- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CRISPR/Cas9-Induced Targeted Mutagenesis of the Moss Physcomitrium patens by Particle Bombardment-Mediated Transformation
Published: Vol 15, Iss 18, Sep 20, 2025 DOI: 10.21769/BioProtoc.5452 Views: 1231
Reviewed by: Diarmuid Seosamh Ó’MaoiléidighAnonymous reviewer(s)
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) system is a widely used programmable nuclease system for gene modification in many organisms, including Physcomitrium patens. P. patens is a model species of moss plants, a basal land plant group, which has been extensively studied from the viewpoint of evolution and diversity of green plant lineages. So far, gene modifications by CRISPR/Cas9 in P. patens have been carried out exclusively by the polyethylene glycol (PEG)-mediated DNA transfer method, in which a transgene (or transgenes) is introduced into protoplast cells prepared from protonemal tissues. However, this PEG-mediated method requires a relatively large amount of transgene DNA (typically 30 µg for a single transformation), consists of many steps, and is time-consuming. Additionally, this PEG-mediated method has only been established in a few species of moss. In the current protocol, we succeeded in CRISPR/Cas9-induced targeted mutagenesis of P. patens genes by making good use of the biolistic method, which i) requires amounts of transgene DNA as low as 5 μg for each vector, ii) consists of fewer steps and is time-saving, and iii) is known to be applicable to a wide variety of species of plants.
Key features
• In this protocol, particle bombardment-mediated gene transfer is used for CRISPR/Cas9-induced mutagenesis in the moss Physcomitrium patens.
• By this application of particle bombardment, a gene can be modified by the CRISPR/Cas9 system much more conveniently with a smaller amount of transgene DNA.
• This protocol is expected to be easily applicable to non-model moss species, some of which have noteworthy traits, such as tolerance to various stresses.
Keywords: Clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) systemBackground
Physcomitrium patens (Physcomitrella patens) is a model species of moss (Bryopsida) that offers many advantages for experimentation, including applicability of gene targeting based on a highly efficient homologous recombination (HR) [1,2]. Bryopsida is a basal land plant group, phylogenetically positioned between green algae and higher land plant groups [3]. Therefore, P. patens has been extensively studied in the research areas of development, growth, and physiology, largely from the evolutionary point of view [1,4,5]. Gene modification by clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) system was also successfully applied to this moss [6–10], enabling easier disruption of multiple genes and creation of marker-free knockouts and knock-ins, among other advantages. For applying the CRISPR/Cas9 system to P. patens, the polyethylene glycol (PEG)-mediated DNA transfer method has been exclusively used for the introduction of sgRNA and Cas9 expression vectors into cells. However, this PEG-mediated method has the following disadvantages: i) it requires higher amounts of DNA (typically 30 μg per sample) to be introduced into cells; ii) it consists of many steps, including generation of protoplasts from protonema cells, and is time-consuming; iii) sometimes it results in, if at a small ratio, generation of polyploid cells by protoplast fusion [11]. On the other hand, the particle bombardment-mediated DNA transfer into cells requires a much lower amount of DNA (~5 µg for each plasmid) and fewer steps with no need to prepare protoplasts, is time-saving, and involves no cell fusion processes [12,13]. Here, we have established a protocol for modifying P. patens genes using the CRISPR-Cas9 system with particle bombardment-mediated DNA transfer. By using this protocol, the CRISPR/Cas9 system will be more readily applicable not only to P. patens but also to other non-model moss species with interesting traits such as tolerance to various environmental stresses.
Materials and reagents
Biological materials
1. Escherichia coli DH5α (Thermo Fisher Scientific, catalog number: EC0112)
2. Moss [Physcomitrium patens (Physcomitrella patens) ssp. patens] [14] (a kind gift from Dr. Mitsuyasu Hasebe, National Institute for Basic Biology, Okazaki)
Note: The culture methods for P. patens are based on the protocols of PhyscoBase (https://moss.nibb.ac.jp/), developed by Dr. M. Hasebe’s laboratory, with some modifications.
1. Plasmids
a. pSCgRNA (sgRNA expression vector) [7,8]
b. pSCOE1-fcoCas9 (Cas9 nuclease expression vector) [7,8]
2. DNA oligos
a. DNA oligonucleotides for constructing sgRNA vectors (the adapter sequences for cloning in the BbsI-cleaved pSCgRNA are underlined)
i. PPR2_crRNA_Fwd2 (5'-AAACTAGGCAAGTTCGGTCAACTC-3')
ii. PPR2_crRNA_Rv2 (5'-TCTGGAGTTGACCGAACTTGCCTA-3')
iii. PPR38_crRNA_fw2 (5'-TCTGTTAGGAGGTCTAACTACGGG-3')
iv. PPR38_crRNA_rv2 (5'-AAACCCCGTAGTTAGACCTCCTAA-3'
Note: These DNA oligonucleotides were purified by reverse-phase column chromatography and purchased from FASMAC.
3. Primers for genotyping of P. patens clones
a. PpPPR2_cr2_seq_fw (5'-CATCTAGTCGTGCCTCATGAGGA-3')
b. PpPPR2_cr2_seq_rv (5'-TCCATTTGCTGAAGCAAGAGTGG-3')
c. PPR38_cr2_seq_fw2 (5'-GCTGCAATGGAGCCCTGTG-3')
d. PPR38_cr2_seq_rv2 (5'-CGTCGCTTGAACAATGGTGAAGG-3')
Reagents
1. 1.0 μm gold microcarriers (Bio-Rad, catalog number: 1652263)
2. 99.5% ethanol (FUJIFILM Wako Pure Chemical Corporation, catalog number: 057-00456)
3. Glycerol (FUJIFILM Wako Pure Chemical Corporation, catalog number: 075-00616)
4. Spermidine (FUJIFILM Wako Pure Chemical Corporation, catalog number:191-13831)
5. Agar (Sigma, catalog number: A6924)
6. Agar (FUJIFILM Wako Pure Chemical Corporation, catalog number: 016-11875)
Note: Use agar from Sigma and FUJIFILM Wako Pure Chemical for culturing moss and E. coli cells, respectively.
7. G418 (G418 sulfate, also known as geneticin) (FUJIFILM Wako Pure Chemical Corporation, catalog number: 078-05961)
8. D-Glucose (FUJIFILM Wako Pure Chemical Corporation, catalog number: 049-31165)
9. MgSO4·7H2O (FUJIFILM Wako Pure Chemical Corporation, catalog number: 131-00405)
10. MgCl2·6H2O (FUJIFILM Wako Pure Chemical Corporation, catalog number: 135-00165)
11. KH2PO4 (FUJIFILM Wako Pure Chemical Corporation, catalog number: 169-04245)
12. KNO3 (FUJIFILM Wako Pure Chemical Corporation, catalog number: 160-04035)
13. FeSO4·7H2O (FUJIFILM Wako Pure Chemical Corporation, catalog number: 098-01085)
14. CuSO4·5H2O (FUJIFILM Wako Pure Chemical Corporation, catalog number: 039-04412)
15. H3BO3 (FUJIFILM Wako Pure Chemical Corporation, catalog number: 027-02192)
16. CoCl2·6H2O (FUJIFILM Wako Pure Chemical Corporation, catalog number: 036-03682)
17. Na2MoO4·2H2O (FUJIFILM Wako Pure Chemical Corporation, catalog number: 196-02472)
18. ZnSO4·7H2O (FUJIFILM Wako Pure Chemical Corporation, catalog number: 264-00402)
19. MnCl2·4H2O (FUJIFILM Wako Pure Chemical Corporation, catalog number: 139-00722)
20. KI (FUJIFILM Wako Pure Chemical Corporation, catalog number: 164-03972)
21. NaCl (FUJIFILM Wako Pure Chemical Corporation, catalog number: 191-01665)
22. Ammonium tartrate (FUJIFILM Wako Pure Chemical Corporation, catalog number: 195042)
23. CaCl2·2H2O (FUJIFILM Wako Pure Chemical Corporation, catalog number: 033-25035)
24. 2-amino-2-hydroxymethyl-1,3-propanediol (FUJIFILM Wako Pure Chemical Corporation, catalog number: 011-20095)
25. Bacto tryptone (ThermoFisher Scientific, catalog number: 211705)
26. Bacto yeast extract (ThermoFisher Scientific, catalog number: 212750)
27. T4 polynucleotide kinase (Takara Bio, catalog number: 2021S)
28. T4 DNA ligase (Nippon Gene, catalog number: 311-00404)
29. BpiI (BbsI) (ThermoFisher Scientific, catalog number: ER1011)
30. NucleoSpin Gel and PCR Clean-Up kit (Macherey-Nagel, catalog number: 740609.50)
31. BigDye Terminator v3.1 Cycle Sequencing kit (ThermoFisher Scientific, catalog number: 4337455)
32. SupreDye Cycle Sequencing kit v3.1 (M&S TechnoSystems, catalog number: 063002)
33. NucleoBond Xtra Midi kit (Macherey-Nagel, catalog number: 740410.50)
Solutions
1. Stock solutions for culture media: Stock solution B (×100), stock solution C (×100), stock solution D (×100), and alternative TES solution (×1000) (see Recipes)
2. Culture media for moss: BCDG agar medium, BCDAT agar medium, BCDATG agar medium, and BCDATG (G418+) agar medium (see Recipes)
3. Culture media for E. coli: LB liquid medium and LB agar plates (see Recipes)
4. Chemicals: 500 mM ammonium tartrate solution, 50 mM CaCl2 solution, 50% (v/v) glycerol solution, 1.0 M Tris-HCl (pH 7.4), and 1.0 M MgCl2 (see Recipes)
5. Reaction buffer (5× annealing buffer) (see Recipes)
Recipes
1. Stock solutions for culture media
Notes: These stock solutions and culture media for the growth of P. patens are based on the above-mentioned PhyscoBase. In the PhyscoBase protocols, there is also “stock solution A,” which is used for the PEG-mediated transformation method.
Stock solution B (100×): 25 g/L MgSO4·7H2O
Stock solution C (100×): 25 g/L KH2PO4 (pH adjusted to 6.5 with 4 M KOH)
Stock solution D (100×): 101 g/L KNO3, 1.25 g/L FeSO4·7H2O
Alternative TES solution (1,000×): 55 mg/L CuSO4·5H2O, 614 mg/L H3BO3, 55 mg/L CoCl2·6H2O, 25 mg/L Na2MoO4·2H2O, 55 mg/L ZnSO4·7H2O, 389 mg/L MnCl2·4H2O, 28 mg/L KI
2. Culture media for moss
BCDG agar medium: 900 mL of purified water, 10 mL of stock solution B, 10 mL of stock solution C, 10 mL of stock solution D, 1 mL of alternative TES solution, 20 mL of 50 mM CaCl2 solution, 10 g of D-Glucose, and 8 g of agar. Fill up to 1,000 mL with water. Autoclave at 121 °C for 20 min and pour into 9.0-cm diameter plastic plates to solidify.
BCDAT agar medium: 900 mL of purified water, 10 mL of stock solution B, 10 mL of stock solution C, 10 mL of stock solution D, 1 mL of alternative TES solution, 20 mL of 50 mM CaCl2 solution, 10 mL of ammonium tartrate solution (500 mM), and 8 g of agar. Fill up to 1,000 mL with water. Autoclave at 121 °C for 20 min and pour into 9.0-cm diameter plastic plates to solidify.
BCDATG agar medium: 10 g/L D-Glucose supplemented to the BCDAT agar medium.
BCDATG (G418+) agar medium: G418 (20 mg/L) supplemented to the BCDATG agar medium.
Note: Use 10 g/L instead of 5 g/L of D-glucose in the original protocol in PhyscoBase.
3. Culture media for E. coli
LB liquid medium: 900 mL of purified water, 10 g of Bacto tryptone, 5 g of Bacto yeast extract, and 10 g of NaCl. Fill up to 1,000 mL with purified water and autoclave at 121 °C for 20 min.
LB agar plates: Supplement LB liquid medium with 12 g of agar (before autoclaving); then, autoclave at 121 °C for 20 min and pour into 9.0-cm diameter plastic plates to solidify.
Note: For agar, use the Sigma and FUJIFILM Wako Pure Chemical products for culturing moss and E. coli cells, respectively.
4. Chemicals
500 mM ammonium tartrate solution: 92.05 g/L ammonium tartrate.
50 mM CaCl2 solution: 7.35 g/L CaCl2.
50% (v/v) glycerol solution: mix equal volumes of sterile purified water and glycerol.
1.0 M Tris-HCl (pH 7.4): 121.1 g/L 2-amino-2-hydroxymethyl-1,3-propanediol; adjust pH to 7.4 with HCl.
1.0 M MgCl2: 203.3 g/L MgCl2·6H2O.
5. Reaction buffer (5× annealing buffer)
50 mL of 1.0 M Tris-HCl (pH 7.4), 35 mL of 1.0 M MgCl2, and 15 mL of purified water.
Laboratory supplies
1. Petri dishes (90 mm × 15 mm) (Rikaken Holdings, catalog number: LT-DS-9015)
2. 1.5 mL microtubes (Watson, catalog number: 131-815C)
3. 0.2 mL 8-strip PCR tubes (SSIbio, catalog number: 3240-00)
4. 15 mL plastic pipette (As One, catalog number: 1-2247-14)
5. Cellophane sheets (Futamura Chemical, catalog number: PL300)
6. 10 μL plastic pipette tips (BMBio, catalog number: 104-Q)
7. 200 μL plastic pipette tips (Watson, catalog number: 110-705C)
8. 1,000 μL plastic pipette tips (Watson, catalog number: 110-706C)
9. Micropore surgical tape (3M, catalog number: 1530-0)
10. Fluorescent lamps (TOSHIBA LIGHTING & TECHNOLOGY CORPORATION, catalog number: FL20SS W/18)
Equipment
1. Plant growth chamber (SANYO, catalog number: MLR-350)
2. Micropipettes (GILSON, catalog number: P2L, P20L, P200L, P1000L)
3. Tweezers (KOWA Forceps Industry, catalog number: K-17)
4. High-speed refrigerated micro-centrifuges (TOMY SEIKO, catalog number: MX-107)
5. Clean bench (Nippon Medical & Chemical Instruments, catalog number: VSF-850A)
6. Thermal cycler (TAKARA Bio, catalog number: TP-600)
7. Block incubator (ASTEC, catalog number: BI-515)
8. Autoclave (TOMY SEIKO, catalog number: BS-245)
9. Agarose gel electrophoresis tanks (TAKARA Bio, catalog number: M-2P)
10. Ultraviolet transilluminator (ATTO, catalog number: AE-6932)
11. Particle delivery system (Tanaka, catalog number: GIE-III)
12. Homogenizer (Microtec nition, catalog number: NS-310E)
Software and datasets
1. SnapGene version 5.1.7 (released July 20, 2020; a subscription license is required to use this software)
2. CRISPR-P 2.0 (released October 2016; http://crispr.hzau.edu.cn/CRISPR2/) [15]
3. CRISPRdirect version 140413 (https://crispr.dbcls.jp/) [16]
4. PPR version 6719c98 (https://ppr.plantenergy.uwa.edu.au/) [17]
Procedure
A. Construction of sgRNA expression vector
1. Prepare the DNA fragments for the target sequence.
a. Select the target sequence for CRISPR RNA (crRNA) as previously described [7,8] by using the software CRISPR-P 2.0 and CRISPRdirect.
b. Design the sense and antisense oligonucleotides so that they form, when they hybridize with each other, double-stranded DNA fragments that contain the target sequence and have adapter sequences for cloning (see “DNA oligos” above) protruding at the 5′ end of each oligonucleotide.
c. Prepare the following mixture in a 1.5 mL microtube:
i. Sense oligonucleotide (100 pmol/µL): 20 μL (2,000 pmol in total)
ii. Antisense oligonucleotide (100 pmol/µL): 20 μL (2,000 pmol in total)
iii. 5× annealing buffer: 10 μL
iv. Add sterile purified water until the total volume reaches 50 μL.
d. Heat the mixture at 95 °C for 5 min using the block incubator.
e. Cool the mixture at room temperature (24–26 °C for 1 h, resulting in the insert DNA solution (40 pmol/µL).
f. Prepare the following mixture in a 1.5 mL microtube for phosphorylation of the insert DNA fragments:
Note: This phosphorylation reaction can be omitted, but we include this step in order to maximize cloning efficiency.
i. Insert DNA fragment solution: 1.25 μL (50 pmol)
ii. 10× T4 polynucleotide kinase buffer: 5.0 μL
iii. ATP (100 mM): 0.5 μL
iv. T4 polynucleotide kinase (10 U/µL): 1.5 μL
v. Add sterile water until the total volume reaches 50 μL.
g. Mix by tapping and centrifuge at 2,000× g for 2 s.
h. Incubate at 37 °C for 30 min, resulting in the phosphorylated insert DNA solution.
i. Heat the mixture at 75 °C for 10 min.
2. Prepare the restriction enzyme-cleaved sgRNA expression vector.
a. Prepare the following mixture in a 1.5 mL microtube for the restriction enzyme reaction.
i. pSCgRNA: 3.0 µg
ii. 10× G buffer: 2.0 μL
iii. BpiI (BbsI) (10 U/µL): 2.0 μL
iv. Add sterile purified water until the total volume reaches 20 μL.
b. Incubate at 37 °C for 5 h.
c. Separate DNA fragments by agarose gel electrophoresis.
d. Excise a gel block containing the cleaved pSCgRNA (3.7 kb).
e. Extract DNA from the gel block using the NucleoSpin Gel and PCR Clean-Up kit (Macherey-Nagel, catalog number: 740609.50).
3. Cloning of the insert DNA fragments into pSCgRNA vector.
a. Prepare the following mixture in a 1.5 mL microtube for the ligation reaction.
i. BpiI (BbsI)-cleaved pSCgRNA: 50 ng
ii. Phosphorylated insert DNA solution: 2.2 μL (2.2 pmol)
iii. 10× ligation buffer: 2.0 μL
iv. T4 DNA ligase: 1.0 μL (500 U/µL)
v. Add sterile purified water until the total volume reaches 20 μL.
b. Incubate at 16 °C for 16 h.
c. Use 5 μL of the ligation reaction mixture for transformation of DH5α-competent cells.
d. Spread the DH5α-competent cells on LB agar plates and incubate the plates at 37 °C for 14–16 h.
e. Pick up colonies and grow each clone in 2 mL of LB liquid medium by shaking at 170 rpm at 37 °C for 14–16 h.
f. Extract plasmid DNA from each clone by using NucleoSpin Plasmid EasyPure.
g. Select the clone that carries the target sequence by sequencing using BigDye Terminator v3.1 Cycle Sequencing Kit or SupreDye Cycle Sequencing Kit v3.1.
B. Transformation of moss cells by particle bombardment
1. Purify sgRNA expression vector (pSCgRNA that carries the target sequence) and Cas9 expression vector (pSCOE1-fcoCas9) [7,8] using NucleoBond Xtra Midi Kit, resulting in DNA solutions (2~3 µg/µL) for transformation of moss cells.
2. Prepare the DNA-coated gold particle suspension for transformation.
a. Prepare the following mixture in a 1.5 mL microtube:
i. 60 mg of gold particles (1.0 µm diameter)
ii. 1 mL of 70% EtOH
b. Vortex vigorously for 5 min.
c. Allow to stand for 15 min at room temperature.
d. Centrifuge at 2,000× g for 2 s and discard the supernatant.
e. Add 1 mL of sterile purified water, vortex vigorously for 1 min, and allow to stand at room temperature for 1 min.
f. Centrifuge at 2,000× g for 2 s and discard the supernatant.
g. Repeat steps B2e–f three additional times.
h. Add 1 mL of 50% (v/v) glycerol solution and vortex vigorously until the gold particles are uniformly suspended (for ~10 s).
Pause point: The resulting solution can be stored at -80 °C.
3. Coating of gold particles with plasmid DNAs for transformation.
a. Prepare the mixture of the gold particle suspension and DNA by adding the following items in the given order:
i. 25 μL of gold particles suspended in 50% (v/v) glycerol solution
ii. 5 µg of each plasmid DNA
iii. 25 μL of 2.5 M CaCl2 solution
iv. 1 μL of 1 M spermidine
v. Add sterile purified water until the total volume reaches 80 μL.
b. Vortex vigorously for 3 min and allow to stand at room temperature (24–26 °C) for 1 min.
c. Centrifuge at 1,900× g for 2 s at 25 °C and discard the supernatant.
d. Add 200 μL of 70% EtOH and suspend by pipetting.
e. Centrifuge at 1,900× g for 2 s at 4 °C and discard the supernatant.
Note: Be careful not to remove gold particles when removing supernatant.
f. Add 200 μL of 99.5% EtOH and suspend by pipetting.
g. Centrifuge at 1,900× g for 2 s at 4 °C and discard the supernatant.
h. Add 30 μL of 99.5% EtOH and suspend by pipetting.
i. Chill on ice.
4. Preparation of moss cells for particle bombardment (Figure 1A).
a. Grow protonema cells on BCDAT or BCDATG agar plates that were laid with cellophane sheets in continuous white light (irradiated by white fluorescent lamps at a fluence rate of 45 μmol m-2·s-1), collect them, grind with a homogenizer (NS-310E), and apply to new agar plates (with cellophane sheets) every 4–7 days.
Note: If the homogenizer (or a similar device) is not available, a mortar and pestle can be used instead to grind the protonemal tissue.
b. Apply the ground protonemata onto BCDG agar plates with no cellophane sheets for the last subculture.
c. Use the protonema cells that have grown for 5 days to form a thick lawn on the BCDG agar plates for particle bombardment.
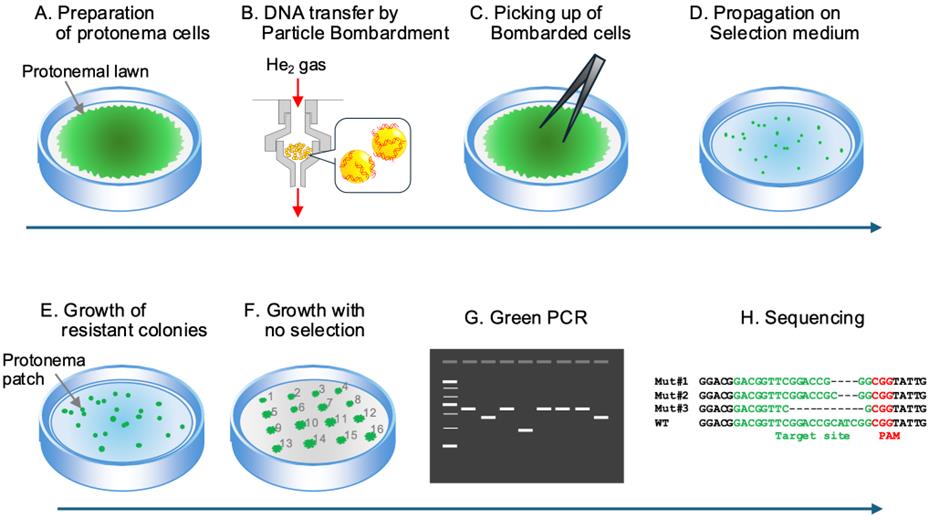
Figure 1. Overview of the protocol for CRISPR/Cas9 mutagenesis of P. patens by particle bombardment-mediated transformation. Proceed with steps from A to H. (A) First, culture protonema cells on agar plates to form the protonemal lawn. (B) Next, bombard the gold particles coated with the sgRNA and CAS9 vector DNAs onto the protonemal lawn. (C) Collect the bombarded portions, grind them into small tissue fragments, and (D) propagate on selection medium. (E) When small patches of resistant cells appear, transfer each of them onto agar plates with no antibiotics. (F) Once colonies with a diameter of 5–10 mm have formed, (G) amplify the target genomic region by green PCR and (H) use the amplified DNA fragments for genotyping by sequencing.
5. Introduction of DNA into the moss protonema cells (Figure 1B).
Note: We use the particle inflow gun system IDERA GIE-III for particle bombardment. Appropriate optimization of conditions is required if other systems are used.
a. Dry a DNA cartridge in the clean bench after sterilizing by immersing it in 70% EtOH.
b. Add 10 μL of the DNA-coated gold particle suspension into the well of the DNA cartridge.
c. Set the DNA cartridge and the agar plate with the protonemal lawn in the barotolerant chamber so that the distance between the cartridge muzzle and the agar surface is ~75 mm.
d. Set the helium exit pressure and the vacuum pressure in the barotolerant chamber as 4.0 kgf/cm2 and 600 mmHg, respectively.
e. Bombard the protonemal lawn with the DNA-coated particle suspension.
Note: Perform the bombardment steps as quickly as possible to avoid contamination of the protonema culture.
f. Remove the DNA cartridge from the chamber and refill it with 10 μL of the DNA-coated gold particle suspension.
g. Set the DNA cartridge and carry out the bombardment using the same conditions, targeting another portion of the protonemal lawn.
h. Repeat the refilling and bombardment processes again (three times of bombardments in total).
6. Selection of G418 resistant clones (Figures 1C–E).
a. Cover the plate and seal with micropore surgical tape.
b. Incubate the plate in the dark at 25 °C for 24 h.
Note: Post-bombardment culture in the dark does not have to be exactly 24 h, but longer cultures gradually lead to cell death and should be avoided.
c. With tweezers, pick up the protonema cells at the positions where plasmid DNAs were bombarded (Figure 2) and put them in a glass tube.
d. Add 4 mL of sterile purified water and grind with the homogenizer (N-310E).
e. Propagate onto two BCDATG (G418+) agar plates that were laid with cellophane sheets.
f. Cover the plate, seal with micropore surgical tape, and incubate at 25 °C in continuous white light (45 μmol m-2 s-1) for ~2 weeks.
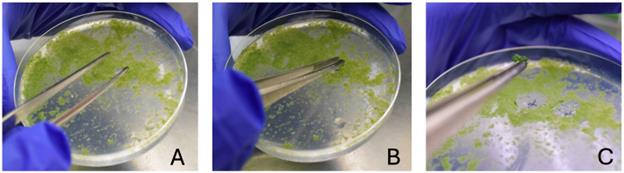
Figure 2. Collection of protonema cells at the positions where plasmid DNAs were bombarded. Proceed with steps from A to C. In (C), a small clump of protonema cells is visible at the tip of the forceps. Collect cells from a circular area with a diameter of approximately 1.5–2 cm.
C. Growth of surviving protonemata to small colonies and genotyping (Figures 1F–H).
1. Using tweezers, pick up surviving colonies and transfer to new BCDAT or BCDATG agar plates with no cellophane sheet.
2. Incubate the plates at 25 °C in continuous white light (45 μmol m-2·s-1) until each protonemal patch has grown to a 5–10 mm diameter colony.
3. Transfer a pinch of fresh protonemata from each colony to PCR tubes and perform green PCR as previously described [8] with an appropriate primer set to amplify a genomic region including the target sequence.
4. After confirming that the green PCR products show a band with the expected size on agarose gel electrophoresis, check the DNA sequences of the PCR products by direct sequencing using BigDye Terminator v3.1 Cycle Sequencing Kit or SupreDye Cycle Sequencing Kit v3.1. Use the reaction mixture of green PCR directly as the sequencing template.
Validation of protocol
By using this protocol, we attempted disruption of two genes, PpPPR_2 (Pp6c16_4940 in Physcomitrium patens v6.1; https://phytozome-next.jgi.doe.gov/info/Ppatens_v6_1) and PpPPR_38 (Pp6c6_14240), encoding two distinct PentatricoPeptide Repeat (PPR) proteins [18]. PPR proteins are nuclear-encoded RNA-binding proteins that are involved in post-transcriptional control of gene expression in chloroplasts and mitochondria [18]. Hattori et al. [19] succeeded in HR-based targeted disruption of PpPPR_38 using PEG-mediated transformation. On the other hand, so far, there have been no reports that studied PpPPR_2. In the construction of the sgRNA expression vectors, two sets of DNA oligonucleotides were used for generating the DNA fragments of the target sequences: PPR2_crRNA_Fwd2 and PPR2_crRNA_Rv2 for PpPPR_2 (designed based on the coding region sequence within the first PPR motif), and PPR38_crRNA_fw2 and PPR38_crRNA_rv2 for PpPPR_38 (based on the coding region sequence upstream from the first PPR motif). PPR motifs were predicted using the website PPR [17]. The following set of primers was used for green PCR and sequencing of the resulting amplified DNA fragments: PpPPR2_cr2_seq_fw and PpPPR2_cr2_seq_rv for PpPPR_2, and PPR38_cr2_seq_fw2 and PPR38_cr2_seq_rv2 for PpPPR_38. We conducted two biologically independent experiments for each gene, the results of which are shown in Table 1.
Table 1. Results of attempts to obtain mutant clones by using the current protocol. Numbers of clones obtained in two independent experiments (Exp. #1 and #2) for each target gene (PpPPR_2 and PpPPR_38) are summarized. Surviving clones: the total number of colonies observed immediately after G418 selection. WT: the number of clones with no difference to the wild-type sequence around the target sequence. FS+: the number of clones with frameshift mutations around the target sequence (supposedly resulting in disruption of gene function). FS- (in) and FS- (del): the number of clones with non-frameshift insertion and deletion of nucleotides, respectively, around the target sequence. Mosaic: the number of clones showing mixed sequences, possibly representing aggregation of cells from two (or more) distinct clones. KO ratio: the ratio of FS+ to Sequenced clones.
| Target gene | Exp. | Surviving clones | Sequenced clones | WT | FS+ | FS- (in) | FS- (del) | Mosaic | KO ratio |
|---|---|---|---|---|---|---|---|---|---|
PpPPR_2 | #1 | > 200 | 46 | 36 | 4 | 1 | 1 | 4 | 8.7% (4/46) |
| #2 | > 200 | 32 | 26 | 4 | 0 | 0 | 2 | 12.5% (4/32) | |
| PpPPR_38 | #1 | > 200 | 26 | 21 | 2 | 0 | 2 | 1 | 7.7% (2/26) |
| #2 | > 200 | 24 | 14 | 3 | 0 | 5 | 2 | 12.5% (3/24) |
The overall average of the ratios of the FS+ clones to sequenced clones is 10.35% (±2.52), indicating that we can modify a given gene with practically enough efficiency by using this protocol. Figure 3 shows the alignment of the sequences around the target sequence of each gene from all the clones in which frameshift mutations occurred. We compared the growth of colonies of the wild-type strain (WT), two clones with a frameshift mutation (FS+), two clones with a non-frameshift mutation (FS-), and two clones with no mutation (m-) (Figure 4). As for the PpPPR_38 gene, the FS+ clones showed small colonies due to retarded growth of caulonemal filaments (Figure 4). The retardation of caulonemal growth was more significant in colonies grown in BCDG or BCDAT agar medium than in those grown in BCDATG agar medium (Figure 4). These phenotypes are in good agreement with those of PpPPR_38 knockout (KO) strains produced by the HR-based method in a previous study [19], corroborating that the frameshift mutations caused functional disruption of PpPPR_38. On the other hand, we did not observe a significant difference between the growth of WT and the FS+ clones of PpPPR_2 (data not shown).
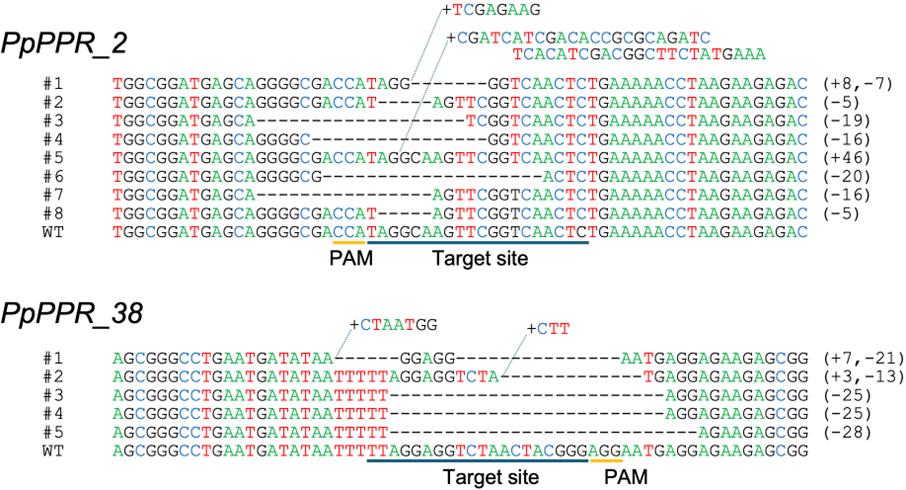
Figure 3. Alignments of the target (and flanking) sequences between the WT strain and the clones with frameshift mutations. The results of experiments conducted for the PpPPR_2 (top) and PpPPR_38 (bottom) genes are presented. Dashes indicate deletions. Sequences starting with a plus indicate insertions. The numbers beginning with plus and minus signs in parentheses indicate the number of nucleotide insertions and deletions, respectively, in the corresponding sequences. PAM and target site sequences are underlined below the alignment for each gene.

Figure 4. Growth of PpPPR_38 mutant strains obtained by this protocol. Growth of the wild-type strain (WT), two deletion mutants with frameshift (FS+#4 and FS+#5), two deletion mutants with no frameshift (FS-#1 and FS-#2), and two surviving clones with no mutation (m-#1 and m-#2) are presented. Colonies of these clones grown on BCDG, BCDAT, or BCDATG agar medium were photographed when they were similar sizes (day 0), and grew for the indicated days (day 7, 14, and 19) under white light irradiation (45 μmol m-2·s-1) at 25 °C.
General notes and troubleshooting
General notes
Depending on the target gene, the presence of glucose and/or ammonium tartrate in the culture medium may affect the efficiency of obtaining clones with frame shifts. Hattori et al. [19] reported that the colonies of the PpPPR_38 KO strain were much smaller than the WT colonies when grown on BCD (which contains no glucose or ammonium tartrate), BCDG, or BCDAT, while there was no significant difference between the KO strain and WT in colony sizes when grown on BCDATG. According to this, we used BCDATG for G418 selection and later culture for PpPPR_38 experiments.
Troubleshooting
Problem 1: Sequencing of the product of green PCR sometimes results in low signal-to-noise ratios and an absence of clearly defined peaks.
Possible cause: Low quality of template DNA.
Solution: Purification of the amplification product using NucleoSpin Gel and PCR Clean-Up kit significantly improves the sequencing results.
Problem 2: The helium gas emitted during bombardment sometimes blows away the target part of protonemata.
Possible cause: Protonemata contain too much water and are easily peeled off the agar surface.
Solution: Culture the protonemal lawn until the water on the agar surface well dries.
Acknowledgments
Contributions of each author: Conceptualization, M.S. and S.A.; Investigation, S.T.; Writing – Original Draft, S.A.; Writing – Review & Editing, S.A., S.T., M.S., and T.N.; Funding acquisition, M.S. and S.A.; Supervision, M.S. and S.A. This work was supported by JSPS KAKENHI Grant Numbers JP23K05146 to M.S. and JP23K05864 to S.A. We also thank Mitsuyasu Hasebe (National Institute for Basic Biology; NIBB) for the kind sharing of the wild-type strain of P. patens and its basic experimental procedures.
Competing interests
The authors declare that they have no competing interests.
References
- Knight, C., Perroud, P.-F. and Cove, D. (2009) The moss Physcomitrella patens. Annual Plant Reviews 36, first ed., Wiley-Blackwell, Oxford. https://doi.org/10.1093/ 2Faob_2Fmcp228
- Rensing, S. A., Goffinet, B., Meyberg, R., Wu, S. Z. and Bezanilla, M. (2020). The Moss Physcomitrium (Physcomitrella) patens: A Model Organism for Non-Seed Plants. Plant Cell. 32(5): 1361–1376. https://doi.org/10.1105/tpc.19.00828
- Bowman, J. L., Floyd, S. K. and Sakakibara, K. (2007). Green Genes—Comparative Genomics of the Green Branch of Life. Cell. 129(2): 229–234. https://doi.org/10.1016/j.cell.2007.04.004
- Rensing, S. A., Lang, D., Zimmer, A. D., Terry, A., Salamov, A., Shapiro, H., Nishiyama, T., Perroud, P. F., Lindquist, E. A., Kamisugi, Y., et al. (2008). The Physcomitrella Genome Reveals Evolutionary Insights into the Conquest of Land by Plants. Science. 319(5859): 64–69. https://doi.org/10.1126/science.1150646
- Rensing, S. A., Goffinet, B., Meyberg, R., Wu, S. Z. and Bezanilla, M. (2020). The Moss Physcomitrium (Physcomitrella) patens: A Model Organism for Non-Seed Plants. Plant Cell. 32(5): 1361–1376. https://doi.org/10.1105/tpc.19.00828
- Lopez-Obando, M., Hoffmann, B., Géry, C., Guyon-Debast, A., Téoulé, E., Rameau, C., Bonhomme, S. and Nogué, F. (2016). Simple and Efficient Targeting of Multiple Genes Through CRISPR-Cas9 in Physcomitrella patens. G3 Genes|Genomes|Genetics 6(11): 3647–3653. https://doi.org/10.1534/g3.116.033266
- Nomura, T., Sakurai, T., Osakabe, Y., Osakabe, K. and Sakakibara, H. (2016). Efficient and Heritable Targeted Mutagenesis in Mosses Using the CRISPR/Cas9 System. Plant Cell Physiol. 57(12): 2600–2610. https://doi.org/10.1093/pcp/pcw173
- Nomura, T. and Sakakibara, H. (2017). Targeted Mutagenesis Using RNA-guided Endonucleases in Mosses. Bio Protoc. 7(12): e2359. https://doi.org/10.21769/bioprotoc.2359
- Collonnier, C., Epert, A., Mara, K., Maclot, F., Guyon‐Debast, A., Charlot, F., White, C., Schaefer, D. G. and Nogué, F. (2016). CRISPR‐Cas9‐mediated efficient directed mutagenesis and RAD51‐dependent and RAD51‐independent gene targeting in the moss Physcomitrella patens. Plant Biotechnol J. 15(1): 122–131. https://doi.org/10.1111/pbi.12596
- Collonnier, C., Guyon-Debast, A., Maclot, F., Mara, K., Charlot, F. and Nogué, F. (2017). Towards mastering CRISPR-induced gene knock-in in plants: Survey of key features and focus on the model Physcomitrella patens. Methods. 103–117. https://doi.org/10.1016/j.ymeth.2017.04.024
- Schween, G., Egener, T., Fritzowsky, D., Granado, J., Guitton, M., Hartmann, N., Hohe, A., Holtorf, H., Lang, D., Lucht, J. M., et al. (2005). Large‐Scale Analysis of 73 329 Physcomitrella Plants Transformed with Different Gene Disruption Libraries: Production Parameters and Mutant Phenotypes. Plant Biol. 7(3): 228–237. https://doi.org/10.1055/s-2005-837692
- Tasaki, E., Hattori, M. and Sugita, M. (2010). The moss pentatricopeptide repeat protein with a DYW domain is responsible for RNA editing of mitochondrial ccmFc transcript. Plant J. 62(4): 560–570. https://doi.org/10.1111/j.1365-313x.2010.04175.x
- Anami, S., Yamashino, T., Suzuki, R., Nakai, K., Sato, K., Wu, B., Ryo, M., Sugita, M. and Aoki, S. (2021). Red light‐regulated interaction of Per‐Arnt‐Sim histidine kinases with partner histidine‐containing phosphotransfer proteins in Physcomitrium patens. Genes to Cells. 26(9): 698–713. https://doi.org/10.1111/gtc.12878
- Ashton, N. W. and Cove, D. J. (1977). The isolation and preliminary characterisation of auxotrophic and analogue resistant mutants of the moss, Physcomitrella patens. Mol Gen Genet. 154(1): 87–95. https://doi.org/10.1007/bf00265581
- Liu, H., Ding, Y., Zhou, Y., Jin, W., Xie, K. and Chen, L. L. (2017). CRISPR-P 2.0: An Improved CRISPR-Cas9 Tool for Genome Editing in Plants. Mol Plant. 10(3): 530–532. https://doi.org/10.1016/j.molp.2017.01.003
- Naito, Y., Hino, K., Bono, H. and Ui-Tei, K. (2014). CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics. 31(7): 1120–1123. https://doi.org/10.1093/bioinformatics/btu743
- Gutmann, B., Royan, S., Schallenberg-Rüdinger, M., Lenz, H., Castleden, I. R., McDowell, R., Vacher, M. A., Tonti-Filippini, J., Bond, C. S., Knoop, V., et al. (2020). The Expansion and Diversification of Pentatricopeptide Repeat RNA-Editing Factors in Plants. Mol Plant. 13(2): 215–230. https://doi.org/10.1016/j.molp.2019.11.002
- Sugita, M. (2022). An Overview of Pentatricopeptide Repeat (PPR) Proteins in the Moss Physcomitrium patens and Their Role in Organellar Gene Expression. Plants 11(17): 2279. https://doi.org/10.3390/plants11172279
- Hattori, M., Miyake, H. and Sugita, M. (2007). A Pentatricopeptide Repeat Protein Is Required for RNA Processing of clpP Pre-mRNA in Moss Chloroplasts. J Biol Chem. 282(14): 10773–10782. https://doi.org/10.1074/jbc.m608034200
Article Information
Publication history
Received: Jun 23, 2025
Accepted: Aug 15, 2025
Available online: Sep 2, 2025
Published: Sep 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Takenaka, S., Sugita, M., Nomura, T. and Aoki, S. (2025). CRISPR/Cas9-Induced Targeted Mutagenesis of the Moss Physcomitrium patens by Particle Bombardment-Mediated Transformation. Bio-protocol 15(18): e5452. DOI: 10.21769/BioProtoc.5452.
Category
Plant Science > Plant molecular biology > Genetic analysis
Molecular Biology > DNA > Mutagenesis
Biological Sciences > Biological techniques > CRISPR/Cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.