)
33. Lithium hydroxide (LiOH) (TCI, catalog number: L0225-25G)
34. Sodium hydroxide (NaOH), 2 M solution (Aldrich, catalog number: 1091361003)
35. Hydrochloric acid (HCl), 1 M solution (Aldrich, catalog number: 1603271003)
36. Trifluoracetic acid (TFA) (Fisher Chemical, catalog number: 10155347)
37. Formic acid (FA) (Fisher Scientific, catalog number: 30280925)
38. Acetonitrile (ACN) (Aldrich, catalog number: 60004-2.5L)
39. Triethylamine (Thermo Scientific Alfa Aesar, catalog number: 11489046)
40. 4-(Dimethylamino)pyridine (DMAP) (Aldrich, catalog number: 8510550100)
41. Deuterated methanol (CD3OD) (Aldrich, catalog number: 444758-10X1ML)
42. Deuterated dimethyl sulfoxide (DMSO-d6) (Aldrich, catalog number: 151874-10X0.75ML)
43. Deuterated chloroform (CDCl3) (Aldrich, catalog number: 151858-10X1ML)
44. Dimethyl sulfoxide (DMSO) (Fischer Scientific, catalog number: 11401611)
45. Lipofectamine 3000 (Thermo Fisher Scientific, catalog number: L3000008)
46. Phenol red-free DMEM (Gibco, catalog number: 31053-028)
47. High glucose DMEM + GlutaMAXTM (Gibco, catalog number: 31966-021)
48. OptiMEM (Thermo Fisher Scientific, catalog number: A4124802)
49. L-glutamine (Thermo Fisher Scientific, catalog number: 25030024)
50. Sodium pyruvate (Thermo Fisher Scientific, catalog number: 11360070)
51. Fetal bovine serum (FBS) (Fisher Scientific, catalog number: 15553681)
Solutions
1. Final peptide cleavage solution (see Recipes)
2. Fmoc-deprotection solution (see Recipes)
3. ivDde-deprotection solution (see Recipes)
4. Dde-deprotection solution (see Recipes)
5. Full DMEM (-) (see Recipes)
6. Full DMEM (-), phenol red-free (see Recipes)
7. Full DMEM (+) (see Recipes)
Recipes
1. Final peptide cleavage solution
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| DCM | 93% v/v | 930 μL |
| TFA | 5% v/v | 50 μL |
| TIS | 2% v/v | 20 μL |
| Total | n/a | 1,000 μL |
Prepare freshly every time. The preparation must be conducted inside a ventilated fume hood.
2. Fmoc-deprotection solution
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| DMF | 80% v/v | 800 μL |
| Piperidine | 20% v/v | 200 μL |
| Total | n/a | 1,000 μL |
Piperidine in solution is stable for up to one month at 4 °C.
3. ivDde-deprotection solution
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| DMF | 95% v/v | 950 μL (12.3 mmol) |
| 60% Hydrazine monohydrate solution | 5% v/v | 50 μL |
| Total | n/a | 1,000 μL |
Prepare freshly every time. The preparation must be conducted inside a ventilated fume hood.
Caution: 60% Hydrazine monohydrate solution is flammable, toxic if swallowed, damages skin and eyes, potentially cancerous, and fatal if inhaled. The reaction must be conducted inside a ventilated fume hood.
4. Dde-deprotection solution
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 1-methyl-2-pyrrolidinone | 8.7 M | 1 mL (10.4 mmol) |
| Imidazole | 0.104 M | 8.51 mg (125 μmol) |
| Hydroxylamine hydrochloride | 0.125 M | 10.4 mg (150 μmol) |
| DCM | 2.61 M | 200 μL (3.13 mmol) |
| Total | n/a | 1,200 μL/0.1 mmol resin |
Can be prepared for multiple reactions and stored for 1 month at 4 °C (without DCM).
Immediately prior to the deprotection, dilute the cocktail 5/1 (v/v) with DCM (1 mL of Dde-deprotection cocktail, 0.2 mL of DCM) for better efficiency of polystyrene-type resin deprotection.
The preparation must be conducted inside a ventilated fume hood.
Caution: 1-methyl-2-pyrrolidinone is a known irritant of skin and eyes, has reproductive toxicity, and damages the lungs upon single exposure. The reaction must be conducted inside a ventilated fume hood.
5. Full DMEM (-)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| High glucose DMEM + GlutaMAXTM | n/a | 495 mL |
| Sodium pyruvate | 1 mM | 5 mL |
| Total | n/a | 500 mL |
Store at 4 °C. The preparation must be conducted under sterile conditions.
6. Full DMEM (-), phenol red-free
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Phenol red-free DMEM | n/a | 490 mL |
| L-glutamine | 2 mM | 5 mL |
| Sodium pyruvate | 1 mM | 5 mL |
| Total | n/a | 500 mL |
Store at 4 °C. The preparation must be conducted under sterile conditions.
7. Full DMEM (+)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Full DMEM (-) | n/a | 450 mL |
| Heat-inactivated sterile-filtered FBS | 10% v/v | 50 mL |
| Total | n/a | 500 mL |
Store at 4 °C. The preparation must be conducted under sterile conditions.
Laboratory supplies
1. Personal protective equipment (the person performing the experiment must wear personal protective equipment, including goggles and a lab coat)
2. Magnetic stirrer (Heidolph, catalog number: 505-30000-00-4) and magnetic stir bars, various sizes (Fisher Scientific, catalog numbers: 16390656, 16380656, 16320666)
3. Oil BM2 viscosity bath media (Fisher Scientific, catalog number: 16020172)
4. pH-indicator strips MColorpHastTM (Merck, catalog number: 1.09535.0001)
5. Thin-layer chromatography POLYGRAM® SIL G/UV254 (Macherey-Nagel, catalog number: 805021)
6. UV lamp to visualize the TLC result (Herolab, catalog number: H468.1)
7. 0.3 mL low adsorption vial for HRMS (Supelco Analytical, catalog number: 29661-U)
8. 0.1 mL vial for LCMS (Carl Roth, catalog number: C518.1) with screwcaps (Carl Roth, catalog number: E161.1) and silicone/PTFE septum (Carl Roth, catalog number: E165.1)
9. NMR tubes with coded CLOSED caps (Bruker BioSpin, catalog number: Z172600)
10. Glassware and supplies: round-bottom flasks (VWR International GmbH, catalog numbers: SCOT241701307, SCOT241703702, SCOT2417011307; sizes: 25, 250, and 500 mL); pointed flasks (DWK Life Sciences, catalog numbers: 241950809, 241951402; sizes: 5 and 25 mL); Schlenk flask (Fisher Scientific, catalog number: 15884894); Erlenmeyer flask (Fisher Scientific, catalog number: 10840861)
11. Vials for storage 20 mL (Carl Roth, catalog number: LC88.1) and screwcaps (Carl Roth, catalog number: LC96.1)
12. Büchner funnel (Fisher Scientific, catalog number: 10771752)
13. Filter paper circles (Whatman, catalog number: 10311809)
14. Septum, mixed sizes (Aldrich, catalog number: Z124400)
15. PTFE filters (FisherbrandTM, catalog number: 11705285)
16. Disposables: plastic syringes (Henke Sass Wolf, catalog numbers: 4020-000V0, 4010-200V0), cannulae (Sterican®, catalog number: 4665643)
17. Microcentrifuge tubes (Eppendorf, catalog number: 15128344)
18. Amber-colored microcentrifuge tubes (Eppendorf, catalog number: 0030121155)
19. Fritted syringe: PP reactor 20 mL with PE frit, disposable (Multisyntech GmbH, catalog number: V200PE100)
20. Fritted syringe: PP reactor 5 mL with PE frit, disposable (Multisyntech GmbH, catalog number: V050PE063)
21. 50 mL Polypropylene centrifuge tubes (CEM Corp., catalog number: 330090)
22. Syringe pressure caps (Sigma-Aldrich, catalog number: Z120979-100EA)
23. µ-Slide 18-well glass bottom dishes (Ibidi, catalog number: 81817)
Equipment
1. Ventilated fume hood for chemistry (Waldner Laboreinrichtungen BmbH & Co.KG, model: SCALA)
2. Dual-manifold Schlenk line (Merck, catalog number: SYNM181004)
3. Cooling traps (Gaßner Glastechnik GmbH, catalog number: 5.3550.70)
4. Vacuum pump (Vacuubrand®, model: RC6)
5. Heat gun (Steinel Professional, model: 3515)
6. Dewar (KGW Isotherm, catalog number: 10558672)
7. Mass spectrometry for characterization of compounds (Bruker, model: maXis IITM ETD-HRMS)
8. 1H NMR for characterization of compounds (Bruker, model: Advance III HD 400 NMR), equipped with a CryoProbeTM
9. HPLC for the purification of compounds (Thermo Scientific, model: UltiMate300 UHPLC)
10. C18 column for small molecule purification [Merck, Ascentis® C18 (5 μm) HPLC Column (25 × 212 nm), catalog number: 5811347-U]
11. C4 column for lipo-peptide purification [HiChrom, Vydac 214TP 10 μm C4 column (22 × 250 mm), catalog number: HI-214TP1022]
12. Liquid chromatography mass spectrometry (LC-MS) (Shimadzu, model: MS20-20)
13. C18 column for LCMS (Merck, TitanTM C18 80 Å 1.9 μm, 2.1 × 50 mm, catalog number: 577122-U)
14. Flash chromatography (Biotage®, model IsoleraTM One with UV-Vis Detector, part number: ISO-1EW)
15. Flash chromatography columns (Supelco®, model: SupelTM Flash Cartridge, 40 g, 40–60 μm silica, catalog number: FSISI040)
16. Rotary evaporator (Heidolph, catalog number: 571-01300-00)
17. Lyophilizer (Christ, model: Alpha 2–4 LDplus)
18. Peptide synthesizer (CEM Corp., model: Liberty BlueTM)
19. H12 resin loader (CEM Corp., catalog number: 909912)
20. 20 L stainless steel bottle with sight tube (CEM Corp., catalog number: 551255)
21. 2 L stainless steel bottle with sight tube (CEM Corp., catalog number: 551240)
22. 1 L safety coated glass bottle (CEM Corp., catalog number: 551330)
23. 500 mL safety coated glass bottle (CEM Corp., catalog number: 551325)
24. 30 mL reaction vessel (CEM Corp., catalog number: 167260)
25. Microscope (Leica, model: SP8 LIGHTNING Super-resolution live cell imaging in multicolor) equipped with white-light laser and FRAP module
26. Power meter (Thorlabs, catalog number: PM400)
27. Microscope slide power meter sensor heads (Thorlabs, catalog number: S170C)
Software and datasets
1. ImageJ (Wayne Rasband and contributors NIH, USA, version: 1.54p), free to use
2. Leica Application Suite X (LAS X) Microscopy Software (Leica, version: 3.5.7.23225), requires a license
3. Excel (Microsoft® Office, version 2108), requires a license
4. GraphPad Prism (Software MacKievTM, version: 10.5.0 (774), May 27, 2025), requires a license
5. Liberty Blue Application Software (CEM Corp., version: 1.50.5913.17379, March 10, 2016)
Procedure
A. Synthesis of dual SLIPT and dual SLIPTNVOC
Section A1 describes the multi-step synthesis to access linker-functionalized TMP with (compound 4) or without photocaging (compound 5). First, TMP is functionalized with an ethyl-protected carboxylic acid linker (Figure 3). Thereafter, the photocaging group may be appended (Figure 6), followed by the removal of the ethyl-protecting group. This results in functionalized TMP derivatives that are ready to couple to the self-localizing lipid-like anchor (compound 9).
Section A2 describes a similar functionalization of HTL (Figure 11), rendering it ready to couple to the self-localizing lipid-like anchor (compound 9). Commercially available, Boc-protected HTL is first de-protected, before appending a linker that terminates in a carboxylic acid.
Section A3 describes the synthesis of the trivalent self-localization motif (Figure 14), as well as the stepwise assembly leading to the final probes dual SLIPT and dual SLIPTNVOC (Figure 15).
Critical: Perform the chemical synthesis steps in a well-ventilated fume hood and obtain chemical safety training before attempting the steps in this protocol. Wear personal protective equipment at all times!
Pause point: The synthesis of compounds (2–7) (steps A1.2–A2.2) can be stopped after completion of the reaction but before the purification, or at any stage of the purification, by simply drying the crude product under reduced pressure (Schlenk line, rotary evaporator) and storing at 4 °C. Prior to resuming any steps of the synthesis, all starting materials and crude products that had been stored at 4 °C should be allowed to reach room temperature prior to opening the storage vials, to prevent condensation of water.
Pause point: The pure products may be stored at 4 °C post-analysis, before using them as starting materials for the next synthetic step.
Use ultrapure water (Millipore) for any aqueous solutions and wash steps. Use synthesis-grade reagents, unless otherwise noted. Prepare solutions at 20 °C. All solutions can be stored at 20 °C, unless otherwise indicated. Diligently follow all waste disposal regulations when disposing of waste.
A1. Synthesis of the linker-functionalized TMP
Caution: All steps (A1.1–5) leading to compounds (4) and (5) need to be conducted in a ventilated fume hood.
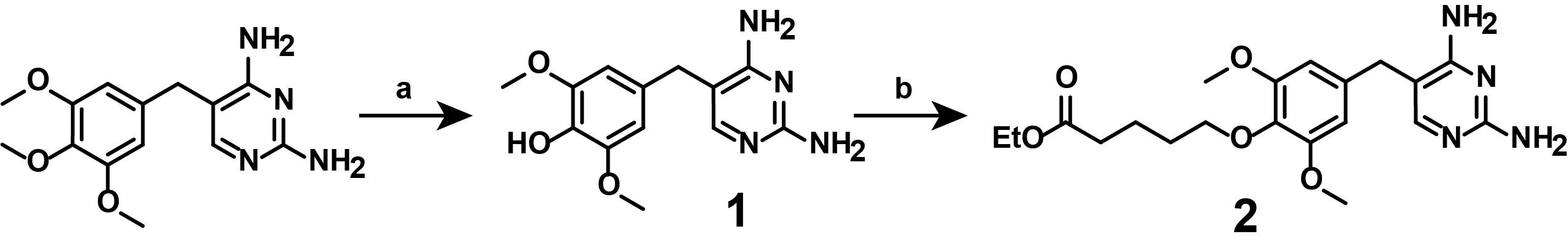
Figure 3. Multi-step overview of the synthesis of ethyl-protected, linker-functionalized TMP (2). (a) 1) 48% HBraq, 95 °C, 18 min; 2) 50% NaOH, room temperature, then 4 °C. (b) ethyl 5-bromopentanoate, Cs2CO3 in DMF, 70 °C, overnight. Synthesize linker-functionalized TMP-COOH (5) and photocaged, linker-functionalized TMP-COOHNVOC (4) manually.
1. Synthesis of compound (1)
Note: This synthetic step describes the mono-demethylation of TMP from the commercially available TMP, as displayed in Figure 4.
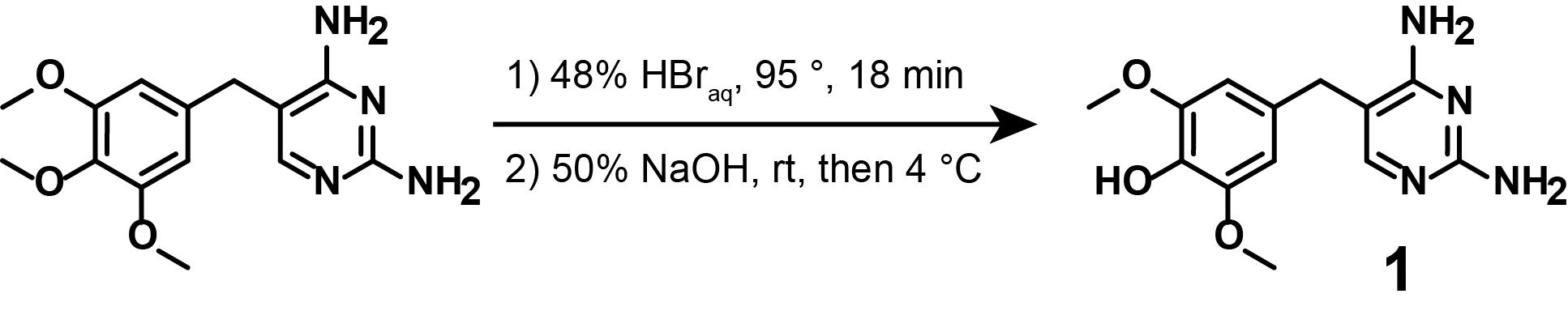
Figure 4. Demethylation of commercially available TMP (1)
a. Prepare an ice bath (ice and water mixture) with a separate magnetic stirrer and a lifting platform for later use. Place the oil bath and magnetic stirrer on a lifting platform. Secure a round-bottom flask (500 mL) using a clamp above the oil bath, such that rapid removal of the heat source by lowering the lifting platform is possible. Equip both the round-bottom flask and the oil bath with a magnetic stirring bar, and insert the thermometer into the oil bath.
b. Pre-heat the oil bath to 95 °C. Then, add the HBraq (48%, 80 mL) into the flask and raise the lifting platform such that the flask reaches the desired temperature.
c. Weigh Trimethoprim (6 g, 20.7 mmol, 1 eq.) and transfer into the flask. Start timing the reaction immediately.
d. After 18 min, remove the heat source and move the flask from the heat source to the prepared ice bath to rapidly cool the reaction mixture to 0 °C. This should require 5 min.
Critical: Do not exceed the specified timing. After demethylation of the central methoxy moiety, the starting material will undergo further demethylation reactions, leading to the doubly demethylated side product. See Troubleshooting problem 3.
e. Add NaOH (2 M, 50 mL) via a syringe over a duration of 3 min while continuously stirring.
Caution: Addition of NaOH solution can cause the reaction mixture to splash up; close the fume hood as far as possible while adding the base dropwise. Place the reaction mixture at 4 °C overnight to allow for precipitation.
f. Filter the precipitate into an Erlenmeyer flask (250 mL) using a Büchner funnel, equipped with filter paper, and wash the precipitate with ice-cold H2O. Transfer the solid residue back into the round-bottom flask.
g. Recrystallize the precipitate in water by heating H2O to a boil and adding successively to the filtered precipitate until fully dissolved.
h. Adjust the pH again (~ pH 7.0) using NaOH solution (2 M), let cool down to room temperature (RT) gradually, then place at 4 °C overnight.
i. Filter the resulting precipitate as before (step A1.1f), wash with ice-cold H2O, and dry under high vacuum.
j. Final product: White crystals, approximately 50% yield.
k. Analytical data: 1H NMR (400 MHz, d-DMSO): δ 7.37 (s, 1H), 6.79 (brs, 2H), 6.49 (s, 1H), 6.30 (brs, 2H), 5.68 (s, 2H), 2.86 (s, 6H), 2.67 (s, 2H). brs = broad singlet. HRMS (ESI): calculated for [M + H] +, 277.1295; found, 277.1298.
2. Synthesis of compound (2)
Note: This synthetic step describes the linker functionalization of mono-demethylated TMP (1), as displayed in Figure 5.
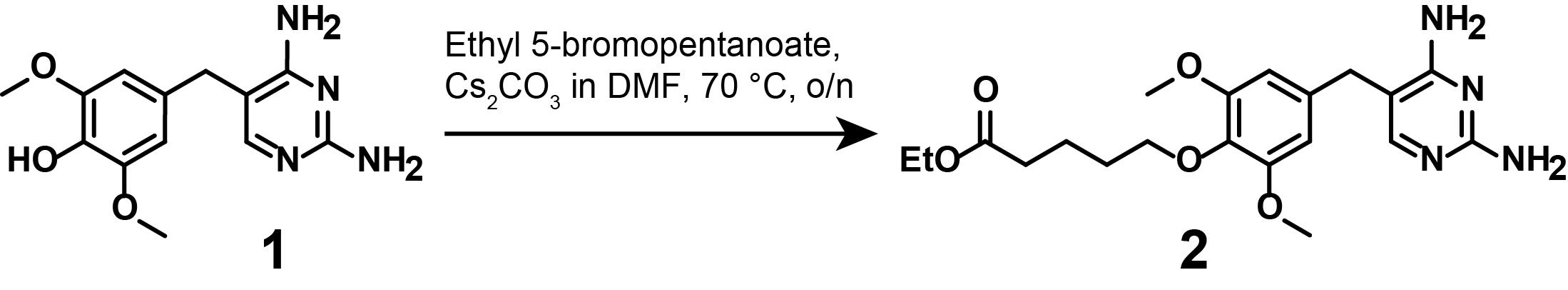
Figure 5. Synthesis of ethyl-protected, linker-functionalized TMP (2)
General notes on Schlenk technique:
The Schlenk technique is a set of glassware and workflow practices for conducting air- or moisture-sensitive reactions under inert gas such as nitrogen or argon. It enables the reaction of reagents without exposure to air and its moisture. Upon assembly of the Schlenk flask, the dual-manifold Schlenk line is attached to the apparatus. In a typical workflow, the apparatus is purged of air via the vacuum pump with all other ports closed. After 5 min, inert gas is added into the apparatus. This workflow is repeated twice more to ensure full removal of any traces of air or moisture. Optionally, a heat gun may be used to evenly heat and thus further dry the Schlenk flask under high vacuum (flame-drying). Addition of solid substances occurs under light reverse flow of inert gas, followed by one more evacuation and inert gas refill cycle. This should be done carefully to avoid disturbing the substance. Addition of liquid substances should be done via syringe and cannula by piercing the septum.
This reaction may be conducted under a regular atmosphere. Use of an inert gas atmosphere is, however, advisable and was reported in the original manuscript [2].
a. Assemble and turn on the dual-manifold Schlenk line with a cold trap between the vacuum manifold and pump. Place the oil bath and magnetic stirrer on a lifting platform. Secure a 25 mL round-bottom flask with a Schlenk adaptor (Schlenk flask) using a clamp. Equip both the Schlenk flask and the oil bath with a magnetic stirring bar and insert the thermometer into the oil bath. Equip the Schlenk flask with a stopcock and a septum. Conduct three cycles of evacuation and inert gas. Use a heat gun to evenly heat the Schlenk flask when under vacuum.
b. Upon cooling down of the Schlenk flask, add compound (1) (1.00 g, 3.62 mmol, 1 eq.) under light reverse flow of inert gas, then evacuate carefully once more, before adding inert gas.
Caution: Evacuate carefully to prevent dispersion of compound (1).
c. Add anhydrous DMF (15 mL) via cannula and heat the Schlenk flask to 70 °C.
d. Upon reaching temperature, add Cs2CO3 (2.36 g, 7.24 mmol, 2.00 eq.) under light reverse flow of inert gas.
e. Heat the mixture to 70 °C before adding ethyl-5-bromovalerate (0.86 mL, 5.43 mmol, 1.50 eq.) by cannula and stir the reaction mixture at 70 °C overnight.
f. Filter the reaction mixture using a Büchner funnel, equipped with filter paper, to remove the excess Cs2CO3, transfer the collected filtrate back into the round-bottom flask, and concentrate the crude product.
g. Purify the product by silica gel chromatography using a gradient of 5%–10% MeOH in DCM.
h. Final product: Brown solid, approximately 70% yield. See Troubleshooting problem 4.
i. Analytical data: 1H NMR (400 MHz, CD3OD): δ 7.52 (s, 1H), 6.53 (s, 2H), 4.14 (q, J = 7.1 Hz, 2H), 3.92 (t, J = 6.0 Hz, 2H), 3.80 (s, 6H), 3.66 (s, 2H), 2.41 (t, J = 7.4 Hz, 3H), 1.88–1.78 (m, 2H), 1.78–1.66 (m, 2H), 1.26 (t, J = 7.1 Hz, 3H). HRMS (ESI): calculated for [M + H]+, 405.2132; found, 405.2131.
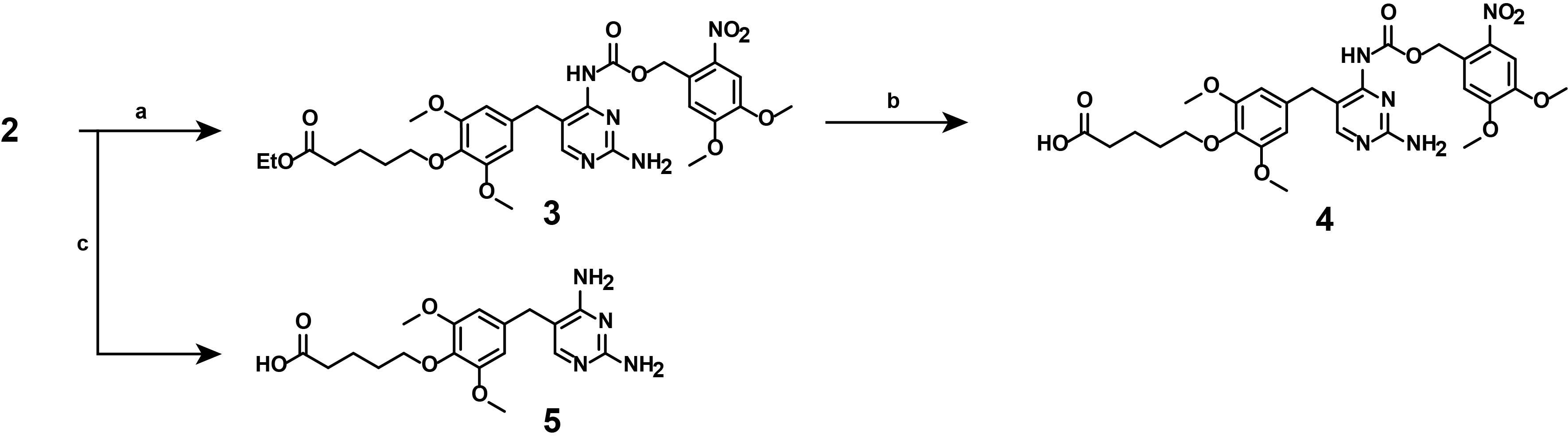
Figure 6. Multi-step overview of the synthesis of linker-functionalized, photocaged TMP (4) and non-photocaged TMP (5). (a) (Nitroveratryl)oxy)chlorocarbamate, DIPEA, DCM, room temperature, 16 h, exclusion of light. (b) 1 M LiOH in THF/H2O, room temperature, 4 h, exclusion of light. (c) 1) 2 M NaOH in MeOH, 45 °C, 1 h; 2) 1 M HCl in H2O.
3. Synthesis of compound (3)
Note: This synthetic step describes the appending of the photocaging moiety onto linker-functionalized TMP (2), as displayed in Figure 7.
Figure 7. Photocaging of ethyl-protected, linker-functionalized TMP (2)
a. Place a magnetic stirrer onto a lifting platform. Secure the round-bottom flask with a clamp. Equip a 25 mL round-bottom flask with a magnetic stirring bar. Add compound (2) (500 mg, 1.24 mmol, 1.00eq.) into the round-bottom flask, then add anhydrous DCM (5 mL), as well as DIPEA (204 μL, 1.24 mmol, 1 eq.), using a syringe.
b. Stir the reaction mixture for 5 min at RT, then darken the room (turn off lights; if possible, close blinds), weigh (nitroveratryl)oxy)chlorocarbamate (342 mg, 1.24 mmol, 1 eq.), and add to the reaction mixture. Cover the reaction flask with aluminum foil to minimize ambient light exposure and stir the reaction overnight at RT.
c. Dilute the reaction mixture with DCM (50 mL) and evaporate the solvent under reduced pressure on a rotary evaporator.
d. Purify the product by silica gel chromatography using a gradient of 0%–10% MeOH in DCM.
e. Final product: Pale-yellow solid, approximately 25% yield. See Troubleshooting problem 5.
f. Analytical data: 1H NMR (400 MHz, CD3OD): δ 8.00 (s, 1H), 7.76 (s, 1H), 7.14 (s, 1H), 6.46 (s, 2H), 5.53 (s, 2H), 4.13 (q, J = 7.1 Hz, 2H), 3.93 (s, 3H), 3.89 (s, 3H), 3.86 (t, J = 4.8 Hz, 2H), 3.83 (s, 2H), 3.73 (s, 6H), 2.40 (t, J = 7.4 Hz, 2H), 1.89–1.76 (m, 2H), 1.76–1.65 (m, 2H), 1.26 (t, J = 7.1 Hz, 3H). HRMS (ESI): calculated for [M + H]+, 644.2562; found, 644.2566.
Critical: The nitroveratryl-protection group can attach to either of the exocyclic amines, resulting in two different regioisomer products. Under the purification conditions described, the desirable regioisomer is recovered from the silica column last. Identification of the two regioisomers can be conducted via 1H, 1H NOESY correlation between the carbamate and the CH2 bridging group (as demonstrated in [14]). Alternatively, the 1H NMR peak pattern between 7.0 and 8.5 ppm may be used to identify the desirable regioisomer (Figure 8).
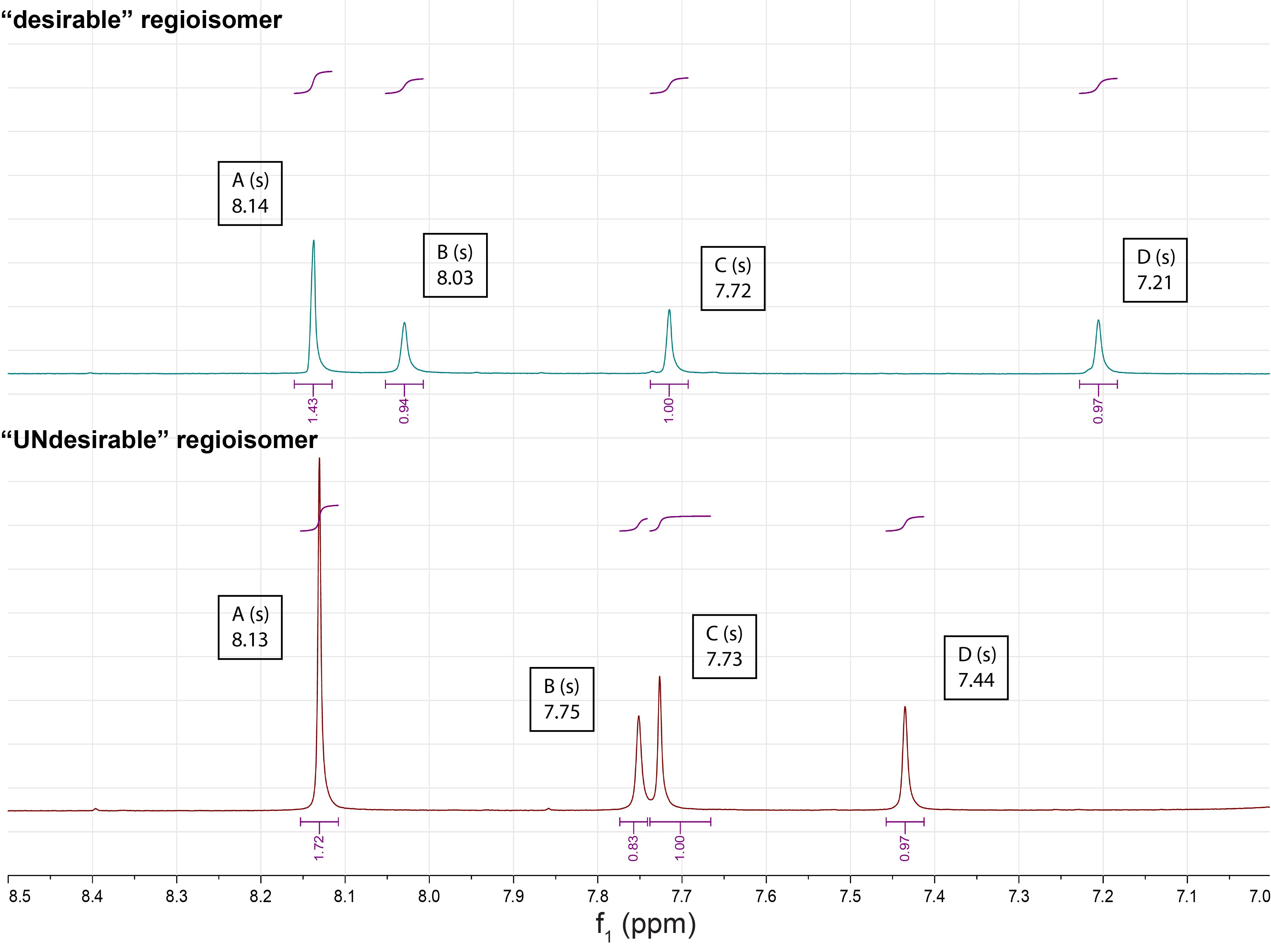
Figure 8. 1H NMR peak pattern of the two regioisomers (desirable, top; undesirable, bottom) as measured in DMSO.
The characteristic shifts of peaks B, C, and D can help identify the desirable regioisomer.
4. Synthesis of compound (4)
Note: This synthetic step describes the saponification of the photocaged, linker-functionalized TMPNVOC (3), as displayed in Figure 9.
Figure 9. Saponification of (3) to lead to linker-functionalized, photocaged TMP (4)
a. Prepare a 1 M LiOH solution. To do so, add 24 mg of LiOH into 1 mL of water and stir until the solution becomes clear.
b. Place a magnetic stirrer underneath a cork ring. Place a 25 mL round-bottom flask into the cork ring and secure it with a clamp. Equip the flask with a magnetic stirring bar. In the 25 mL round-bottom flask, dissolve compound (3) (70 mg, 109 μmol, 1 eq.) in THF (1.4 mL) and H2O (1.4 mL). Cover the flask with aluminum foil to minimize ambient light exposure.
c. Add 1 M LiOH solution (280 μL) and stir the reaction at RT for 4 h.
d. Dilute the reaction with H2O (15 mL) and adjust to pH 3 by the addition of 1 M HCl.
e. Darken the room.
f. Equip the separatory funnel with a stopcock and hang it in the fume hood. To extract the crude product, close the stopcock, add 30 mL of ethyl acetate first, then add the diluted reaction mixture to the hanging separatory funnel. Close the separatory funnel with an appropriate plug and shake vigorously, securing the plug by hand.
Caution: Mixing the organic and aqueous phases may generate overpressure in the separatory funnel. To relieve it, invert the funnel to allow air to collect near the stopcock, then slowly vent away from your face and body.
g. Close the stopcock once more, turn the separatory funnel right side up, and hang it by clamp. Allow approximately 5 min for the organic and aqueous phase to separate.
Critical: The upper (pale yellow) phase contains the crude product; the lower phase contains the residual reactants.
h. Cover the beaker with aluminum foil.
i. Place an Erlenmeyer flask (50 mL) underneath the separatory funnel. Carefully separate the aqueous phase into the Erlenmeyer flask, then close the stopcock, exchange the capturing vessel for the 250 mL beaker, and collect the organic phase.
j. Pour the captured aqueous phase into the separatory funnel once more and add 30 mL of fresh ethyl acetate. Repeat the extraction process two more times, combining the organic phases.
k. Dry the combined organic layers over anhydrous MgSO4 until no further clumping is observed upon addition.
l. Filter the organic layer into a 250 mL round-bottom flask using a glass funnel and filter paper.
m. Rinse the filter paper with ethyl acetate.
n. Evaporate the solvent under reduced pressure on a rotary evaporator.
o. Resuspend the crude product in minimal DMSO, filter through a 0.45 μm PTFE filter, and purify it via reverse-phase C18 HPLC, using a linear gradient of 20%–70% acetonitrile in H2O (0.1% TFA).
p. Final product: Pale yellow solid, approximately 70% yield. See Troubleshooting problem 6.
q. Analytical data: 1H NMR (400 MHz, CD3OD): δ 8.00 (s, 1H), 7.76 (s, 1H), 7.14 (s, 1H), 6.46 (s, 2H), 5.53 (s, 2H), 4.13 (q, J = 7.1 Hz, 2H), 3.93 (s, 3H), 3.89 (s, 3H), 3.86 (t, J = 4.8 Hz, 2H), 3.83 (s, 2H), 3.73 (s, 6H), 2.40 (t, J = 7.4 Hz, 2H), 1.89–1.76 (m, 2H), 1.76–1.65 (m, 2H), 1.26 (t, J = 7.1 Hz, 3H). HRMS (ESI): calculated for [M + H]+, 616.2249; found, 616.2257.
5. Synthesis of compound (5)
Note: This synthetic step describes the saponification of the constitutively active, linker-functionalized TMP (2), as displayed in Figure 10.
Figure 10. Saponification of (2) to lead to linker-functionalized, non-photocaged TMP (5)
a. Place a magnetic stirrer underneath a cork ring. Place a 25 mL round-bottom flask into the cork ring and secure it with a clamp. Equip the flask with a magnetic stirring bar. Weigh compound (2) (200 mg, 0.45 mmol, 1 eq.) and add to the round-bottom flask; then, add MeOH (2 mL) and 2 M NaOH aq. (50 μL, 1.35 mmol, 3.00 eq.).
b. Stir the reaction mixture at RT for 1 h before evaporating the solvent under reduced pressure on a rotary evaporator.
c. Redissolve the product in H2O (10 mL), then bring to pH 4 with 1 M HCl aq. to precipitate the product.
d. Filter the precipitate using a Büchner funnel and wash the precipitate with ice-cold H2O.
e. Final product: White solid, approximately 30% yield.
f. Analytical data: 1H NMR (400 MHz, CD3OD): δ 7.32 (s, 1H), 6.52 (s, 2H), 3.94 (t, J = 5.9 Hz, 2H), 3.79 (s, 6H), 3.64 (s, 2H), 2.27 (t, J = 7.1 Hz, 2H), 1.80–1.66 (m, 4H). HRMS (ESI): calculated for [M + H]+, 377.1819; found, 377.1823.
A2. Synthesis of the linker-functionalized HTL

Figure 11. Multi-step overview of the synthesis of linker-functionalized HTL (7). (a) 50% (v/v) TFA in DCM, room temperature, 1 h. (b) Glutaric anhydride, Triethylamine, DMAP in DCM, room temperature, 1 h.
1. Synthesis of compound (6)
Note: This synthetic step describes the deprotection of the commercially available Boc-protected HTL, as displayed in Figure 12.

Figure 12. Deprotection of commercially available Boc-protected HTL
a. Place a magnetic stirrer underneath a cork ring. Place a 25 mL pointed flask into the cork ring and secure it with a clamp. Equip the flask with a magnetic stirring bar. Add DCM (2 mL) into the pointed flask, weigh Boc-protected HTL (100 mg, 309 μmol, 1 eq.), and add it to the solvent.
Critical: Because Boc-protected HTL is highly viscous, it should be added directly into the reaction flask after the appropriate volume of solvent has been introduced, to facilitate its transfer and ensure complete dissolution.
b. Add TFA (1.032 mL, 14 mmol, 45 eq.) by syringe and allow to stir at RT for 1 h.
Caution: TFA produces toxic gases when in contact with moist air. Open and handle TFA only in the ventilated fume hood.
c. Dilute the crude product with H2O (2 mL).
d. Extract the product by addition of ethyl acetate (4 mL) to the aqueous solution, with vigorous stirring for 2 min, before phase separation and collection of the organic (upper) fraction by Pasteur pipette.
e. Repeat the extraction twice more. Combine the collected organic fractions into a 25 mL round-bottom flask.
f. Evaporate the solvent under reduced pressure on a rotary evaporator.
g. Final product: Pale yellow oil, quantitative yield.
h. Analytical data: 1H NMR (400 MHz, DMSO-d6): δ 7.65 (s, 4H), 3.63 (t, J = 6.6 Hz, 2H), 3.60–3.54 (m, 4H), 3.53–3.48 (m, 2H), 3.38 (t, J = 6.6 Hz, 2H), 2.96 (t, J = 5.3 Hz, 2H), 1.76–1.65 (m, 2H), 1.49 (q, J = 7.0 Hz, 2H), 1.45–1.26 (m, 4H). HRMS (ESI): calculated for [M + H] +, 224.1412; found, 224.1406.
2. Synthesis of compound (7)
Note: This synthetic step describes the linker functionalization of deprotected HTL-amine (6), as displayed in Figure 13.
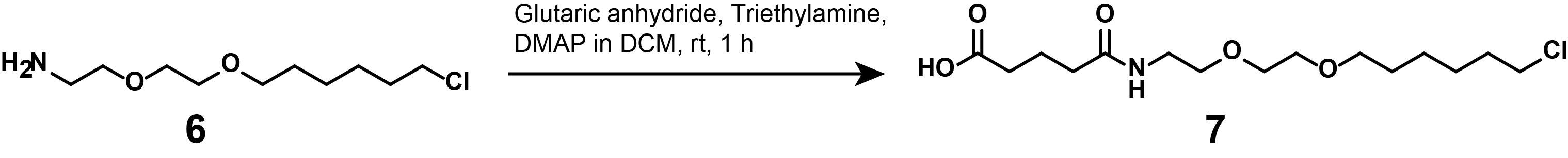
Figure 13. Synthesis of linker-functionalized HTL (7)
a. Place a magnetic stirrer underneath a cork ring. Place a 25 mL round-bottom flask into the cork ring and secure it with a clamp. Equip the flask with a magnetic stirring bar. Add anhydrous DCM (2.5 mL) into the round-bottom flask, weigh, and add compound (6) (50 mg, 223 μmol, 1 eq.).
b. Add triethylamine (155 μL, 1.12 mmol, 5 eq.) into the reaction flask and stir for 5 min at RT.
Caution: Do not breathe in glutaric anhydride.
c. Add glutaric anhydride (25.5 mg, 223 μmol, 1 eq.) and 4-dimethylaminopyridine (24.6 mg, 201 μmol, 0.9 eq.) and stir the reaction for 1 h, before adding 1 N HCl (18 mL) to neutralize.
d. Extract the product via ethyl acetate in a separatory funnel. Equip the separatory funnel with a stopcock and hang it into the fume hood. Close the stopcock, add 30 mL of ethyl acetate first, and then add the diluted reaction mixture to the hanging separatory funnel. Close the separatory funnel with an appropriate plug and shake vigorously, securing the plug by hand.
Caution: Mixing the organic and aqueous phases may generate overpressure in the separatory funnel. To relieve it, invert the funnel to allow air to collect near the stopcock, then slowly vent away from your face and body.
e. Close the stopcock once more, turn the separatory funnel right side up, and hang it by clamp. Allow approximately 5 min for the organic and aqueous phase to separate.
Critical: The upper (pale yellow) phase contains the crude product; the lower phase contains the residual reactants.
f. Place an Erlenmeyer flask (50 mL) underneath the separatory funnel. Carefully separate the aqueous phase into the Erlenmeyer flask, then close the stopcock, exchange the capturing vessel for a 250 mL round-bottom flask, and collect the organic phase.
g. Pour the captured aqueous phase into the separatory funnel once more and add 30 mL of fresh ethyl acetate. Repeat the extraction process two more times, combining the organic phases.
h. Evaporate the solvent under reduced pressure on a rotary evaporator.
i. Final product: Orange oil, quantitative yield.
j. Analytical data: 1H NMR (400 MHz, CDCl3): δ 6.19 (s, 1H), 3.61 (s, 4H), 3.58 – 3.44 (m, 8H), 2.45 (dt, J = 9.2, 6.8 Hz, 4H), 2.33 (t, J = 7.0 Hz, 2H), 2.03–1.93 (m, 2H), 1.84–1.72 (m, 2H), 1.64 (dt, J = 14.2, 6.9 Hz, 2H), 1.51–1.42 (m, 2H), 1.42–1.33 (m, 2H). HRMS (ESI): calculated for [M + H]+, 338.1729; found, 338.1723.
A3. Synthesis of the trivalent self-localization motif
The synthesis of the localization motif up to compound (8) can be done automatically by a peptide synthesizer (Liberty BlueTM, CEM Corporation) or manually (see, for example, [15]). The following describes the synthesis on a 0.1 mmol scale as conducted on a peptide synthesizer.
1. Synthesis of compound (8)
Note: These synthetic steps describe the assembly of the lipid-like localization motif intermediate (8), as displayed in Figure 14, via an automated peptide synthesizer using the commercially available amino acid building blocks (Reagents no. 3–8) and the commercially available resin (Reagents no. 9).
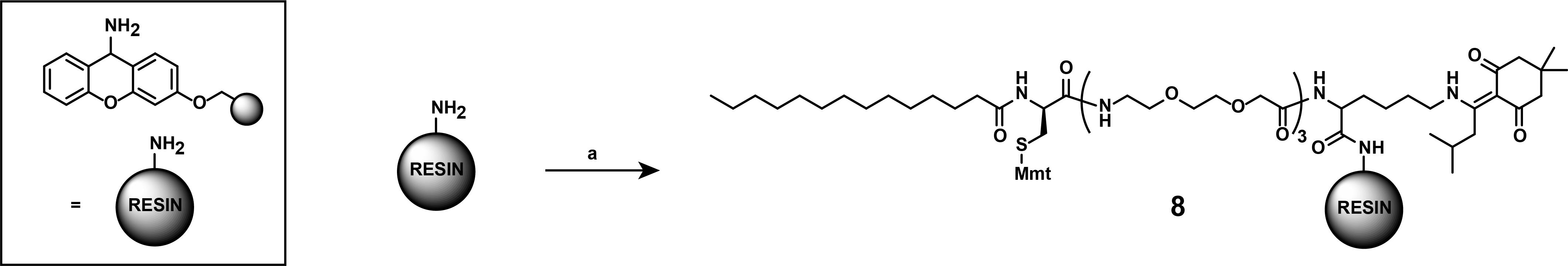
Figure 14. Synthesis of the self-localization motif (8). (a) All steps were conducted using a Liberty BlueTM peptide synthesizer (CEM Corp.). Fmoc-deprotections 20% piperidine in DMF. Coupling conditions: mixture of N-terminally Fmoc-protected amino acid, or myristic acid (0.2 M), Oxyma (base, 1 M), DIC (1 M) in DMF. Building blocks (used in this order): Fmoc-Lys(ivDde)-OH, Fmoc-8-amino-3,6-dioxaoctanoid-acid, Fmoc-D-Cys(Mmt)-OH, and Myristic acid. All couplings of amino acids were in single coupling steps and followed by deprotection. Coupling of Fmoc-D-Cys(Mmt)-OH was conducted at 50 °C. Myristic acid was coupled twice to ensure full conversion.
Notes:
1. Use peptide synthesis-grade DMF as the solvent for all solutions. DMF should not be used if older than six months. Undissolved amino acids should be stored at -20 °C for no longer than 1 year. Amino acids or Oxyma in solution are stable for two weeks. DIC and Piperidine in solution are stable for up to one month.
2. Compound (8) is generated from Carboxy- to “Amino”-terminus and will be cleaved off the resin under the generation of an amide.
3. Coupling settings:
Deprotection: 10 mL of 20% piperidine, 100 s, 90 °C
Wash: 4 × 10 mL of DMF
Coupling: 1 M DIC, 0.5 M Oxyma, and 0.2 M protected amino acid, 265 s, 90 °C
Caution: Coupling of Fmoc-D-Cys(Mmt)-OH should be conducted at 50 °C to prevent racemisation.
Caution: Double coupling, instead of single coupling, is recommended for the hydrophobic building block myristic acid. A final deprotection post-coupling of all building blocks is superfluous.
a. Weigh the reagents listed in Table 1 into the indicated vessels.
Note: The peptide synthesizer automates the calculation of reagent usage. Add an excess of approximately 1 mL of solvent to the calculated usage of amino acids to prevent the machine from running dry. Adjust the quantity of building blocks (amino acids and myristic acids) as indicated below to maintain a final concentration of 0.2 M.
b. Shake until fully dissolved.
Note: Weigh the resin last and pre-swell it in 1 mL of DMF for 5 min at RT before starting the reaction. The resin is not pre-loaded with an amino acid but is Fmoc-protected. Thus, initial deprotection is followed by washing and coupling of the first amino acid.
c. Assemble the machine (amino acids in solution, 50 mL centrifuge tubes; activator and activator base in solution, safety coated glass bottles; deprotection reagent (piperidine) in solution, stainless steel bottle; resin in solution, 50 mL centrifuge tube; transfer solvent, safety coated glass bottle; main wash, stainless steel bottle), ensuring a tight seal on all bottles and tubes.
Table 1. Reagents used for solid-phase peptide synthesis
| Compound | Quantity | Concentration | Solvent volume (DMF) | Vessel |
|---|---|---|---|---|
| Main wash (DMF) | N/A | N/A | 1 L | 20 L stainless steel bottle with sight tube (for main wash) |
| Transfer solvent (DCM 50%) | 100 mL | 50% v/v | 100 mL | 1 L safety coated glass bottle (for main wash) |
| Fmoc-Lys(ivDde)-OH | 575 mg | 0.2 M | 5 mL | 50 mL centrifuge tube |
| Fmoc-8-amino-3,6-dioxaoctanoic acid | 848 mg | 0.2 M | 11 mL | 50 mL centrifuge tube |
| Fmoc-D-Cys(Mmt)-OH | 616 mg | 0.2 M | 5 mL | 50 mL centrifuge tube |
| Myristic acid | 365 mg | 0.2 M | 8 mL | 50 mL centrifuge tube |
| Piperidine | 12 mL | 20% v/v | 48 mL | 2 L stainless steel bottle with sight tube (for deprotection) |
| DIC | 3.1 mL | 1 M | 16.9 mL | 1 L safety coated glass bottle (for activator/activator base) |
| Oxyma | 5.86 g | 1 M | 20 mL | 500 mL safety coated glass bottle (for activator/activator base) |
| Sieber amide resin | 133 mg | N/A | 10 mL | 50 mL centrifuge tube |
Note: Post synthesis, the peptide-decorated resin is flushed into the resin loader in a suspension of DMF and DCM.
d. Weigh the fritted PP reactor. To do so, gently agitate the resin and transfer to the fritted PP reactor. Allow the solvent to drain, and then flush the remaining resin into the PP reactor using DCM.
e. Rinse the resin thoroughly with DMF, then DCM.
f. Monitor the purity of the reaction by removing up to 4 resin beads from the crude product and transferring to a 1.5 mL microcentrifuge tube.
g. Prepare the final peptide cleavage solution.
h. Add 10 μL of freshly prepared final peptide cleavage solution to the microcentrifuge tube and wait for 5 min for the peptide to be cleaved off the resin.
Note: This occurs with a vibrant yellow coloring of the reaction mixture that abates over time.
i. Dilute 1 μL of peptide dissolved in the final peptide cleavage solution in 100 μL of ACN and measure the crude product’s mass via LC-MS.
Note: LC-MS is used to follow the consumption of starting materials or to ascertain the completion of a reaction. To monitor the reaction via LC-MS, dilute an aliquot of the crude product in ACN (1:100). Transfer the sample into an LC-MS vial and cap and insert it into the LC-MS machine. A typical method separates the sample via a gradient of 10%–90% ACN [with 0.1% (v/v) FA] in water [with 0.1% (v/v) FA] over 6 min with a flow rate of 0.2 mL/min. If the detection mode selected is ESI+, one can search the resulting chromatogram for the mass-to-charge ratio (m/z) of the mono- or double-protonated species.
Pause point: Compound (8) and all following resin-coupled intermediates can be stored by washing thoroughly with DMF, then DCM, and drying the functionalized resin under high vacuum inside the fritted reactor without the pressure cap. This can be conducted either with the Schlenk line, with the PP reactor placed in a round-bottom flask, or in a desiccator. Thereafter, the resin is easy to handle for aliquotation, as it is less sticky, and can be stored at 4 °C. Post drying, determine the mass of the peptide-decorated resin by weighing the peptide-containing fritted PP reactor for the purpose of aliquotation.
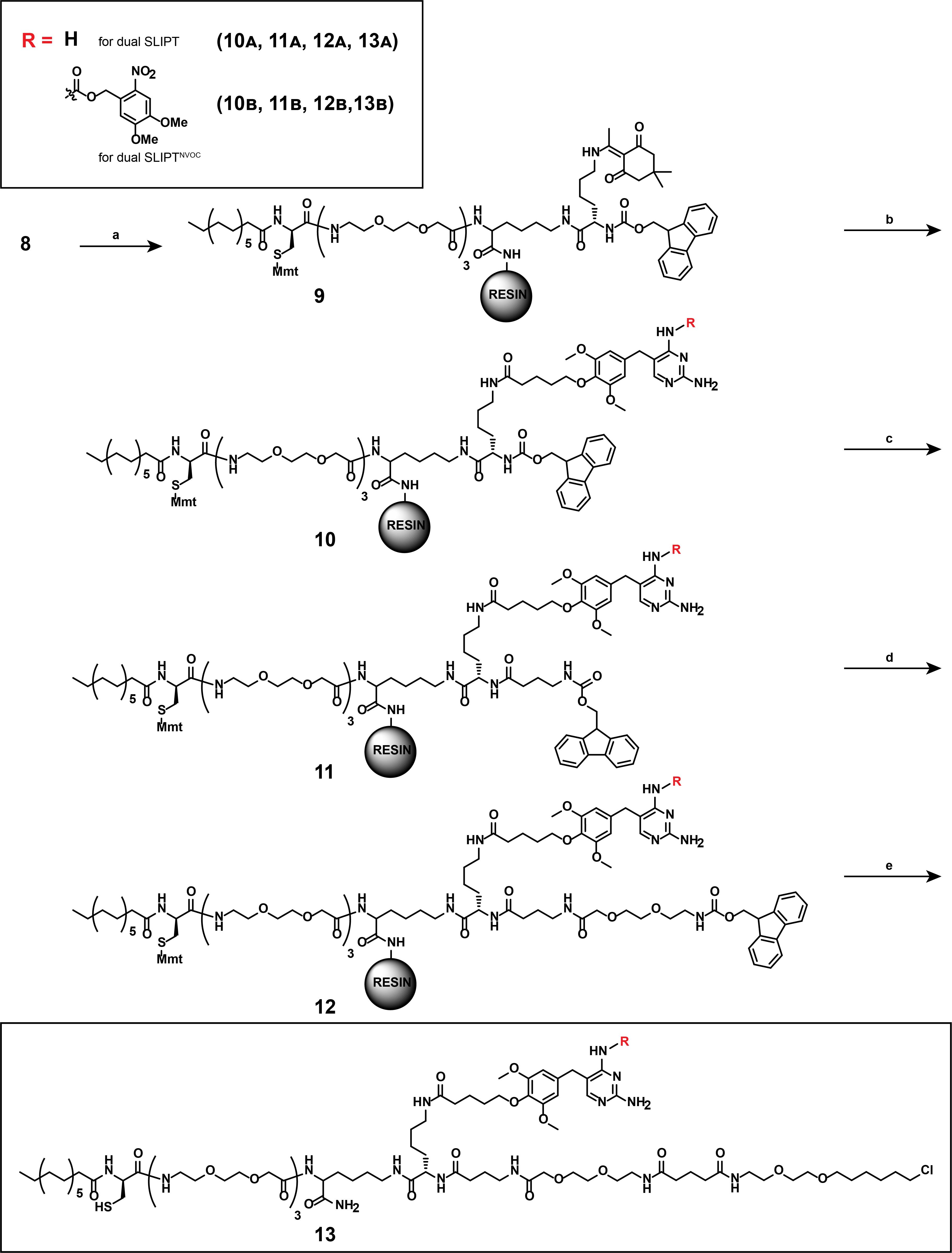
Figure 15. Multi-step overview of the synthesis of dual SLIPT (13A) and dual SLIPTNVOC (13B). (a) 1) 5% Hydrazine in DMF, room temperature (RT), 1 h. 2) Fmoc-Lys(Dde)-OH, DIPEA, PyBOP in DCM/DMF, RT, 2 h. (b) 1) Hydroxylamine hydrochloride, imidazole, 1-methyl-2-pyrrolidone, RT, 5 h, 60%; 2) for (13A): TMP-COOH (5), DIPEA, PyBOP in DCM/DMF, RT, 2 h. For (13B): TMPNVOC-COOH (4), DIPEA, PyBOP in DCM/DMF, RT, 2 h. (c) 1) 20% piperidine in DMF, RT, 20 min; 2) Fmoc-4-aminobutyric acid, HATU in DCM/DMF, RT, 2 h. (d) 1) 20% piperidine in DMF, RT, 20 min; 2) Fmoc-8-amino-3,6-dioxaoctanoic acid, DIPEA, HATU in DCM/DMF, RT, 2 h. (e) 1) 20% piperidine in DMF, RT, 20 min; 2) HTL-COOH (7), DIPEA, PyBOP in DCM/DMF, RT, 2 h; 3) 5% (v/v) TFA, 2% (v/v) TIS, in DMF, RT, 15 min.
Notes:
1. Conduct the synthesis of compounds 9, 10A/B, 11A/B, 12A/B, and 13A/B manually on the Sieber amide resin.
2. Prior to chemical modification of the peptide, the resin needs to be pre-swelled in DMF/DCM (1:1) for at least 5 min. Before conducting the reaction, the resin should be gently drained via the plunger without crushing the resin. Reactants may be added via pipetting to the PP reactor when closed with a pressure cap and the plunger removed.
3. The resin must be thoroughly washed with DMF (10 times the reaction volume) after every reaction, before draining the PP reactor.
Critical: For couplings to succeed, the carboxylic acid-containing compound must be weighed first and added to a separate 5 mL pointed flask. To this, add solvent (1 mL/0.02 mmol peptide), containing DIPEA (4 eq.), before addition of HATU, or PyBOP (1.8 eq.) as indicated. This reaction must stir at RT for at least 5 min until a visible color change occurs, indicating the activation of the deprotected carboxylic acid. Only then may the reagents be added to the pre-swelled peptide-decorated resin.
2. Synthesis of compound (9)
Note: This synthetic step describes the ivDde-deprotection of the localizing motif (8), before describing the appending of the branching moiety [Fmoc-Lysine(Dde)-OH], as displayed in Figure 16. These steps are part of both the synthesis of dual SLIPT and dual SLIPTNVOC.
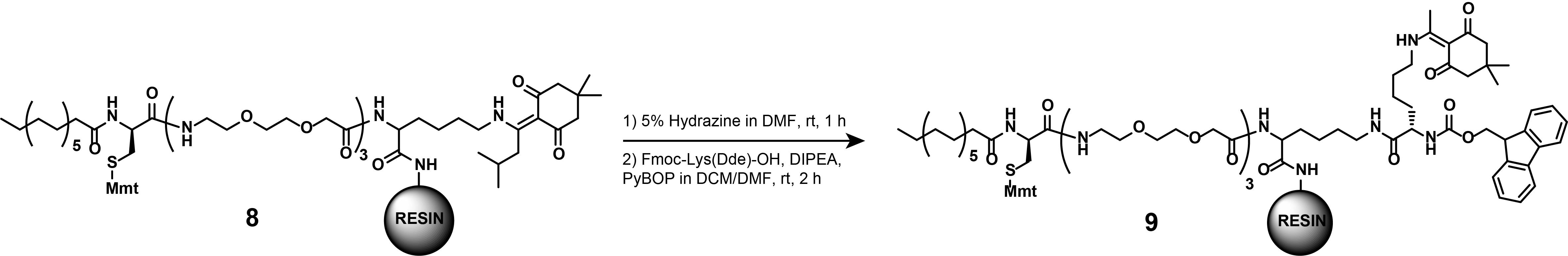
Figure 16. Synthesis of compound (9)
a. Prepare the ivDde-deprotection solution.
Caution: 60% Hydrazine monohydrate solution is explosive and toxic. The preparation of this solution must be conducted in a ventilated chemical fume hood. Avoid contact with the skin and eyes of both the 60% Hydrazine monohydrate solution and the ivDde-deprotection solution under all circumstances. Avoid breathing in the fumes of 60% Hydrazine monohydrate solution or those from the ivDde-deprotection solution.
b. Remove the plunger from the PP reactor and close the PP reactor with the pressure cap.
c. Add the ivDde-deprotection solution (2 mL) to compound (8) to cleave off the ivDde-protection group from the ε-amine.
d. Seal the PP reactor with parafilm and gently agitate at RT for 1 h.
e. Drain and wash the deprotected resin thoroughly with DMF.
f. Place a magnetic stirrer underneath a cork ring. Place a 5 mL pointed flask into the cork ring and secure it with a clamp. Equip the flask with a magnetic stirring bar. To couple Fmoc-Lys(Dde)-OH to the deprotected peptide (8), weigh Fmoc-Lys(Dde)-OH (106.5 mg, 0.2 mmol, 2 eq.) to the pointed flask.
g. Add 50% (v/v) DCM in DMF solution (2 mL) to dissolve Fmoc-Lys(Dde)-OH. Add DIPEA (70 μL, 0.4 mmol, 4 eq.) by pipette. Weigh PyBOP (68.4 mg, 180 μmol, 1.8 eq.) and add to the reaction mixture.
h. Stir vigorously at RT for 5 min or until a change in color from yellow to orange is observed.
i. Remove the plunger from the PP reactor and close the PP reactor with a pressure cap.
j. Transfer the activated amino acid to the pre-swelled resin (8) by pipetting. Seal the PP reactor with parafilm and gently agitate at RT for 2 h.
k. Drain and wash the resin thoroughly with DMF.
l. Monitor the efficiency of the reaction as described above by separating a fraction of the resin and cleaving the crude off the resin, and analyze via LC-MS.
3. Synthesis of compound (10A)
Notes:
1. These synthetic steps describe the Dde-deprotection of (9) followed by the appending of the linker-functionalized TMP (5) to the localization motif (9), as displayed in Figure 17. This step is part of the synthesis of dual SLIPT. To append the TMP- and HTL-headgroups, a sequential deprotection of the crude product (9) must occur.
2. The Dde-protection group is quasi-orthogonal to the Fmoc-protection group and can be deprotected selectively with acceptable yields (60%) while maintaining the N-α-Fmoc protection.
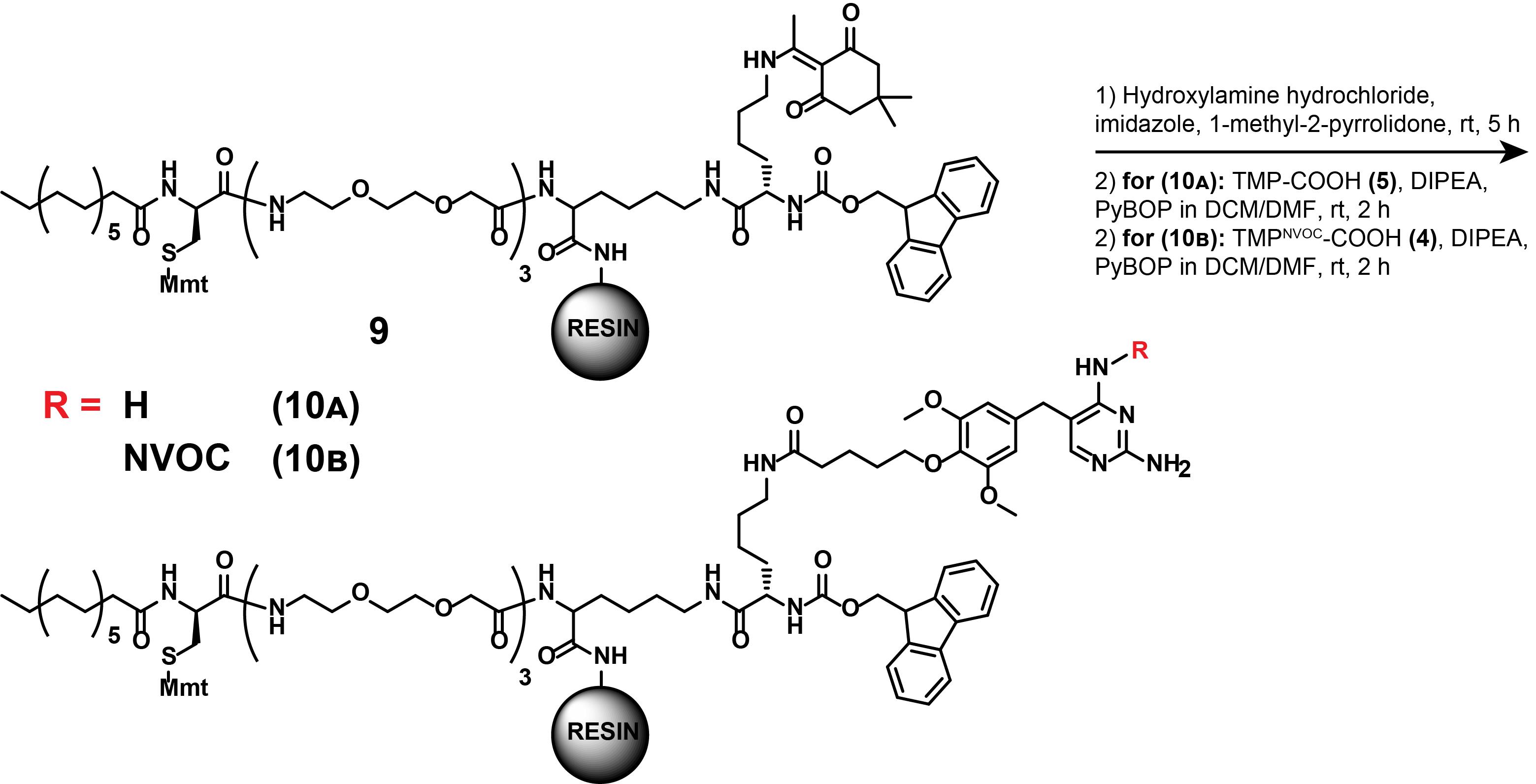
Figure 17. Synthesis of compounds (10A) and (10B)
a. Remove the plunger from the PP reactor and close the PP reactor with a pressure cap.
b. Prepare the Dde-deprotection solution (1,200 μL) and transfer it to (9)-coupled, pre-swelled resin by pipetting. Seal the PP reactor with parafilm and gently agitate at RT for 5 h.
Caution: The preparation of the Dde-deprotection solution must be conducted in a ventilated fume hood. 1-Methyl-2-pyrrolidinone is a known irritant (skin, eye, respiratory tract) and has reproductive toxicity. Change gloves immediately after preparation inside the ventilated fume hood.
c. Drain and wash thoroughly with DMF.
d. Monitor the efficiency of the reaction as described above, by separating a fraction of the resin and cleaving the crude off the resin, and analyze via LC-MS.
e. Split the peptide-decorated resin into 5 fractions of equal mass for chemical diversification and transfer each fraction into its individual 5 mL PP reactor with frit. Each fraction thereafter contains 0.02 mmol of the peptide decorated resin.
f. Place a magnetic stirrer underneath a cork ring. Place a 5 mL pointed flask into the cork ring and secure it with a clamp. Equip the flask with a magnetic stirring bar. To couple (5) to the Dde-deprotected resin (9), weigh compound (5) (15.1 mg, 40 μmol, 2 eq.) and add 50% (v/v) DCM in DMF (1 mL) into the pointed flask. Add DIPEA (14 μL, 80 μmol, 4 eq.) and PyBOP (18.7 mg, 36 μmol, 1.8 eq.) and stir vigorously for 5 min at RT until the suspension of the white solid turns clear.
g. Remove the plunger from the PP reactor and close the PP reactor with a pressure cap.
h. Pipette the activated compound (5) onto the Dde-deprotected, pre-swelled resin (9). Seal the PP reactor with parafilm and gently agitate at RT for 2 h.
i. Drain and wash the crude product thoroughly with DMF.
j. Monitor the efficiency of the reaction as described above, by separating a fraction of the resin, cleaving the crude off the resin, and analyzing via LC-MS.
4. Synthesis of compound (10B)
Note: These synthetic steps describe the Dde-deprotection of compound (9) followed by the appending of the linker-functionalized, photocaged TMPNVOC (4) to the localization motif (9), as displayed in Figure 17. This step is part of the synthesis of dual SLIPTNVOC.
Perform the synthesis of compound (10B) in analogy to compound (10A).
Difference (step A3.3f): Weigh compound (4) (24.6 mg, 40 μmol, 2 eq.) instead of compound (5). Perform all steps in reaction vessels covered with aluminum foil to prevent exposure to ambient light. The activation of the carboxylic acid is complete when the pale-yellow solution turns a deep orange.
Critical: Conduct all subsequent steps following compound (10B) in an aluminum foil-covered PP reactor.
5. Synthesis of compounds (11A) and (11B)
Note: These synthetic steps describe the Fmoc-deprotection of (10A) and (10B) and subsequent extension of the other linker arm, as displayed in Figure 18. These steps are part of both dual SLIPT and dual SLIPTNVOC.
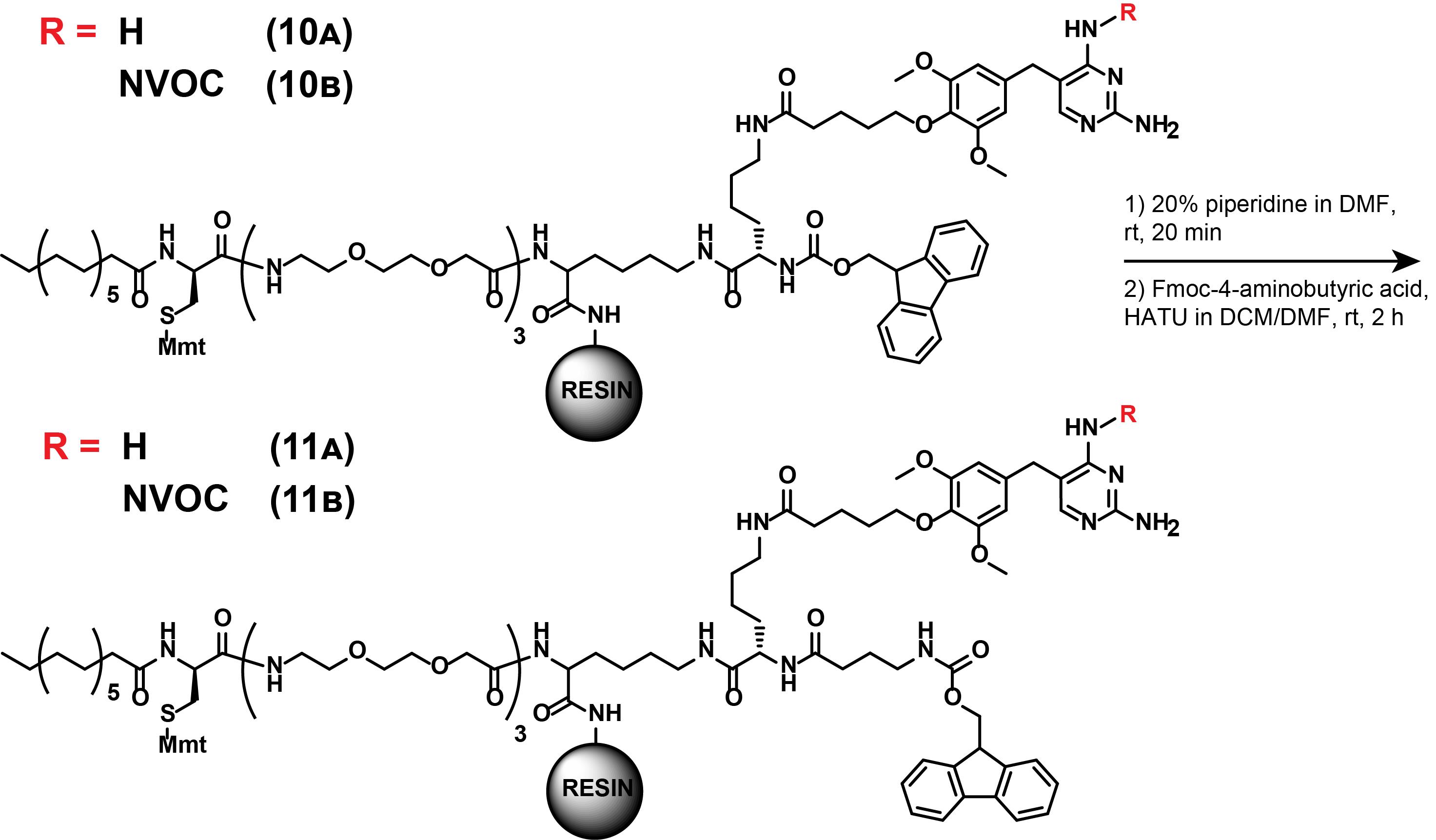
Figure 18. Synthesis of (11A) and (11B)
a. Remove the plunger from the PP reactor and close the PP reactor with a pressure cap.
b. Prepare the Fmoc-deprotection solution and pipette onto the pre-swelled resin (10A) or (10B). Seal the PP reactor with parafilm and gently agitate at RT for 10 min.
c. Drain and wash thoroughly with DMF and monitor the efficiency of the reaction as described above, by separating a fraction of the resin, cleaving the crude off the resin, and analyzing via LC-MS.
d. Place a magnetic stirrer underneath a cork ring. Place a 5 mL pointed flask into the cork ring and secure it with a clamp. Equip the flask with a magnetic stirring bar. Weigh Fmoc-4-aminobutyric acid (13 mg, 40 μmol, 2 eq.) into the pointed flask and add 50% (v/v) DCM in DMF (1 mL). Add DIPEA (14 μL, 80 μmol, 4 eq.) and HATU (13.7 mg, 36 μmol, 1.8 eq.) and stir vigorously for 5 min at RT until a visible color-shift from yellow to orange occurs.
e. Remove the plunger from the PP reactor and close the PP reactor with a pressure cap.
f. Pipette the activated Fmoc-4-aminobutyric acid onto the Fmoc-deprotected (10A) or (10B), and gently agitate at RT for 2 h.
g. Drain and wash thoroughly with DMF.
h. Monitor the efficiency of the reaction as described above, by separating a fraction of the resin, cleaving the crude off the resin, and analyzing via LC-MS.
6. Synthesis of compounds (12A) and (12B)
Note: These synthetic steps describe the Fmoc-deprotection and subsequent further extension of the other linker arm of the intermediates (11A) and (11B), as displayed in Figure 19. These steps are part of both dual SLIPT and dual SLIPTNVOC.
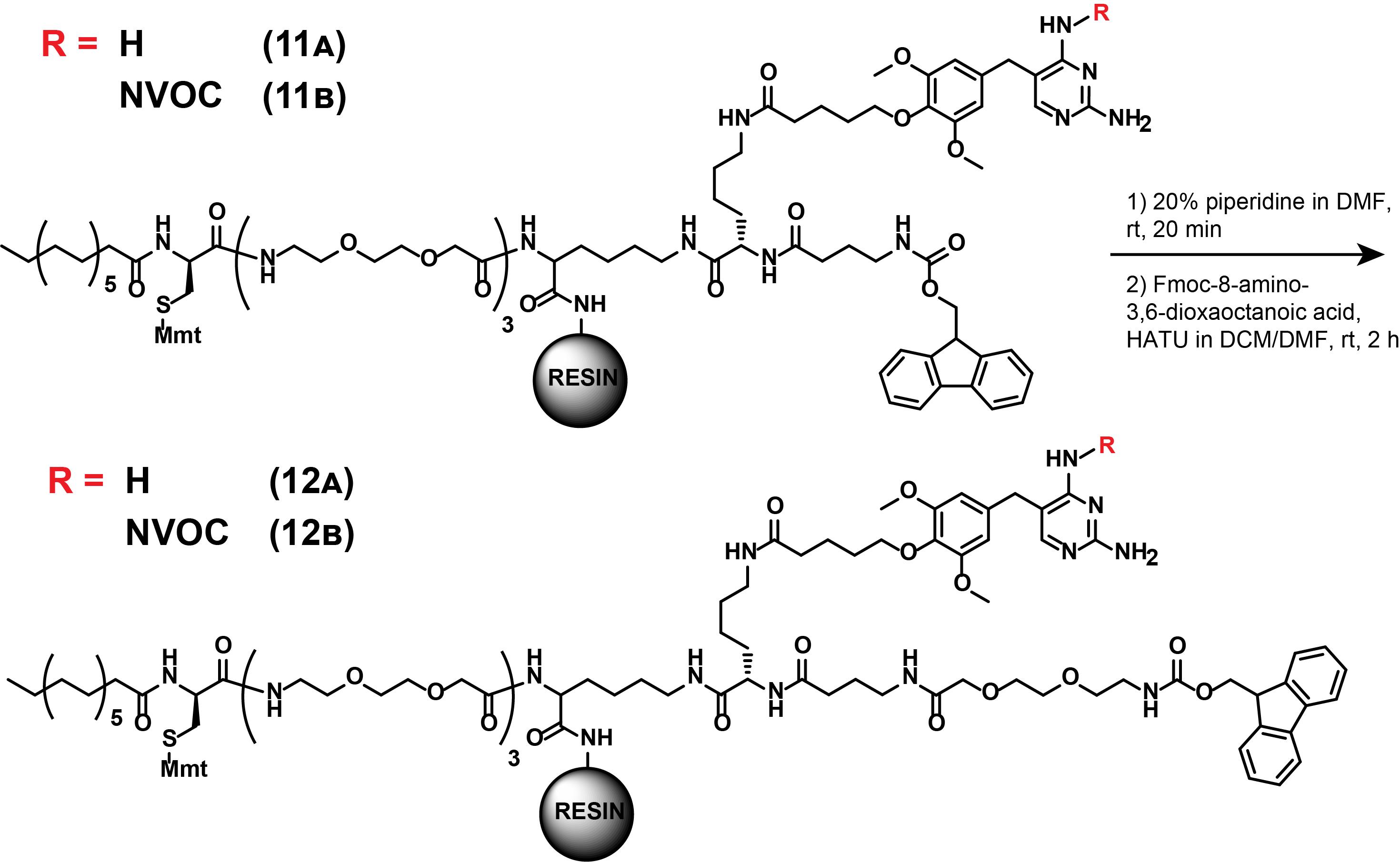
Figure 19. Synthesis of (12A) and (12B)
a. Remove the plunger from the PP reactor and close the PP reactor with a pressure cap.
b. Prepare the Fmoc-deprotection solution and pipette onto the pre-swelled resin (11A), or (11B). Seal the PP reactor with parafilm and gently agitate at RT for 10 min.
c. Drain and wash thoroughly with DMF and monitor the efficiency of the reaction as described above, by separating a fraction of the resin, cleaving the crude off the resin, and analyzing via LC-MS.
d. Place a magnetic stirrer underneath a cork ring. Place a 5 mL pointed flask into the cork ring and secure it with a clamp. Equip the flask with a magnetic stirring bar. Weigh Fmoc-8-amino-3,6-dioxaoctanoic acid (15.4 mg, 40 μmol, 2 eq.) into the pointed flask and add 50% (v/v) DCM in DMF (1 mL). Add DIPEA (14 μL, 80 μmol, 4 eq.) and HATU (13.7 mg, 36 μmol, 1.8 eq.) and stir vigorously for 5 min at RT, until a visible color-shift from yellow to orange occurs.
e. Remove the plunger from the PP reactor and close the PP reactor with a pressure cap.
f. Pipette the activated Fmoc-8-amino-3,6-dioxaoctanoic acid onto the Fmoc-deprotected (11A) or (11B) and gently agitate at RT for 2 h.
g. Drain and wash thoroughly with DMF.
h. Monitor the efficiency of the reaction as described above, by separating a fraction of the resin, cleaving the crude off the resin, and analyzing via LC-MS.
7. Synthesis of dual SLIPT (13A)
Note: These synthetic steps describe the final steps to access constitutively active dual SLIPT, as displayed in Figure 20. First, Fmoc-deprotection of (12A) is described, before linker-functionalized HTL is appended.
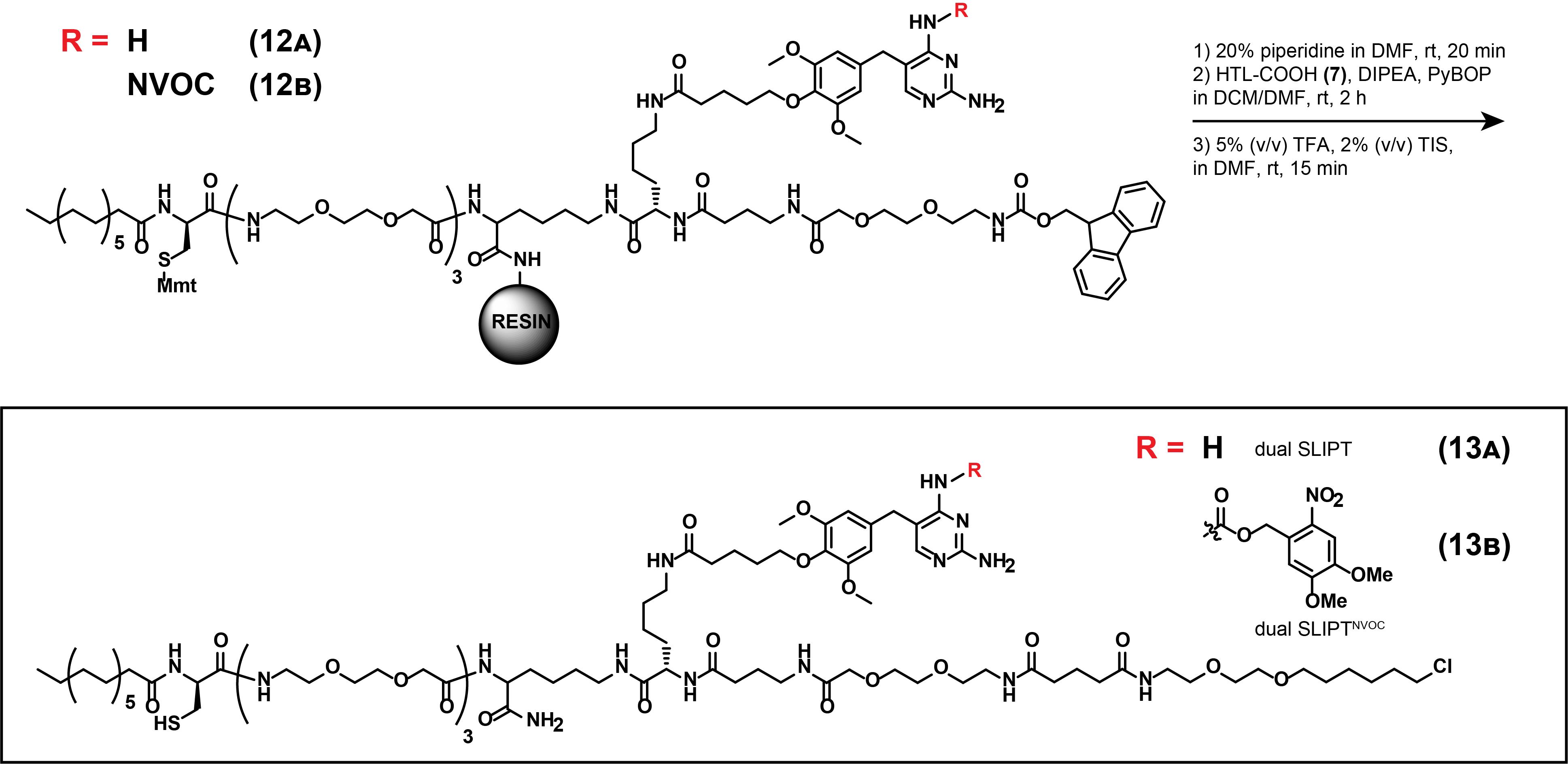
Figure 20. Synthesis of dual SLIPT (13A) and dual SLIPTNVOC (13B)
a. Remove the plunger from the PP reactor and close the PP reactor with a pressure cap.
b. Prepare the Fmoc-deprotection solution and pipette onto the pre-swelled resin (12A) or (12B). Seal the PP reactor with parafilm and gently agitate at RT for 10 min.
c. Drain and wash thoroughly with DMF and monitor the efficiency of the reaction as described above, by separating a fraction of the resin, cleaving the crude off the resin, and analyzing via LC-MS.
d. Place a magnetic stirrer underneath a cork ring. Place a 5 mL pointed flask into the cork ring and secure it with a clamp. Equip the flask with a magnetic stirring bar. Weigh (7) (13.5 mg, 40 μmol, 2 eq.) into the pointed flask and add 50% (v/v) DCM in DMF (1 mL). Add DIPEA (14 μL, 80 μmol, 4 eq.) and PyBOP (18.7 mg, 36 μmol, 1.8 eq.) and stir vigorously for 5 min at RT, until a visible color-shift from pale yellow to deep yellow occurs.
e. Remove the plunger from the PP reactor and close the PP reactor with a pressure cap.
f. Pipette the activated (7) onto the Fmoc-deprotected (12A) or (12B), and gently agitate at RT for 2 h.
g. Drain and wash thoroughly with DMF.
h. Monitor the efficiency of the reaction as described above, by separating a fraction of the resin, cleaving the crude off the resin, and analyzing via LC-MS.
i. Prepare the final peptide cleavage solution.
j. Remove the plunger from the PP reactor, close it with a pressure cap, and pipette the cleavage solution onto the crude product coupled to the resin. Seal the PP reactor with parafilm and gently agitate at RT for 15 min.
Note: The final peptide cleavage solution contains the final crude product.
k. Collect the cleaved-off crude product in a 20 mL vial.
l. Repeat the peptide cleavage as described above and combine the crude product in the 20 mL vial.
m. Wash the resin thoroughly with DCM and combine the wash fraction with the remaining crude product.
n. Dry the crude product under reduced pressure using a rotary evaporator.
o. Re-dissolve the crude product in minimal 55% ACN in H2O, filter the solution (PTFE filters, 0.45 μm) prior to injection, and purify on HPLC, using a linear gradient of 45%–70% ACN (0.1% FA) in H2O (0.1% FA) over 45 min and a C4-column. Read out the presence of the product via absorption at λ = 280 nm.
p. Combine the product fractions in 50 mL Falcon tubes and dry by lyophilization. To do so, evaporate ACN from the solvent mixture by rotary evaporation until the final ACN concentration is approximately 10%, before freezing the Falcon tubes containing the purified product in liquid nitrogen. Then, before allowing to thaw, partially unscrew the cap and place the Falcons into the lyophilizer.
q. Final product: White solid (13A), approximately 1% yield.
r. Analytical data: HRMS (ESI): calculated for [M+3H]3+, 644.0378; found, 644.0385.
8. Synthesis of dual SLIPTNVOC (13B)
Note: These synthetic steps describe the final steps to access photocaged dual SLIPTNVOC. First, Fmoc-deprotection of (12B) is described, before linker-functionalized HTL is appended. Perform the synthesis of (13B) in analogy to (13A) (steps A3.7 a–n).
Caution: Minimize exposure to ambient light in all reaction and purification steps.
a. Re-dissolve the crude product in minimal 75% ACN in H2O, filter the solution (PTFE filters, 0.45 μm) prior to injection, and purify on HPLC, using a linear gradient of 55%–65% ACN (0.1% FA) in H2O (0.1% FA) over 45 min and a C4-column. Read out the presence of the product via absorption at λ = 280 nm.
b. Combine the product fractions in 50 mL Falcon tubes, and dry by lyophilization, as described for (13A) (step A3.7p).
c. Final product: Pale yellow solid, approximately 1% yield.
d. Analytical data: HRMS (ESI): calculated for [M+3H]3+, 723.7188; found, 723.7187.
B. Quantitation and preparation of stock solutions
DMSO stock solutions for subsequent cell experiments can be prepared by accurately weighing at least 10 mg of the pure, dry product and calculating the concentration based on its molecular weight. In case of low amounts of obtained, pure product (13A) and (13B), quantitation needs to be conducted via quantitative NMR. For this purpose, add a known molar quantity of DMSO into the NMR sample. In a 1H NMR, conducted in CD3OD, the 6 protons of DMSO result in a singlet peak at δ = 2.65 ppm. This signal can be integrated, divided by six to equal the signal that one DMSO-proton would give, and compared to an integrated signal of the product (singlet at δ = 7.23 ppm). To determine the signal of the product, a proton signal in the aromatic region is chosen, ideally, for ease of identification and integration. The molar quantity of the product can then be determined by the following equation:
With I indicating the integral value for one proton of the product, or the integral value of the DMSO signal, and n denoting the molar quantity.
The amount of DMSO added should be approximately equal to the expected quantity of product.
1. Dissolve the lyophilized purified product in approximately 250 μL of CD3OD and transfer into an NMR tube.
2. Add 5 μL of DMSO into an Eppendorf containing 95 μL of CD3OD and vortex.
3. Of this dilution, add 1 μL into the NMR tube.
Note: The NMR sample now contains V(DMSO) = 0.05 μL, which is equivalent to 0.704 μmol of DMSO.
4. Measure the 1H NMR.
5. Recover the entire NMR sample by transferring into a 20 mL vial. Rinse the NMR tube thoroughly to recover the entirety of the purified product.
6. Dry the purified product under reduced pressure using a rotary evaporator.
7. To prepare a 1 mM stock solution, dilute the purified product in the appropriate volume of DMSO for storage at 4 °C.
The volume of DMSO (µL) needed to prepare a 1 mM stock solution can be determined by the following equation:
With n denoting the molar quantity of the product determined by quantitative NMR, and V denoting the volume of DMSO to add.
8. Aliquot the stock solution into 500 μL amber-colored microcentrifuge tubes to minimize freezing and thawing cycles of the product. Store at -20 °C for long-term storage, and at 4 °C for daily use.
C. Cell experiments: inducing dimerization of any two POIs at the plasma membrane using dual SLIPT and dual SLIPTNVOC
Notes:
1. iK6eDHFR and HOB are the functional variants of the protein tags eDHFR and HaloTag7 (HT7), respectively. iK6eDHFR and HOB, contrary to eDHFR and HT7, contain positive charges near the binding cleft that point toward the inner leaflet of the plasma membrane. Thus, electrostatic attraction with the negatively charged inner leaflet is enabled, which has been found to aid PM recruitment of the protein tags [2,3].
Linkers: Between iK6eDHFR and the functional protein (EGFP, or other POIs), a short, amino acid linker is ideal. No linker has been found by the authors to be necessary between HOB and the functional protein (mScarlet, or other POIs). cDNA can be found at the typical plasmid repositories (Addgene numbers can be found in the Biological materials list) and modified with the traditional cloning strategies.
2. This section explains how to perform the dual SLIPT & dual SLIPTNVOC-induced dimerization of two fluorescent POIs at the PM using transient transfection. Establishment of stable cell lines is, however, advisable to maintain comparable expression levels between cells.
3. This section describes the post-hoc visualization of PM-recruited POIs, using dual SLIPT or dual SLIPTNVOC. If visualization under observation is desired, replace the FBS-containing medium [full DMEM (+)] in step C8 with phenol red-free full DMEM (-), lacking probe. With the culture dish on the microscope, introduce a 2× diluted probe solution instead (step C9). Full DMEM (-) is used to minimize probe absorption by FBS.
4. The microscopy experiments in this section are conducted in µ-Slide dishes (Ibidi); however, any microscopy-compatible dishes or multi-well plate may be used. The 18-well µ-Slide dishes (Ibidi) can accommodate approximately 200 μL of medium. The growth area per well is 0.34 cm2. Scale accordingly.
1. Prepare mini-prepped stocks of the plasmids and dilute to approximately 100 ng/μL.
2. Thaw HeLa cells and keep them in culture in full DMEM (+) for at least 2 passages.
3. Seed 5 × 104 cells in 100 μL of full DMEM (+) per well into the µ-Slide dish when cells are around 70%–80% confluent. Allow cells to adhere for at least 8 h.
4. Thaw the plasmids at RT.
5. Transfect the cells using Lipofectamine 3000. To do so, generate solution A and B in separate microcentrifuge tubes, as indicated in Table 2. Scale the transfection mixtures according to the number of wells to transfect. Upon assembly of solutions, vortex both solutions and incubate them in separate microcentrifuge tubes for 5 min at RT. Thereafter, mix them together by pipetting vigorously and incubate at RT for 10–15 min.
Note: The amount given here for cDNA refers to the absolute amount of plasmid DNA per well. If co-transfection with more than one plasmid is conducted, add 100 ng of the combined plasmids per well.
Table 2. Preparation of the transfection mix
| Reagent | Quantity (per well) | |
| Solution A | OptiMEM | 10 μL |
| cDNA | 100 ng, 1 μL | |
| P3000TM | 0.2 μL | |
| Solution B | OptiMEM | 10 μL |
| LipofectamineTM 3000 | 0.2 μL |
6. Pipette the transfection mix into the wells by gently ejecting the volume toward the side of the well. Mix gently by repeated aspiration and ejection. Incubate for 6 h.
7. Dilute dual SLIPT or dual SLIPTNVOC stock 1:100 with phenol red–free full DMEM (-) per well. Vortex vigorously to mix. Warm to 37 °C.
8. Aspirate the medium carefully, wash the cells once with phenol red–free full DMEM (-), then replace the medium with 100 μL of the 100× diluted probe (dual SLIPT or dual SLIPTNVOC) in phenol red–free full DMEM (-). Incubate overnight.
9. Acquire images on a confocal microscope (60×, oil immersion, 1.4 NA). If dual SLIPT (13A) was used (steps C7–8), the signal of the two POIs, attached to iK6eDHFR and HOB, should overlap and be localized at the plasma membrane. If dual SLIPTNVOC (13B) was used (steps C7–8), the signal of the HOB-attached POI should be localized at the plasma membrane, and the iK6eDHFR-attached POI should be localized cytosolically. If dual SLIPTNVOC (13B) was used (steps C7–8), proceed to step 10.
Note: To induce cellular phenomena reliant on PM recruitment of the two POIs, a nicely distinguishable fluorescent signal at the PM is not necessary and varies between cell types and seeding densities. If a clear PM-localized fluorescent signal is desired, as in Figure 21, however, an increase in seeded cell density may be appropriate, as tight seeding reduces the cell spread and accumulates PM-localized signal in the z-axis. See Troubleshooting problem 7.
10. To dimerize the two POIs with high spatiotemporal precision using dual SLIPT NVOC, irradiate a defined region of interest with 405 nm light.
Notes:
1. The absorption maximum of the nitroveratryl-protection group is at 365 nm (see [16]). However, the photoprotection can be cleaved off with reasonable efficiency with the more widely implemented 405 nm (blue light) laser. In the original work [2], this was implemented using a FRAP module.
2. Photocleavage efficiency depends on multiple factors, including laser type, alignment, and operational time; thus, specifying exact laser settings is not broadly applicable. In our setup, effective cleavage was achieved with 10 s irradiation at 0.26 mW measured at the sample plane using a slide-mounted power meter.
11. Acquire post-irradiation images immediately after performing the photocleavage. The signal of the iK6eDHFR-attached POI signal should now also be localized at the PM in the previously irradiated region of interest.
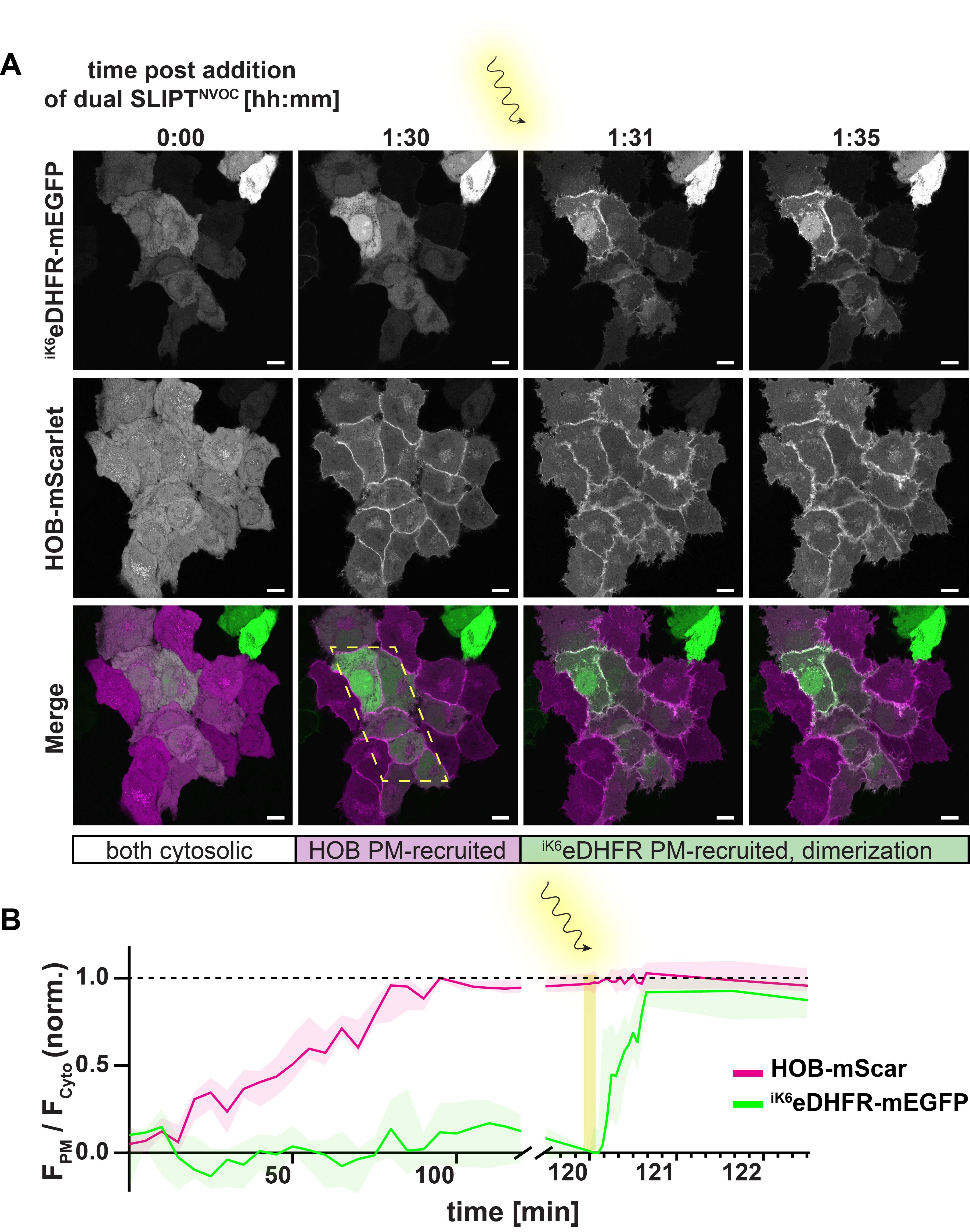
Figure 21. Validation of dimerization of iK6eDHFR- and HOB-tagged proteins of interest (POIs) at the plasma membrane (PM). (A) Example images depicting PM recruitment of HOB-mScarlet (magenta) and iK6eDHFR-mEGFP (green) in response to incubation of HeLa cells with 10 μm dual SLIPTNVOC. Scale bars, 10 μm. Prior to incubation with dual SLIPTNVOC, both iK6eDHFR- and HOB-tagged POIs are cytosolic. After approximately 1.5 h post-incubation with 10 μm of dual SLIPTNVOC, HOB-tagged POIs are maximally PM-recruited, whereas iK6eDHFR-tagged POIs remain cytosolic. Irradiation with 405 nm laser uncages the trimethoprim (TMP) headgroup, which enables immediate co-recruitment of iK6eDHFR-tagged POIs and dimerization of the two PM-recruited POIs at the irradiated area (yellow dotted area). (B) Time-course of PM recruitment of HOB-mScarlet (magenta) and iK6eDHFR-mEGFP (green) in response to incubation of HeLa cells (n = 7 cells) with 10 μm of dual SLIPTNVOC, as measured by the normalized ratio of PM-localized signal and cytosolic signal. Yellow shading indicates the timing of the irradiation and consequent photouncaging of the TMP-headgroup moiety.
Data analysis
Image processing and initial analysis should be conducted using Fiji (ImageJ). Data collection and calculations can be done in Excel, and graph generation can be done with GraphPad Prism or similar programs.
The raw data should consist of two (or more) imaged channels and a time course.
Note: This protocol describes the quantification of the PM-localized signal by generating a mask of the PM-translocated fluorescent proteins. In an alternative strategy, a membrane stain can be used to identify the positioning of the plasma membrane. If done in this manner, follow steps 1–3, and use the membrane stain to generate the mask (steps 4–5) instead.
1. Assemble the raw data into a hyperstack and save as TIFF (File → Save As → Tiff…).
2. Segment the multi-cell image into individual images, depicting individual cells, and save as TIFF, enumerated. Ensure that potential shifts in x,y are manually corrected by aligning the cell nuclei. If a membrane stain is used, this signal may serve as additional orientation.
3. Split the hyperstack into the two individual color channels (Image → Color → Split Channels) and save the resulting time courses individually, denoting their respective channel (“C1”, “C2”).
4. Isolate the post PM-translocation image for the purpose of generating a mask (Image → Duplicate; select the desired time point under Range, making sure to deselect Duplicate stack), smooth twice (Process → Smooth), and threshold the image (Image → Adjust → Threshold) to only include the pixels of the top 2% signal intensity. Click apply to generate the mask (adjust to top 2% signal intensities → Apply). This will result in a mask including regions of interest (ROIs) at the plasma membrane, and excluding signals everywhere else (Figure 22, “C1 mask”).
5. Save this mask as TIFF, noting the corresponding channel, and repeat for the other color channel.
6. Close all images.
7. Open the mask for one of the channels, as well as the corresponding time course. Concatenate the mask and the time course (Image → Stacks → Tools → Concatenate; select open as 4D image). Save the resulting superimposed mask and time course as TIFF.
8. Add the ROIs, as identified by the mask, to the ROI Manager (Analyze → Analyze Particles; select Display results, Clear results, Add to Manager, and Overlay). Select Yes in response to the question “Process all n images?”, with n being the number of time points in the stack. The ROI manager will now appear as a new window.
Note: The Analyze Particles function allows for the exclusion of data points that fall below a certain size [Size (µm2)] or roundness threshold (Circularity). The circularity threshold is not relevant to this analysis and should therefore remain set as (0.00–1.00), but the size threshold may be useful to discriminate against disconnected ROIs. Keep these settings constant between channels and cells.
9. Without selecting any individual ROIs, go to the first time point in the data stack, and select Measure in the ROI Manager to measure the signal intensity of every ROI for that selected time point.
10. The Results window will appear as a new window. Extract the data from the Results window and transfer to Excel.
11. Return to Fiji and repeat steps 9 and 10.
12. Iteratively measure the ROI-localized intensities for all time points and collect the measured data in Excel, separating the individual time points in columns.
13. Return to the first time point of the stack, select the Polygon selections tool, and trace the cytoplasm of the cell, excluding the signal at the plasma membrane.
14. Measure the signal intensity of the created polygon (the measurement will appear in the Results window) and transfer it to Excel, assigning it to the measured ROI-localized intensities for the respective time point.
15. Divide all ROI-localized intensities for each time point by the measured cytosolic signal for that time point. This will result in an FPM/FCyto ratio (median value) for every individual cell at every individual time point, as displayed in Figure 22. Plotted against time, this results in a trace depicting an individual cell’s response over time.
Note: This data can be further processed by normalizing to the pre-incubation FPM/FCyto ratio or spreading between the minimum and maximum mean value to result in data as displayed in Figure 21B. To result in an average response over time for multiple cells, repeat the aforementioned processing for multiple cells. Transfer the resulting FPM/FCyto traces for each cell into GraphPad Prism and combine the individual cell’s responses to the perturbation to plot an average response (median) and its variability (interquartile range) against time post-incubation.
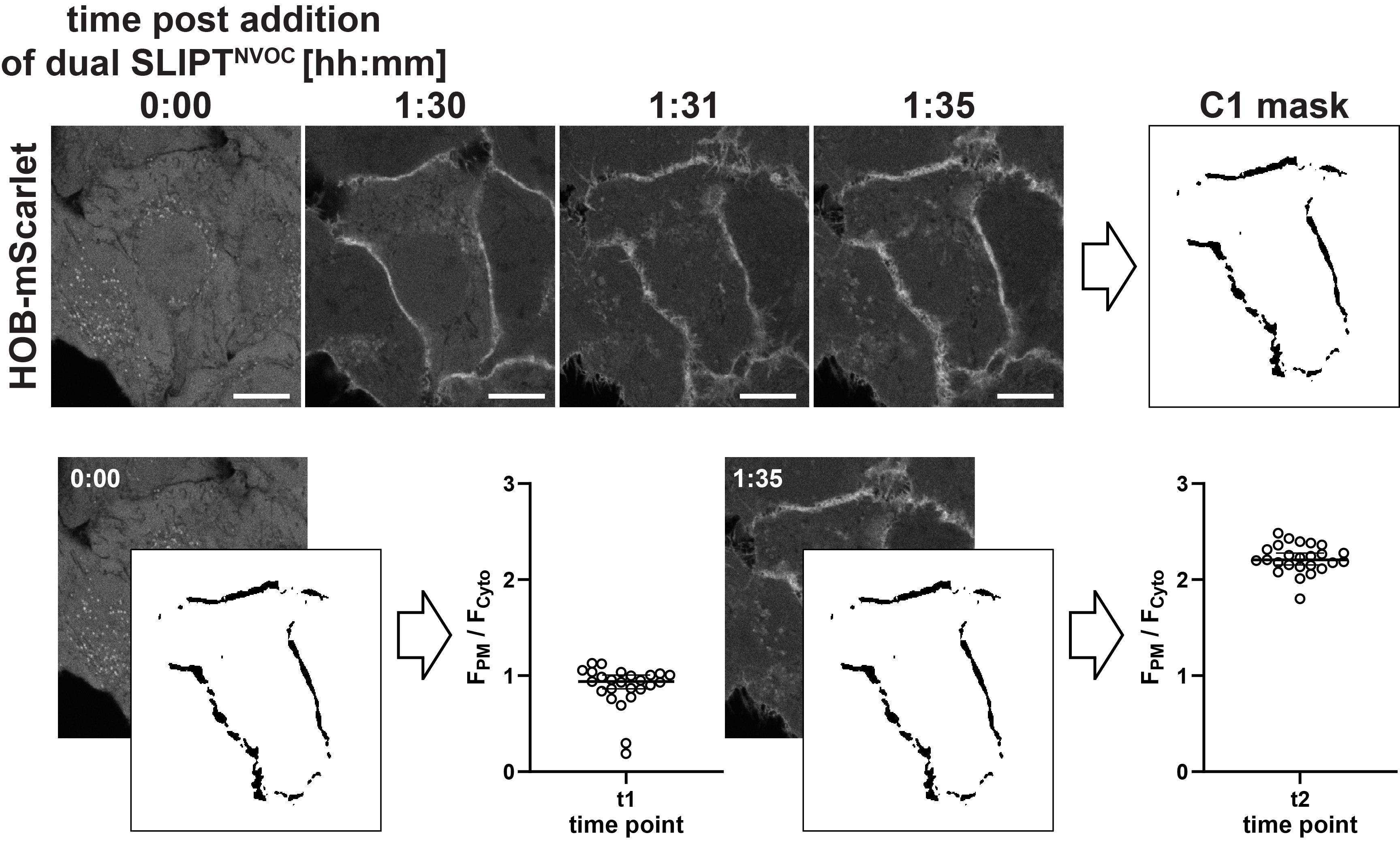
Figure 22. Example data analysis. The raw data is segmented into individual cells, from which masks for each channel are generated (“C1 mask”) that contain several regions of interest (ROIs). The mask is then superimposed with each time point and used to determine the mean plasma membrane (PM)-localized signal for each time point and ROI. The resulting values are then divided by the mean cytosolic signal intensity at every time point to result in the FPM/FCyto ratio for that time point (shown are the individual intensity ratios for every ROI, the median intensity, and the 95% CI). The median ratios of the analyzed cells can be used to plot the FPM/FCyto ratio against time, and normalized to the [0,1] interval, as done in Figure 21B. Scale bars, 10 μm.
Validation of protocol
This protocol (or parts of it) has been used and validated in the following research article(s):
Bayer et al. [2]. Dual SLIPT–A Lipid Mimic to Enable Spatiotemporally Defined, Sequential Protein Dimerization. ACS Chemical Biology. (Figures 1–20 and Figure 21B)
Bayer et al. [17]. The effect of plasma membrane recruitment on SH3BP2 function in lamellipodial regulation-an investigation using tailor-made lipid-mimics. Inaugural Dissertation.
General notes and troubleshooting
General notes
1. Use of dual SLIPT and dual SLIPTNVOC has been successfully demonstrated in HeLa, HeLa Kyoto FlipINTM, Cos-7, and NIH3T3 FlipINTM cells. In HEK cells, however, addition of dual SLIPT has not yet resulted in efficient PM translocation of iK6eDHFR- and HOB-tagged POIs. Co-incubation with efflux inhibitors such as Verapamil has not been attempted. For this reason, no definitive statement can be made as to whether the lack of PM recruitment is caused by impaired ability of dual SLIPT to permeate the membrane, increased efflux activity in HEK cells, or other causes.
2. Dual SLIPT has been successfully demonstrated at probe concentrations between 0.1 and 20 μm. If fast and quantitative responses to the probe are desired, we recommend using probe concentrations between 5 and 10 μm. In case the experimenter wishes to emulate PM recruitment via rare lipid species, low concentrations, down to 0.1 μm of dual SLIPT, or dual SLIPTNVOC-mediated subcellular dimerization may be used. In the original publication [2], a probe concentration-independent mean distance between POIs was verified via FRET.
Troubleshooting
At least some chemical training is advisable to complete the synthetic section safely. If unavailable, contact the corresponding author to provide the final probes dual SLIPT or dual SLIPTNVOC.
Problem 1: The peptide synthesizer reports an error message prior to starting.
Possible cause: Prior to starting the reaction, the peptide synthesizer will check for leakage. The reaction will not commence before all vessels are sealed tightly enough to withstand the system’s pressure during the reaction.
Solution: Re-screw all bottles as tightly as possible. Ensure that no cap sits wonkily on the bottles.
Problem 2: LC-MS analysis of lipopeptides (8–12) yields the mass of the deprotected species, despite the deprotection not having been conducted yet.
Possible cause: The cleavage from the resin to retrieve samples for LC-MS analysis has been observed to deprotect Fmoc-protected species. The cause for this has not been determined.
Solution: Assume the resin-coupled lipopeptide is still Fmoc-protected. Proceed with the Fmoc-deprotection as in the protocol. Do another test reaction post-Fmoc-deprotection. The deprotected species should appear cleanly in the LC-MS.
Problem 3: Demethylated TMP is difficult to separate from impurities (starting material, side products).
Possible cause: Demethylation of TMP may yield both the mono- and the double-demethylated product. With increasing reaction duration, the fraction of the double-demethylated side product increases. A too short reaction duration, on the other hand, results in minimal double-demethylated side product but also limits product formation.
Solution: Err on the longer side of reaction time. Trimethoprim (starting material) is difficult to separate from the product by recrystallisation, whereas the double-demethylated side product is easier to separate due to its increased hydrophilicity. Conduct a small-scale test reaction to gauge the optimal reaction duration to facilitate this.
Problem 4: Formation of multi-alkylated side products.
Possible cause: Alkylation of the exocyclic amines occurs.
Solution: Purify the product via HPLC [10%–50% ACN (0.1% TFA) in H2O (0.1% TFA)] after an initial purification via flash column chromatography with the reported conditions.
Problem 5: No product formation is observed. The major side product is found to be an aldehyde-adduct ([M+H]+ 433.2087).
Possible cause: Prior purification with HPLC resulted in formate-salt formation of the starting material (2).
Solution: Exchange the anion for a chloride anion by the addition of 2 eq. 2 M HCl to the purified starting material (2).
Critical: Too many equivalents (such as 6 eq.) will lead to further demethylation of the TMP methoxy moieties. Too little acid will lead to the incomplete exchange of the anions.
Problem 6: The compound is insoluble, or the saponification does not proceed.
Possible cause: The “undesirable” regioisomer will not dissolve in the conditions indicated in this protocol.
Solution: Double-check the identification of the “desirable” regioisomer.
Problem 7: Pronounced cell death when seeding more than 5 × 104 cells in 100 μL of full DMEM (+) per well in order to clearly visualize the PM recruitment of iK6eDHFR- or HOB-tagged POIs.
Possible cause: Overseeding and resulting cell death.
Solution: We seeded up to 7.5 × 104 cells in 100 μL of full DMEM (+) per well and imaged up to 3 days later. The feasibility of seeding such high densities, however, is dependent on multiple factors, including passage number. Reduce the number of cells seeded and possibly image the plasma membrane with a Z-stack, followed by a max projection to retain distinguishability of the PM-localized fluorescent signal.
Problem 8: Cell movement post-incubation of the final probes, which leads to a shifting localization of the PM.
Possible cause: Cells are not seeded tightly enough.
Solution: Increase cell seeding density.
Acknowledgments
Conceptualization, K.V.B.; Investigation, K.V.B., Writing—Original Draft, K.V.B.; Writing—Review & Editing, K.V.B. & R.W.; Funding acquisition, K.V.B. & R.W.; Supervision, K.V.B. & R.W. K.V.B. acknowledges funding from the Studienstiftung des deutschen Volkes and the Max Planck Society. R.W. acknowledges funding from the Max Planck Society and the Deutsche Forschungsgemeinschaft DFG (SFB/TRR 186). Open access funded by Max Planck Society. This protocol was first described and validated in Bayer et al. 2025. The approach was developed based on the original SLIPT tool by Tsukiji et al. [18].
Competing interests
The authors declare no conflicts of interest.
References
- Ishida, M., Watanabe, H., Takigawa, K., Kurishita, Y., Oki, C., Nakamura, A., Hamachi, I. and Tsukiji, S. (2013). Synthetic Self-Localizing Ligands That Control the Spatial Location of Proteins in Living Cells. J Am Chem Soc. 135(34): 12684–12689. https://doi.org/10.1021/ja4046907
- Bayer, K. V., Taeb, M., Koch, B., Yoshimura, S. H. and Wombacher, R. (2025). Dual SLIPT–A Lipid Mimic to Enable Spatiotemporally Defined, Sequential Protein Dimerization. ACS Chem Biol. 20(5): 1038–1047. https://doi.org/10.1021/acschembio.4c00856
- Suzuki, S., Nakamura, A., Hatano, Y., Yoshikawa, M., Yoshii, T., Sawada, S., Atsuta-Tsunoda, K., Aoki, K. and Tsukiji, S. (2022). A chemogenetic platform for controlling plasma membrane signaling and synthetic signal oscillation. Cell Chem Biol. 29(9): 1446–1464.e10. https://doi.org/10.1016/j.chembiol.2022.06.005
- Koßmann, K. J., Ziegler, C., Angelin, A., Meyer, R., Skoupi, M., Rabe, K. S. and Niemeyer, C. M. (2016). A Rationally Designed Connector for Assembly of Protein‐Functionalized DNA Nanostructures. ChemBioChem. 17(12): 1102–1106. https://doi.org/10.1002/cbic.201600039
- DeRose, R., Miyamoto, T. and Inoue, T. (2013). Manipulating signaling at will: chemically-inducible dimerization (CID) techniques resolve problems in cell biology. Pflugers Arch - Eur J Physiol. 465(3): 409–417. https://doi.org/10.1007/s00424-012-1208-6
- Voß, S., Klewer, L. and Wu, Y. W. (2015). Chemically induced dimerization: reversible and spatiotemporal control of protein function in cells. Curr Opin Chem Biol. 28: 194–201. https://doi.org/10.1016/j.cbpa.2015.09.003
- Idevall-Hagren, O. and De Camilli, P. (2014). Manipulation of Plasma Membrane Phosphoinositides Using Photoinduced Protein–Protein Interactions. Methods Mol Biol. 109–128. https://doi.org/10.1007/978-1-4939-0470-9_8
- Hannanta-Anan, P., Glantz, S. T. and Chow, B. Y. (2019). Optically inducible membrane recruitment and signaling systems. Curr Opin Struct Biol. 57: 84–92. https://doi.org/10.1016/j.sbi.2019.01.017
- Maltas, J., Reed, H., Porter, A. and Malliri, A. (2020). Mechanisms and consequences of dysregulation of the Tiam family of Rac activators in disease. Biochem Soc Trans. 48(6): 2703–2719. https://doi.org/10.1042/bst20200481
- Cherfils, J. and Zeghouf, M. (2013). Regulation of Small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 93(1): 269–309. https://doi.org/10.1152/physrev.00003.2012
- Rohatgi, R., Ho, H. y. and Kirschner, M. W. (2000). Mechanism of N-Wasp Activation by Cdc42 and Phosphatidylinositol 4,5-Bisphosphate. J Cell Biol. 150(6): 1299–1310. https://doi.org/10.1083/jcb.150.6.1299
- Wu, H. D., Kikuchi, M., Dagliyan, O., Aragaki, A. K., Nakamura, H., Dokholyan, N. V., Umehara, T. and Inoue, T. (2020). Rational design and implementation of a chemically inducible heterotrimerization system. Nat Methods. 17(9): 928–936. https://doi.org/10.1038/s41592-020-0913-x
- Schultz, C. and Brügger, B. (2023). Chemical Tools for Lipid Cell Biology. Acc Chem Res. 56(9): 983–983. https://doi.org/10.1021/acs.accounts.3c00225
- Zhou, C., He, H. and Chen, X. (2024). Photoactivatable Nanobody Conjugate Dimerizer Temporally Resolves Tiam1‐Rac1 Signaling Axis. Adv Sci. 11(11): e202307549. https://doi.org/10.1002/advs.202307549
- Nakamura, A., Oki, C., Sawada, S., Yoshii, T., Kuwata, K., Rudd, A. K., Devaraj, N. K., Noma, K. and Tsukiji, S. (2020). Designer Palmitoylation Motif-Based Self-Localizing Ligand for Sustained Control of Protein Localization in Living Cells and Caenorhabditis elegans. ACS Chem Biol. 15(4): 837-843. https://doi.org/10.1021/acschembio.0c00014.
- Yoshii, T., Oki, C. and Tsukiji, S. (2022). A photoactivatable self-localizing ligand with improved photosensitivity for chemo-optogenetic control of protein localization in living cells. Bioorg Med Chem Lett. 72: 128865. https://doi.org/10.1016/j.bmcl.2022.128865
- Bayer, K. V. (2025). The effect of plasma membrane recruitment on SH3BP2 function in lamellipodial regulation - an investigation using tailor-made lipid-mimics. Dr. rer. nat. Inaugural Dissertation. University of Heidelberg.
- Ohira, S., Nakamura, A., Terai, K. and Tsukiji, S. (2025). Development and Recent Advances in SLIPT‐PM: A Chemogenetic Platform for Manipulating Signaling at the Plasma Membrane. ChemBioChem. 26(16): e202500327. https://doi.org/10.1002/cbic.202500327
Article Information
Publication history
Received: Jul 30, 2025
Accepted: Sep 16, 2025
Available online: Oct 17, 2025
Published: Nov 5, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Bayer, K. V. and Wombacher, R. (2025). Lipid-Mediated Sequential Recruitment of Proteins Via Dual SLIPT and Dual SLIPTNVOC in Live Cells. Bio-protocol 15(21): e5489. DOI: 10.21769/BioProtoc.5489.
Category
Biochemistry > Lipid > Lipid-protein interaction
Biochemistry > Protein > Interaction > Protein-lipid interaction
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.