- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Cost-Effective and Reproducible Preparation of mRNA-Loaded Lipid Nanoparticles Using a Conventional Laboratory-Scale Microfluidic Assembly System
Published: Vol 15, Iss 18, Sep 20, 2025 DOI: 10.21769/BioProtoc.5458 Views: 1498
Reviewed by: David PaulNidhi MenonAnonymous reviewer(s)

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
This protocol describes a standardized and economically accessible method for synthesizing mRNA-encapsulated lipid nanoparticles using routine laboratory equipment, including precision syringe pumps, Y-shaped glass microfluidic chips, and silicone tubing. Designed to address the cost and accessibility limitations of commercial microfluidic platforms, the system achieves performance metrics comparable to high-end devices while reducing equipment costs by 90%. By systematically optimizing hydrodynamic parameters (total flow rate: 12 mL/min; lipid-to-aqueous phase ratio: 3:1), the protocol enables consistent production of lipid nanoparticles with key quality attributes: high mRNA encapsulation efficiency (≥ 80%), narrow particle size distribution (100–120 nm, polydispersity index ≤ 0.2), and excellent storage performance (≥ 7 days at 4 °C ).
Key features
• A low-cost mRNA@LNPs synthesis method is developed using common lab equipment, cutting costs by 90% while matching commercial systems' performance.
• The LNP synthesis platform allows researchers to flexibly adjust hydrodynamic parameters to screen various lipid formulations.
Keywords: Laboratory preparationGraphical overview
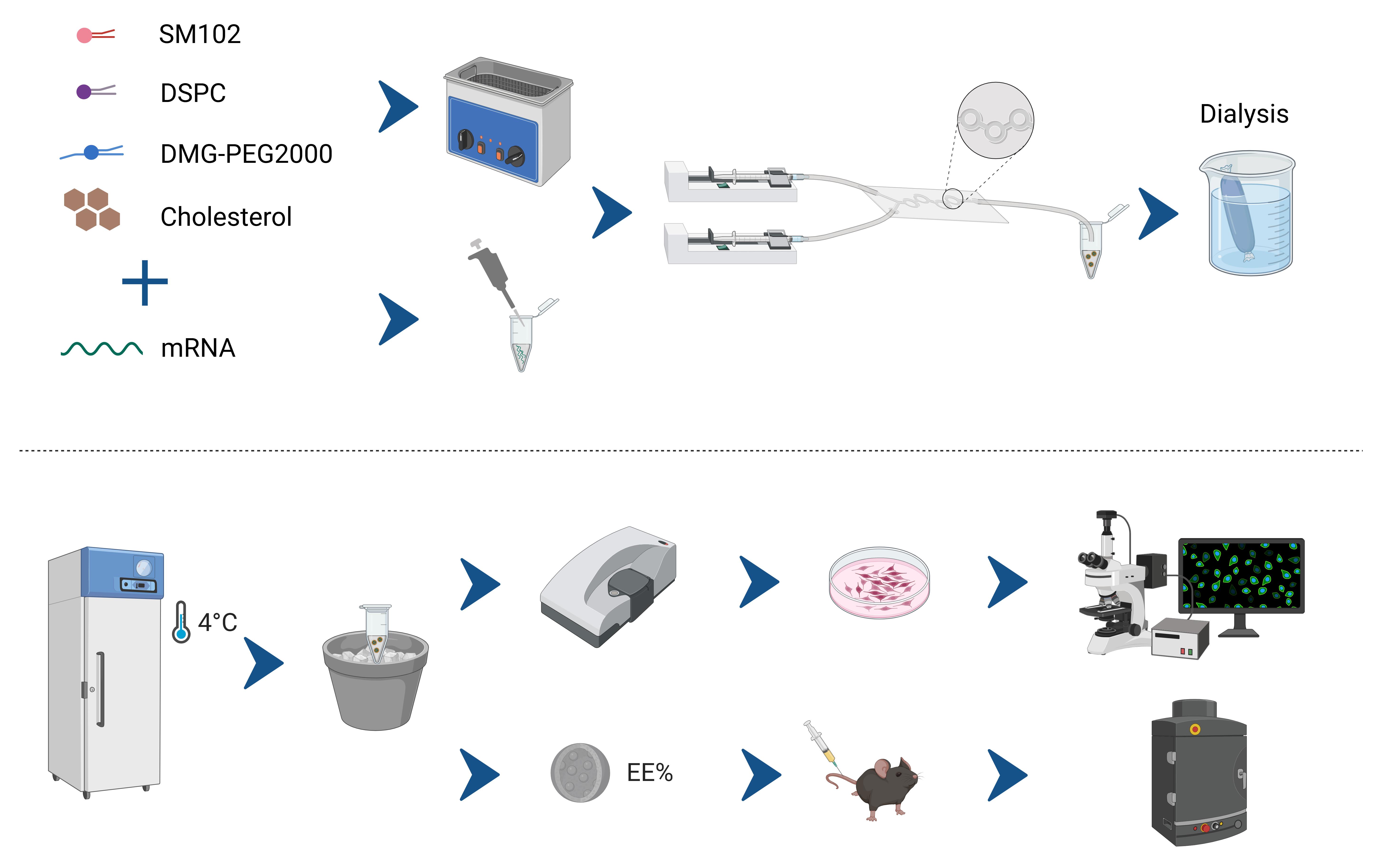
Schematic of the cost-effective microfluidic workflow for preparing mRNA-loaded lipid nanoparticles (mRNA@LNPs). The four lipids—SM102, DSPC, DMG-PEG2000, and cholesterol—are dissolved in anhydrous ethanol, while mRNA is dissolved in citrate buffer (4 °C). Both solutions are infused into a Y-junction glass microchip at a 3:1 (organic: aqueous) flow ratio. After on-chip self-assembly, the crude LNPs are dialyzed against PBS (4–6 h, 4 °C) and stored at 4 °C for up to one week. Downstream quality control includes RiboGreen-based encapsulation efficiency (EE%) determination and functional validation via in vitro and in vivo transfection assays.
Background
Lipid nanoparticles (LNPs) serve as core delivery vehicles for mRNA vaccines and gene therapy products. The scalability and economic viability of LNP production technologies govern both fundamental research and clinical applications [1]. While commercial microfluidic systems (e.g., FluidicLab LNP-S1, NanoAssemblr®) enable reproducible LNP synthesis through precision fluid control, their prohibitive costs (~$20,000–40,000 USD) and proprietary architectures pose significant constraints for academic laboratories [2]. Compared with the ~$35,000 USD price tag of commercial platforms such as the NanoAssemblr® Ignite, our complete system can be assembled for ~$4,000 USD—roughly one-tenth of the cost [3].
These limitations are most pronounced during multiparametric formulation screening and preclinical scaling, as evidenced during the COVID-19 pandemic, which exposed critical shortcomings in LNP technology for equitable vaccine development [4,5]. Current open-source alternatives (bulk mixing) reduce capital expenses but fail to achieve pharmaceutical standards, typically demonstrating compromised encapsulation efficiency (< 60%), broad size dispersity (PDI > 0.25), and inadequate batch-to-batch reproducibility [6,7].
To address these challenges, we developed a modular LNP synthesis platform using standard laboratory equipment that reengineers microfluidic principles through precision syringe pumps coupled with customized Y-junction glass chips. Compared with the commercial systems available on the market, our assembly method can also achieve efficient mRNA packaging by coordinating technical details and optimizing parameters. The encapsulation efficiency exceeds 80%, which is superior to the existing low-cost methods [8]. The prepared nanoparticles have a uniform particle size (80–120 nm, with a particle size distribution index of ≤ 0.2), and these particles have a long colloidal stability (they can remain stable for 7 days at 4 °C), which could meet WHO requirements for nucleic acid delivery systems [9,10]. This accessible platform facilitates the basic mRNA delivery system and vaccine research in laboratories with limited funds.
Materials and reagents
Biological materials
1. C57 mice, aged 8–10 weeks (SPF, Beijing Vital River Laboratory Animal Technology Co., Ltd.)
2. 293T cells (ATCC EY-X0525, China)
Reagents
1. SM102 (AVT, catalog number: O02010)
2. DSPC (AVT, catalog number: S01005)
3. Cholesterol (AVT, catalog number: O01001)
4. PEG2000-DMG (AVT, catalog number: O02005)
5. Anhydrous ethanol (Nanjing Chemical Reagent CO., Ltd, catalog number: C0691510123)
6. Firefly Luciferase (LUC) mRNA (N1-Me-Pseudo UTP, 1 mg/mL) (Vazyme, catalog number: DD4511)
7. eGFP mRNA (N1-Me-Pseudo UTP, 1 mg/mL) (Vazyme, catalog number: DD4503)
Note: If the laboratory prepares mRNA by itself, please refer to items 6 or 7 for quality control, which involves using the commercialized mRNA products from Vazyme, Jiangsu, China.
8. Citrate buffer (Absin, Shanghai, China, catalog number: abs9283)
9. 1× phosphate-buffered saline (PBS) (Servicebio, catalog number: G4250)
10. 1× DMEM basic (Thermo Fisher Scientific, catalog number: 6125052)
11. D-Luciferin potassium salt (Absin, catalog number: abs42075819)
12. Quant-iTTM RiboGreen® RNA Assay kit (Thermo Fisher Scientific, catalog number: R11490)
13. 20× TE buffer (Thermo Fisher Scientific, catalog number: T11493)
14. Triton X-100 (Beyotime, catalog number: ST797)
Solutions
1. 1× TE buffer (see Recipes)
2. 1% Triton X-100 buffer (see Recipes)
Recipes
1. 1× TE buffer
Dilute 20× TE buffer with reverse osmosis (RO) water (18.2 MΩ·cm, Millipore system, USA) to 1× concentration.
2. 1% Triton X-100 buffer
Dilute Triton X-100 with 1× TE buffer to a final concentration of 1%.
Laboratory supplies
1. Amicon® Ultra-15 centrifugal filters (MilliporeSigma, catalog number: UFC903096)
2. Slide-A-LyzerTM G2 dialysis cassettes (Thermo Fisher Scientific, catalog number: 66003)
3. 1.5 mL nuclease-free microcentrifuge tubes (Nest, catalog number: 615601)
4. 1 mL Luer-Lock syringes (Health Beauty, Shanghai, China)
5. 96-well cell culture plates (Nest, catalog number: 725031)
Caution: Ensure that all laboratory supplies are RNase-free to prevent RNA degradation.
Equipment
1. High-precision dual-syringe pump system (Wuhan Jianmi Zhikong Technology Co., Ltd, model: JSP0-02-1B)
2. Custom Y-junction glass microfluidic chip (Wuhan Jianmi Zhikong Technology Co., Ltd, model: JM LNP-B02-100/200, https://www.mifluidic.com/)
3. Chip fixture (https://www.taobao.com)
3. PTFE tubing assembly (Microfluidics & Lab-on-Chip Inc., Hubei, China)
4. Zetasizer Nano ZS (Malvern Panalytical, model: ZEN3600)
5. TEM system (Thermo Fisher Scientific, model: FEI Talos F200S G2)
6. AniView SE (Guangzhou Biolight Biotechnology Co., Ltd, Guangzhou, China)
7. Analytical balance (Jingqi Co., Ltd, model: FA2204T)
Procedure
A. Lipid phase preparation
1. Stock solution preparation: According to the required amount for the experiment, use centrifuge tubes of the appropriate size without nuclease (for example, we used 1.5 mL sterile enzyme-free centrifuge tubes, prepared 1 mL of storage solution, and divided it into 10 portions, which can be used 10 times). Weigh the lipid powder on an analytical balance and dissolve it in anhydrous ethanol according to the ratio specified in Table 1.
Caution: Store the working solution at -20 °C and use it within six months. After dispensing, you should seal the tube openings with sealing film.
Table 1. 10× Moderna LNP formulation [11] (12 mM total lipids)
| Reagent | Molar ratio (%) | Concentration (mg/L) |
| SM102 | 50 | 4261.08 |
| DSPC | 10 | 948.18 |
| Cholesterol | 38.5 | 1786.32 |
| PEG2000-DMG | 1.5 | 451.68 |
2. Dissolving and dispensing: Vortex for 30 s or until the lipids are completely dissolved in 1 mL of anhydrous ethanol. Then, aliquot the solution into ten 100 μL portions and store them at -20 °C.
Caution: Dissolve the lipids completely in anhydrous ethanol; you can mix them by pipetting or by vortexing.
3. Working solution preparation: Dilute one 1× solution with 900 μL of anhydrous ethanol to obtain the final 1× working concentration for production; thoroughly mix, use a 1-mL Luer Lock syringe with a needle to aspirate the solution, then discard the needle and place the syringe on ice until ready for use.
Caution: When using a 1-mL Luer Lock syringe with a needle for aspiration, place the angled end of the syringe downward and slowly withdraw it. At the same time, gradually tilt the microcentrifuge tube slightly to avoid forming bubbles. After the aspiration is completed, remove the needle and use the screw connection to link the syringe and the microfluidic chip. This is to prevent loosening and leakage during the operation. After the aspiration is finished, invert the syringe and gently tap it to expel the bubbles. Ensure that as few bubbles as possible are formed.
B. Aqueous phase preparation
1. Prepare mRNA working solution: Add 100 μL of 1 mg/mL eGFP mRNA stock to 900 μL of pre-chilled citrate buffer (4 °C) to obtain 1 mL of 1 mg/mL mRNA working solution. Carefully draw the solution into a 1 mL Luer-Lock syringe fitted with a needle for aspiration, then remove the needle and place the syringe on ice for immediate use.
Caution: Pre-chill the citrate buffer to 4 °C before mixing; maintaining a low temperature minimizes RNA degradation and preserves mRNA integrity.
2. Prepare a blank control: Draw 1 mL of pre-chilled citrate buffer alone into a separate 1 mL Luer-Lock syringe and keep on ice.
C. Equipment preparation
1. System assembly: Mount the dual-syringe pump on the rail and position the slider. Load two 1 mL Luer-lock syringes with anhydrous ethanol, then secure each syringe to the pump via its Luer-lock fitting (Figure 1A).
2. Chip connection: The custom Y-junction glass microfluidic chip was designed for typical LNP dimensions with two inlets and one outlet (Figure 1B, C). Cut two 15 cm lengths of silicone tubing (ID 0.76 mm, OD 1.65 mm). Connect one end of each tube to a syringe and the other end to the corresponding inlet of the reusable Y-junction glass microchip and tighten all Luer-lock fittings finger-tight to prevent leaks. Attach a 5 cm silicone tube to the chip outlet and direct it to a waste container (Figure 1D).
Caution: After each use, rinse the chip with anhydrous ethanol and fill it up to maintain its reusability.
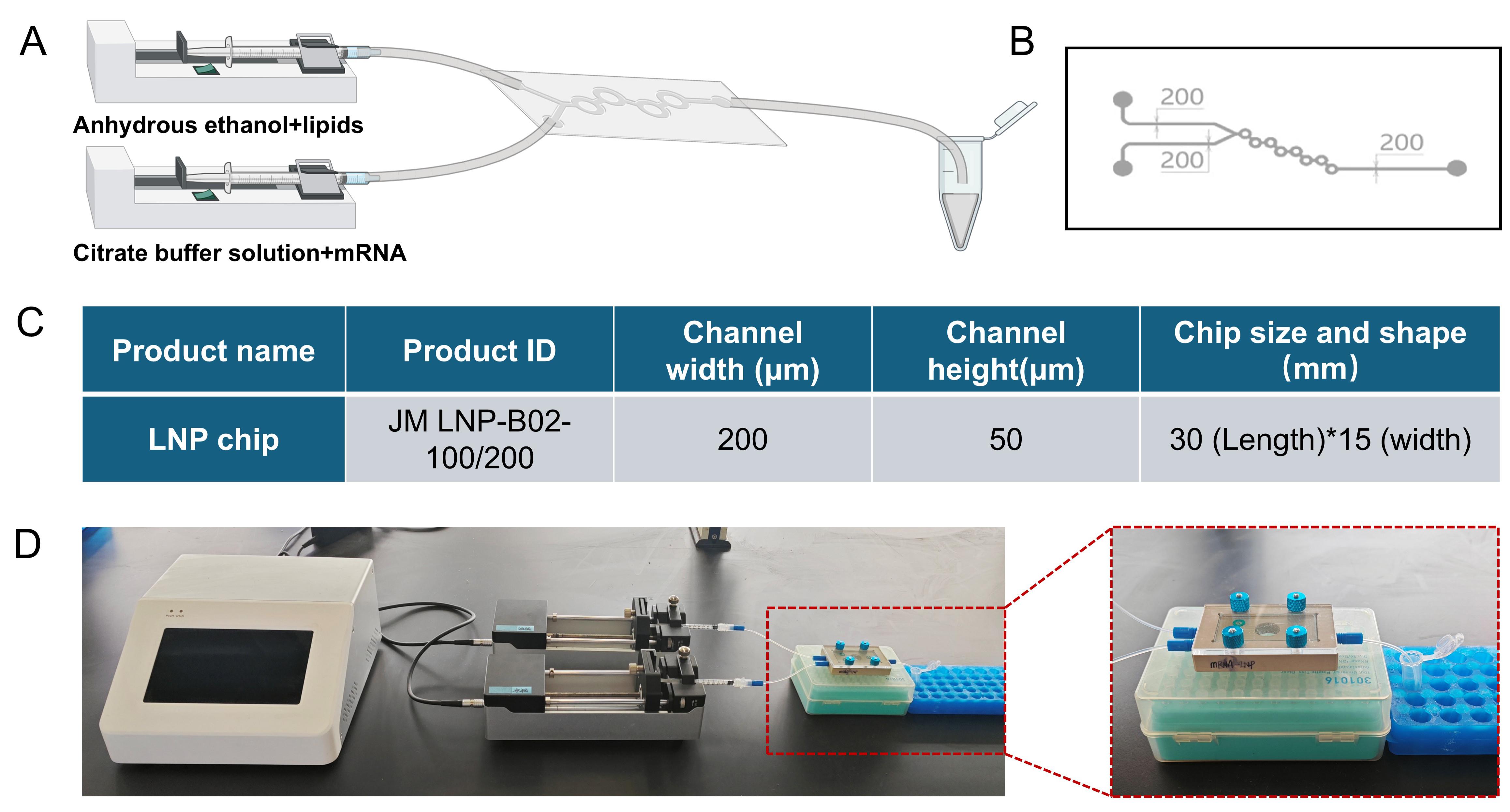
Figure 1. Comprehensive schematic of the low-cost microfluidic setup for synthesizing mRNA-loaded lipid nanoparticles (mRNA@LNPs). (A) Schematic diagram of microfluidic equipment. The lipid phase (dissolved in anhydrous ethanol) and the aqueous phase (containing mRNA dissolved in citrate buffer) are injected into the microfluidic chip and finally into the centrifuge tube. (B) Detailed design of the microchannel within the microfluidic chip. (C) Specifications of the custom Y-junction glass microfluidic chip. The table lists the product name, product ID (JM LNP-B02-100/200), channel width (200 μm), channel height (50 μm), and chip size and shape (30 mm in length and 15 mm in width). (D) The actual setup in the laboratory. Part of the chip fixture is highlighted in a red box.
3. Priming and flushing: Start the syringe pump at 1 mL/min and gently tap the chip to dislodge air bubbles. Once the channels are completely filled and bubble-free, stop the pump and discard the effluent. Repeat the flush once more.
Caution: The method of connecting the chip and the tube can be flexibly selected according to the materials available in the laboratory. Whether to use a chip fixture is not necessary either. The important point is that there should be no leakage, and the interface method should be as simple as possible for better results.
D. LNP production protocol
1. Flow parameter setup: Reset the syringe pump rail and install. Lipid phase syringe: set flow rate to 3 mL/min. Aqueous phase syringe: set flow rate to 9 mL/min. Total flow rate: 12 mL/min [3:1 organic phase (lipid and ethanol): aqueous ratio (water)].
2. Priming and collection: Start the syringe pump with the 1 mL Luer-Lock syringes securely attached via the Luer-Lock connection and rapidly tap the microfluidic chip to dislodge bubbles. Discard the initial 0.2 mL of effluent to clear system dead volume and collect the next 0.7 mL in a new 1.5 mL microcentrifuge tube.
Caution: Secure the outlet silicone tubing against the tube bottom to prevent mRNA@LNP products during chip tapping and foam formation at air–liquid interfaces, as mechanical stress can disrupt the lipid bilayer structure, reducing encapsulation efficiency and particle size uniformity, while foam can introduce air bubbles that further compromise LNP integrity and yield.
3. Post-processing: Store mRNA@LNP products at 4 °C and avoid jolts and vibrations.
4. Dialyze against PBS (4–6 h, 4 °C) with Slide-A-LyzerTM cassettes (20 kDa MWCO).
E. Quality control
E1. Encapsulation efficiency assay
1. Dilute the fluorescent reagent stock solution 200-fold with 1× TE buffer.
2. Dilute mRNA@LNP samples (100 μL) with the corresponding buffer.
3. Total mRNA group: Add 100 μL of 1× TE buffer containing 1% Triton X-100.
4. Free mRNA group: Add 100 μL of 1× TE buffer.
5. Transfer samples to a black-walled, clear-bottom 96-well plate.
6. Dilute RNA standards to generate a low-concentration-range standard curve (for free mRNA quantification). Add standards to the 96-well plate.
7. Add 100 μL of diluted fluorescent reagent to each well containing mRNA@LNP samples or RNA standards.
8. Place the 96-well plate in a microplate reader. Shake for 1 min and then incubate for 4 min.
9. Measure fluorescence intensity at 488 nm excitation/520 nm emission.
10. For detailed steps on calculating the encapsulation rate, please refer to the standard protocol of the Quant-iTTM RiboGreen® RNA Assay kit.
E2. Particle size and zeta potential analysis
1. Use a Malvern Zetasizer Nano ZS dynamic light scattering (DLS) system.
Caution: To clean the disposable cuvettes, first use RO water for ultrasonic cleaning for 5 min, then rinse with anhydrous ethanol. Finally, gently blot dry with lint-free lens paper (do not wipe).
E3. In vitro transfection of 293T cells with eGFP@LNP (lipid nanoparticles encapsulating eGFP mRNA)
1. Culture 293T cells in a 96-well plate, with a seeding density of 3 × 104 cells per well, and incubate for 48 h.
2. Aspirate the culture medium and dilute eGFP@LNP or blank LNP (lipid nanoparticles encapsulating citrate buffer) to ~1 μg per well in 100 μL of fresh medium. Add the mixture directly into the cells without any other operations.
3. Perform fluorescence microscopy imaging and measure the fluorescence intensity using a microplate reader at 24 and 48 h.
E4. In vivo transfection of C57 mice with LUC@LNP (lipid nanoparticles encapsulating LUC mRNA) [12,13]
1. Select 8–10-week-old C57 mice and divide them into groups of three for the experimental group (injected with LUC mRNA@LNP) and the control group (injected with empty LNP). Administer injections via the tail vein, with each mouse receiving 150 μL, and wait for 6 h.
2. Prepare a 15 mg/mL D-luciferin potassium salt solution using 1× PBS.
3. Turn on the in vivo imaging system and allow it to warm up for imaging.
4. Administer 200 μL of the D-luciferin potassium salt solution (recommended dose of 150 mg/kg) to the mice via intraperitoneal injection.
Caution: Catch the mice with their heads facing downward to shift the internal organs forward, preventing the injection from entering the viscera.
5. Place the injected mice in an anesthesia chamber with 2.5% isoflurane and an oxygen flow rate of 2 L/min.
Caution: Gently turn the anesthetized mice to ensure complete anesthesia.
6. After 10 min, when the bioluminescent signal stabilizes, place the mice in the imaging chamber for imaging.
Caution: Put the mice with their abdomens facing up and their heads oriented toward the isoflurane outlet to ensure they remain anesthetized.
7. After imaging, euthanize the mice by cervical dislocation and collect the heart, liver, spleen, lung, and kidney for tissue imaging.
Data analysis
During the process in which the injection pump concurrently delivers the oil phase and aqueous phase into the glass microchip, the chip is intermittently tapped. The initial waste LNP and the final collected LNP are depicted in Figure 2A. The initial waste liquid appears relatively clear and bright, with no obvious bubbles, suggesting that it contains a significant amount of residual anhydrous ethanol from the initial cleaning of the microchip. In contrast, the final collected LNP exhibits a large number of bubbles and an "oily" texture. Based on subsequent experimental observations, the presence and quantity of bubbles can serve as a rudimentary indicator of LNP quality.
As shown in Figure 2B, transmission electron microscopy (TEM) imaging reveals that the particle size of mRNA@LNP is relatively uniform, with a mean diameter of approximately 100 nm and a distinct boundary of the lipid bilayer-like structure. Figure 2C indicates that the average particle size of the empty LNP without encapsulation is 105.5 nm, while it decreases to 102.8 nm after encapsulating eGFP. Three independent transfection experiments were conducted to verify the encapsulation efficiency. Comparing the particle size and zeta potential measurements with those of LNPs obtained from microfluidic devices reported in the literature, it is evident that the quality of our low-cost encapsulation method is not compromised [14].
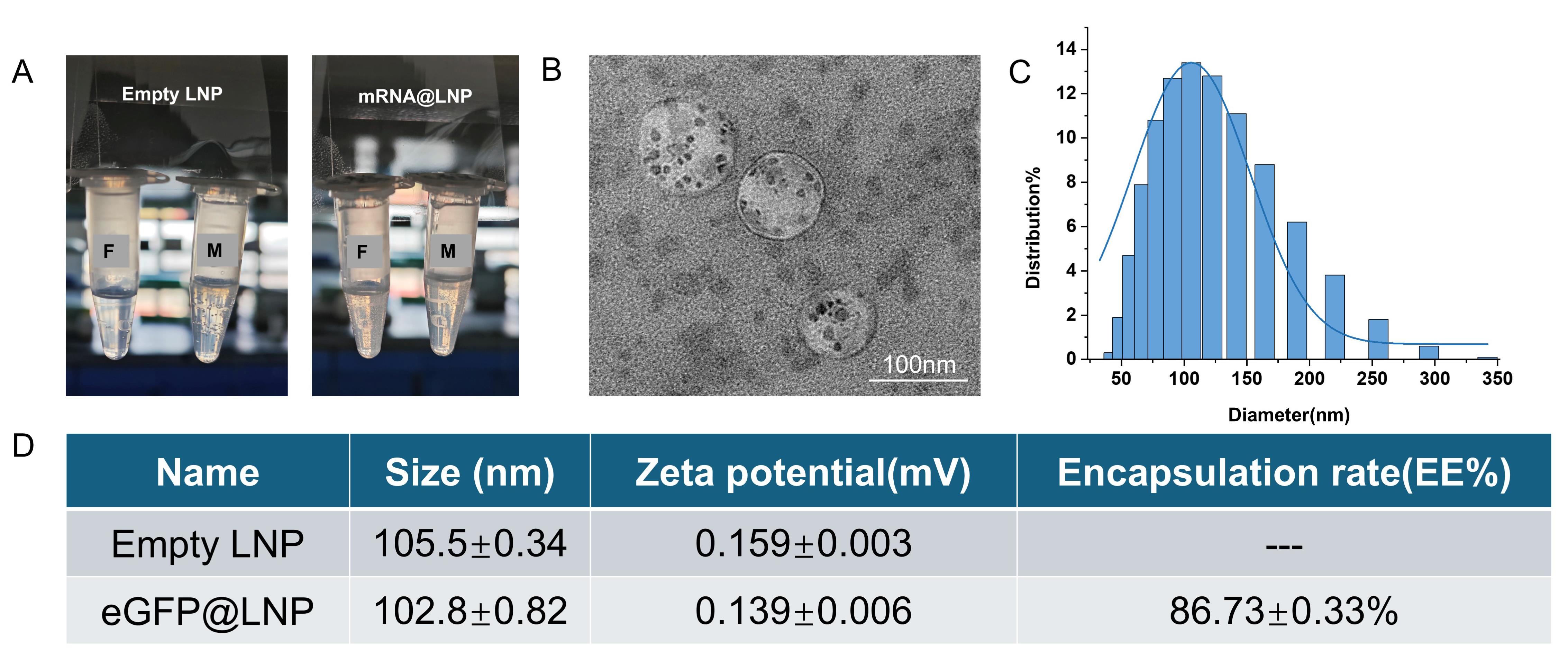
Figure 2. Characterization of mRNA@LNP. (A) Normal production of empty LNP and mRNA@LNP using a syringe pump. F indicates the initially discharged mRNA@LNP product; M indicates the middle mRNA@LNP product. For a 1 mL solution, the initial fraction (discarded when the aqueous syringe has dispensed 0.4 mL) is removed, and the middle fraction is used for experiments. The syringe pump is stopped when the aqueous phase has 0.1 mL remaining. (B) TEM imaging results of lipid nanoparticles (LNPs) after encapsulation of mRNA. Scale bar: 100 nm. (C) Particle size distribution. (D) Particle size and zeta potential measurements of empty LNP and mRNA@LNP after encapsulation. Three independent experiments were conducted, and the average value was taken.
Validation of protocol
In the in vitro experiments, 293T cells and eGFP mRNA were used for testing. LNP samples stored at 4 °C for 0, 3, and 6 days were transfected into 293T cells without any additional transfection reagents or physical methods, and their expression levels were assessed at 24 and 48 h post-transfection. As shown in Figure 3A, the mRNA exhibited robust expression across all storage time points, with no significant reduction in fluorescence intensity observed at 48 h. Meanwhile, using LUC mRNA@LNP, intravenous injection was performed, and the distribution and expression of LNP were monitored 6 h later using an in vivo imaging system. As expected, the mRNA@LNP primarily accumulated in the liver, consistent with the design of the LNP. Further verification through post-mortem tissue fluorescence detection confirmed normal mRNA expression.
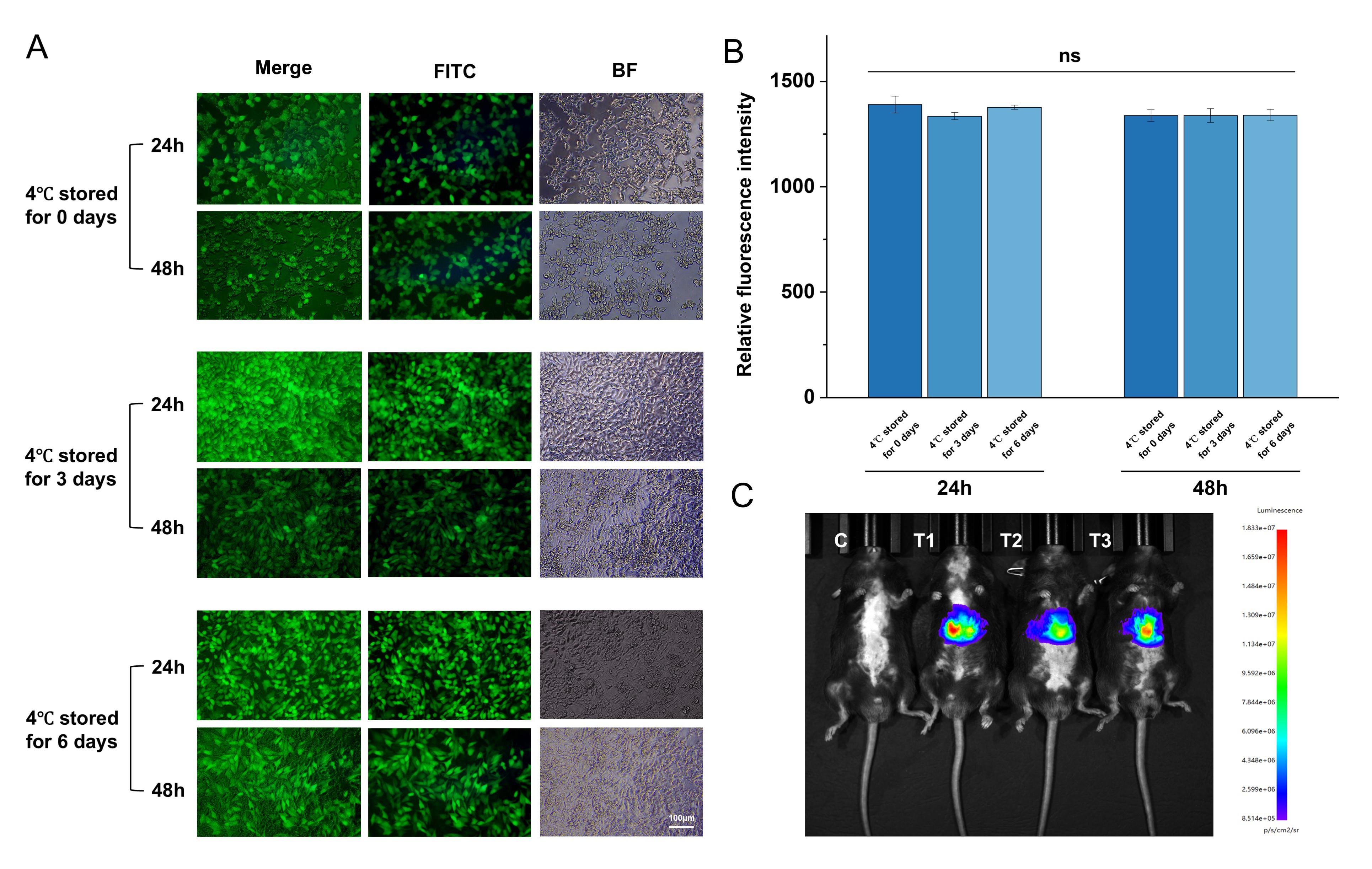
Figure 3. In vitro and in vivo transfection results of mRNA@LNP. (A) Expression efficiency of eGFP-mRNA@LNP stored at 4 °C for 0, 3, and 6 days, assessed by eGFP expression in 293T cells at 24 and 48 h. Scale bar: 100 μm. (B) Relative fluorescence intensity. Quantified eGFP expression (Statistical analysis: t-test, mean fluorescence intensity ± SD, n = 3) remained stable over 0–6 days of storage at 4 °C. (C) In vivo expression of LUC-mRNA@LNP after intravenous injection into mice, evaluated 6 h post-injection. C is control, T1, T2, and T3 are the test groups.
General notes and troubleshooting
General notes
1. Temperature control in winter: Pay attention to the temperature of lipids and citrate buffer, especially in winter. Both phases should be equilibrated to room temperature (25 °C) for optimal results. Elevated temperatures during the preparation process can lead to significant mRNA degradation, while lower temperatures may result in incomplete dissolution of phospholipids, thereby reducing encapsulation efficiency and compromising the final outcome.
2. Storage of mRNA@LNP: Immediately store the prepared mRNA@LNP at 4 °C (or place it on ice). Do not store mRNA@LNP at -20 °C or in metal ice buckets. Sudden temperature changes can cause the LNP to swell and rupture, leading to failure in subsequent experiments.
3. All mRNA handling, LNP formulation, and chip priming steps should be carried out on ice or in a 4 °C cold room using RNase-free consumables.
Troubleshooting
1. Appearance of mRNA@LNP solution: If the mRNA@LNP solution appears turbid during preparation, the experiment is likely to have failed. It is recommended to identify the cause and not proceed further. However, if the solution shows stratification with a clear lower layer and white foam on the surface, the foam can be gently removed with a pipette tip without affecting subsequent experiments. Avoid disturbing the system after preparation.
2. In vivo imaging: If strong autofluorescence from the mouse fur interferes with imaging, depilate the mouse before imaging to reduce background fluorescence.
3. Tail vein injection: During tail vein injection, the injection speed should be slow. Rapid injection or using cold liquid can cause sudden death in mice.
4. Inconsistent size/PDI: Recalibrate flow rates; ensure tubing connections are tight; check that anhydrous ethanol is fresh and lipid stock is fully dissolved.
5. Low encapsulation efficiency: Increase lipid/mRNA ratio or verify RNA integrity on agarose gel.
6. Chip channel blockage: Flush immediately with anhydrous ethanol followed by RO water; sonicate for 5 min if necessary.
Acknowledgments
This study was funded by the National Natural Science Foundation of China (No. 8247010744), National Key Research Project of MOST (2023YFA0915000), and Wenzhou Institute of the University of Chinese Academy of Sciences (WIUCASQD2022020).
The following figures were created using BioRender: Graphical overview, Created in BioRender. Li, C. (2025) https://BioRender.com/5wixvam.
Ethical considerations
This protocol for animal care and use was approved by the Wenzhou Institute, University of Chinese Academy of Sciences (Wenzhou Institute of Biomaterials & Engineering) (WIUCAS24030512).
Competing interests
The authors declare that they have no competing interests.
References
- Niazi, S. K. and Magoola, M. (2024). Advancing Therapeutic and Vaccine Proteins: Switching from Recombinant to Ribosomal Delivery—A Humanitarian Cause. Int J Mol Sci. 25(23): 12797. https://doi.org/10.3390/ijms252312797
- Vogelaar, A., Marcotte, S., Cheng, J., Oluoch, B. and Zaro, J. (2023). Use of Microfluidics to Prepare Lipid-Based Nanocarriers. Pharmaceutics. 15(4): 1053. https://doi.org/10.3390/pharmaceutics15041053
- Morbioli, G. G., Speller, N. C., Cato, M. E. and Stockton, A. M. (2021). An automated low-cost modular hardware and software platform for versatile programmable microfluidic device testing and development. Sens Actuators, B. 346: 130538. https://doi.org/10.1016/j.snb.2021.130538
- Fang, E., Liu, X., Li, M., Zhang, Z., Song, L., Zhu, B., Wu, X., Liu, J., Zhao, D., Li, Y., et al. (2022). Advances in COVID-19 mRNA vaccine development. Signal Transduction Targeted Ther. 7(1): 94. https://doi.org/10.1038/s41392-022-00950-y
- Labouta, H. I., Langer, R., Cullis, P. R., Merkel, O. M., Prausnitz, M. R., Gomaa, Y., Nogueira, S. S. and Kumeria, T. (2022). Role of drug delivery technologies in the success of COVID-19 vaccines: a perspective. Drug Deliv Transl Res. 12(11): 2581–2588. https://doi.org/10.1007/s13346-022-01146-1
- Suwanpitak, K., Huanbutta, K., Weeranoppanant, N., Sriamornsak, P., Panpipat, C. and Sangnim, T. (2024). Optimization of Lipid-Based Nanoparticles Formulation Loaded with Biological Product Using A Novel Design Vortex Tube Reactor via Flow Chemistry. Int J Nanomed. 19: 8729–8750. https://doi.org/10.2147/ijn.s474775
- Lindsay, S., Hussain, M., Binici, B. and Perrie, Y. (2025). Exploring the Challenges of Lipid Nanoparticle Development: The In Vitro–In Vivo Correlation Gap. Vaccines. 13(4): 339. https://doi.org/10.3390/vaccines13040339
- Khairnar, S. V., Pagare, P., Thakre, A., Nambiar, A. R., Junnuthula, V., Abraham, M. C., Kolimi, P., Nyavanandi, D. and Dyawanapelly, S. (2022). Review on the Scale-Up Methods for the Preparation of Solid Lipid Nanoparticles. Pharmaceutics. 14(9): 1886. https://doi.org/10.3390/pharmaceutics14091886
- Knezevic, I. (2009). Stability evaluation of vaccines: WHO approach. Biologicals. 37(6): 357–359. https://doi.org/10.1016/j.biologicals.2009.08.004
- Markova, N., Cairns, S., Jankevics-Jones, H., Kaszuba, M., Caputo, F. and Parot, J. (2021). Biophysical Characterization of Viral and Lipid-Based Vectors for Vaccines and Therapeutics with Light Scattering and Calorimetric Techniques. Vaccines. 10(1): 49. https://doi.org/10.3390/vaccines10010049
- Zhang, X., Li, Y. and Zhou, Z. (2024). Lipid Nanoparticle-Based Delivery System—A Competing Place for mRNA Vaccines. ACS Omega. 9(6): 6219–6234. https://doi.org/10.1021/acsomega.3c08353
- El-Mayta, R., Padilla, M. S., Billingsley, M. M., Han, X. and Mitchell, M. J. (2023). Testing the In Vitro and In Vivo Efficiency of mRNA-Lipid Nanoparticles Formulated by Microfluidic Mixing. J Visualized Exp. 191: e3791/64810. https://doi.org/10.3791/64810
- Fei, Y., Yu, X., Liu, P., Ren, H., Wei, T. and Cheng, Q. (2024). Simplified Lipid Nanoparticles for Tissue‐ And Cell‐Targeted mRNA Delivery Facilitate Precision Tumor Therapy in a Lung Metastasis Mouse Model. Adv Mater. 36(48): e202409812. https://doi.org/10.1002/adma.202409812
- Li, M., Guo, N., Song, G., Huang, Y., Wang, L., Zhang, Y. and Wang, T. (2023). Type II Toxin–Antitoxin Systems in Pseudomonas aeruginosa. Toxins. 15(2): 164. https://doi.org/10.3390/toxins15020164
Article Information
Publication history
Received: Jun 17, 2025
Accepted: Aug 12, 2025
Available online: Sep 3, 2025
Published: Sep 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Li, Y., Wu, M. and Zhao, R. (2025). Cost-Effective and Reproducible Preparation of mRNA-Loaded Lipid Nanoparticles Using a Conventional Laboratory-Scale Microfluidic Assembly System. Bio-protocol 15(18): e5458. DOI: 10.21769/BioProtoc.5458.
Category
Molecular Biology > Nanoparticle > Plan-derived nanoparticles
Biochemistry > Lipid > Lipid isolation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.