- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Verification of N-Linked Glycosylation of Proteins Isolated from Plant or Mammalian Cell Lines Using PNGase Enzyme
(*contributed equally to this work) Published: Vol 15, Iss 18, Sep 20, 2025 DOI: 10.21769/BioProtoc.5444 Views: 2007
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2025
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
N-glycosylation is a ubiquitous post-translational modification (PTM) that regulates protein folding, stability, and biological function. Accurate identification and validation of N-glycosylation are therefore critical for understanding how glycosylation modulates protein activity. Here, we present a robust workflow for analyzing protein N-glycosylation in both animal and plant systems using peptide-N4-(N-acetyl-β-glucosaminyl) asparagine-amidase A and F (PNGase A and PNGase F). After enzymatic cleavage of the asparagine-linked N-glycans, samples are analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting (WB) to detect shifts in apparent molecular weight (MW) indicative of deglycosylation. Key steps include denaturing the protein to expose glycosylation sites, optimizing buffer conditions for PNGase F and A treatment, and comparing glycosylated vs. deglycosylated forms by electrophoretic mobility. A troubleshooting guide addresses common challenges, including incomplete deglycosylation and low transfer efficiency during WB, offering practical solutions to ensure reliable results. This protocol provides researchers with a standardized, cost-effective framework for investigating protein N-glycosylation in diverse systems, from cell lysates to purified proteins, in both animal and plant models.
Key features
• This method employs standard biochemical techniques, such as enzymatic digestion and SDS-PAGE, making it highly accessible and relatively quick to perform.
• Successful deglycosylation results in a detectable downward shift in protein migration on an SDS-PAGE gel. This shift provides a visually confirmable indication of N-glycan removal.
• Prior to engaging in mass spectrometry analyses, this approach serves as a rapid, cost-effective preliminary screening or validation tool for assessing N-glycosylation status.
• Broad system compatibility: PNGase F and A enzymes are active in mammalian, plant, and microbial systems, allowing reliable N-glycosylation assessment regardless of sample origin.
Keywords: Enzymatic assayGraphical overview
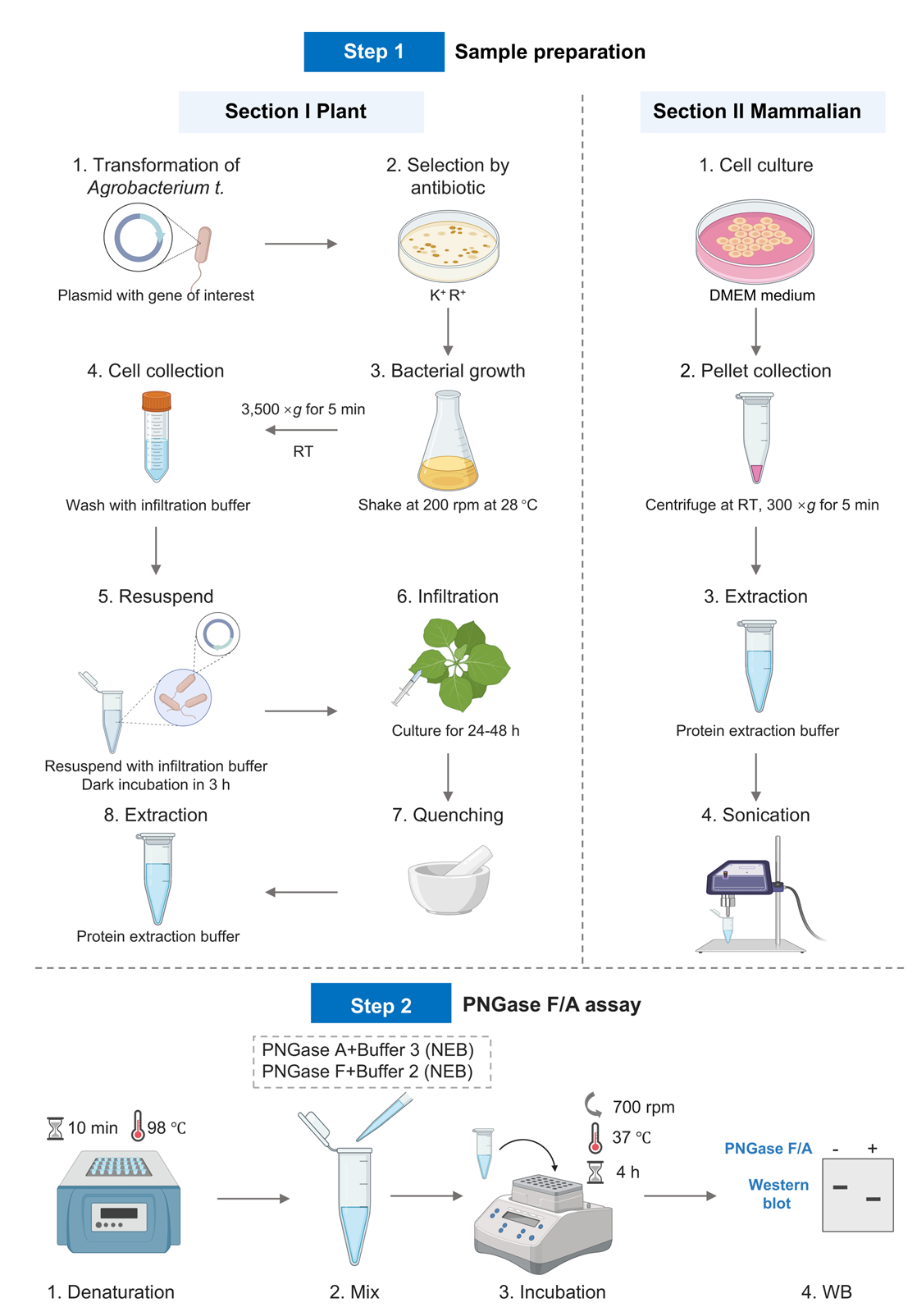
Background
N-linked glycosylation is a major post-translational modification (PTM) in nature, where oligosaccharide chains covalently attach to the amide nitrogen of asparagine (Asn) residues within the classic Asn-X-Ser/Thr (X ≠ Pro) sequence in nascent polypeptides [1]. This modification regulates protein folding, glycan-dependent quality control processes in the endoplasmic reticulum (ER), protein stability, and protein–protein interactions [2]. N-glycosylation is a ubiquitous PTM, with at least 2,000 N-glycosylation sites identified in model organisms such as Arabidopsis thaliana, Drosophila melanogaster, and zebrafish [3], though this number has likely been underestimated.
N-glycosylation plays a crucial role in protein function, trafficking, and immune responses in plants. In Arabidopsis, mutation of N-glycosylation sites in KORRIGAN1 (KOR1) disrupts proper protein folding, Golgi trafficking, and root development [4]. N-glycosylation also regulates the localization of SnRK2.2/2.3 and the brassinosteroid receptor BRI1 [5, 6] and modulates ligand binding of vacuolar sorting receptors such as AtVSR1 [7]. In the EFR receptor, mutation of asparagine at position 143 (Asn143) abolishes ligand-binding capacity, thereby impairing immune responses [8]. In the MIK2 receptor, N-glycosylation at Asn 410 is critical for its interaction with BAK1 [9,10]. In rice, N-glycosylation is essential for the function of the ER-resident lectin OS9 in ER-associated degradation (ERAD) [11]. In wheat, N-glycosylation is pivotal for TaCERK-mediated defense against wheat yellow mosaic virus infection [12]. Despite the identification of thousands of N-glycosylated proteins through omics approaches, functional studies of N-glycosylation on individual plant proteins remain in the early stages, largely due to the limited availability of robust methods for validating N-glycan modifications.
The initial steps of N-glycosylation are highly conserved between plants and animals [2]. However, due to the presence of unique modifications in the plant-specific N-glycosylation pathway, particularly the incorporation of β1,2-xylose and α1,3-fucose residues into the core structure, there are distinct differences in the verification of N-glycan modifications on plant vs. animal proteins [13]. PNGase F, an amidase, can hydrolyze the β-aspartyl glycosylamine bond between the innermost N-acetylglucosamine (GlcNAc) and asparagine, releasing the intact oligosaccharide and converting the Asn to aspartic acid [14]. Unlike endoglycosidases (e.g., Endo H) that cleave within the core glycan, PNGase F can remove all N-linked glycans, making it the gold standard for complete deglycosylation [15]. Its activity requires the protein to be denatured to expose glycosylation sites that may be obscured by the tertiary structure. This enzyme is indispensable in studies where N-glycosylation needs to be precisely validated, such as distinguishing glycosylation isoforms in SDS-PAGE or mapping glycosylation sites by mass spectrometry. PNGase F removes all complex, hybrid, and high-mannose-type glycans and is widely used for N-glycan cleavage in mammalian systems [16]. However, its activity is limited by the presence of α1,3-core fucose in the glycan, which is common in plant systems. In contrast, PNGase A, derived from Oryza sativa, can hydrolyze N-glycans on glycoproteins/peptides regardless of the presence of xylose or fucose (α1-3 or α1-6) (Table 1) [17]. This renders PNGase A the gold standard for complete removal of N-glycans from plant-derived proteins.
Table 1. PNGase F and PNGase A
| Enzyme | PNGase F | PNGase A |
|---|---|---|
| Source | Elizabethkingia spp | Oryza sativa |
| Core specificity | High-mannose-type, complex-type, and hybrid-type without α1-3 core fucosylated glycans | Cleaves α1-3 and α1-6 core fucosylated and high-mannose-type glycans |
| Optimal pH | 7.5 | 4.5–5.5 |
| Compatibility | Mammalian system | Mammalian, plant, and insect systems |
In addition to PNGase F and A, various other enzymes target specific glycan structures or linkage types. For example, endoglycosidases (Endo H/F/S) cleave within the core glycan, leaving a single GlcNAc residue [18]. A comparative overview of key glycan-cleaving enzymes is provided in Table 2, sourced from [18] and the NEB website. In some cases, sequential digestion with enzyme combinations (e.g., sialidase + PNGase F) can enhance the efficiency of glycan removal from highly modified proteins. In this study, a detailed protocol has been established, with PNGase F and PNGase A as the core analytical tools. Specifically, the N-glycosylation of exogenously transformed plant proteins is detected using the plant system, while the N-glycosylation of endogenous animal proteins is examined via the animal system.
Table 2. Enzymes for glycan cleavage
| Enzyme | Source | Substrate specificity | Optimal pH | Key applications |
| Endo H | Streptomyces plicatus | Cleaves high-mannose and hybrid N-glycans | 5.5 | ER-resident glycoprotein analysis |
| Endo F1/F2/F3 | Elizabethkingia spp. | F1: High mannose | F1: 5.5 | Glycoengineering antibody glycosylation patterns |
| F2: Bi-antennary | F2: 4–6 | |||
| F3: Tri-antennary | F3: 5.5 | |||
| Endo S | Streptococcus pyogenes | Cleaves N-glycans on IgG Fc region | 5.5 | Antibody glycan profiling without full digestion |
| O-Glycosidase | Enterococcus faecalis | Removes core 1 O-linked glycans (Galβ1-3GalNAc) | 5.5–7.0 | O-glycan validation (e.g., mucins) |
| Sialidase | Clostridium perfringens | Hydrolyzes α2-3/6/8/9-linked sialic acids | 5.0–6.0 | Desialylation for glycan accessibility studies |
Materials and reagents
Biological materials
1. Nicotiana benthamiana
2. Agrobacterium tumefaciens strain GV3101
3. HEK 293T cell line
Reagents
1. Plant expression vector pCAMBIA2301
2. Rifampicin (Rif) (Sigma, catalog number: R3501)
3. Kanamycin (Kana) (Sigma, catalog number: 420311)
4. DMEM (Gibco, catalog number: 11995065)
5. FBS (Gibco, catalog number: A5256701)
6. Penicillin/streptomycin (Gibco, catalog number: 15140122)
7. Trypsin-EDTA (Gibco, catalog number: 25200056)
8. MES (Sigma, catalog number: M8250)
9. MgCl2 6H2O (Sigma, catalog number: M2393)
10. Acetosyringone (AS) (Sigma, catalog number: D134406)
11. PNGase F (New England Biolabs, catalog number: P0705S)
12. PNGase A (New England Biolabs, catalog number: P0707S)
13. Sodium dodecyl sulfate (SDS) (Sigma, catalog number: L4390)
14. Sodium chloride (NaCl) (Sigma, catalog number: S7653)
15. Potassium chloride (KCl) (Sigma, catalog number: P5405)
16. Disodium hydrogen phosphate (Na2HPO4·12H2O) (Sigma, catalog number: 106579)
17. Potassium dihydrogen phosphate (KH2PO4) (Sigma, catalog number: 104873)
18. Dithiothreitol (DTT) (Sigma, catalog number: D0632)
19. Nonidet-40 (NP-40) (Sigma, catalog number: I8896)
20. Methanol (Sigma, catalog number: 34860)
21. Chloroform (Sigma, catalog number: 319988)
22. 4%–20% gradient SDS-PAGE gel (LabLead, catalog number: P42012)
23. Protein ladder (Thermo Scientific, catalog number: 26616)
24. 5× loading buffer (LabLead, catalog number: G2527)
25. Tris (Aladdin, catalog number: T105287)
26. Glycine (Aladdin, catalog number: G432934)
27. BSA (Aladdin, catalog number: B265993)
28. Primary antibodies: Anti-SEL1L (ABclonal, catalog number: A12073), anti-Myc (MBL, catalog number: M047-3), anti-actin for plants (Abmart, catalog number: M20009), and anti-actin for animals (Beyotime, catalog number: AF0003)
29. Secondary antibodies: HRP-conjugated anti-mouse IgG (Cell Signaling, catalog number: 7076S) and HRP-conjugated anti-rabbit IgG (Cell Signaling, catalog number: 7074S)
30. Liquid nitrogen
31. 10% glycerol (Coolaber, catalog number: SL9280)
32. ECL substrate (Millipore, catalog number: WBKLS0050)
33. Tryptone (Aladdin, catalog number: T139519)
34. Yeast extract (Aladdin, catalog number: Y278690)
Solutions
1. Antibiotic stock solutions (see Recipes)
2. LB liquid medium (see Recipes)
3. Induction buffer (see Recipes)
4. Infiltration buffer (see Recipes)
5. 1× PBS (see Recipes)
6. Protein extraction buffer (see Recipes)
7. Denaturing buffer (1 M DTT) (see Recipes)
8. 10% NP-40 (see Recipes)
9. Transfer buffer (see Recipes)
10. Cell culture medium (see Recipes)
Recipes
1. Antibiotic stock solutions
| Antibiotic | Stock concentration | Quantity (per mL) | Storage |
|---|---|---|---|
| Rif | 50 mg/mL (liquid) | 50 mg | -20 °C, protected from light |
| Kana | 50 mg/mL (liquid) | 50 mg | -20 °C |
2. LB liquid medium
| Reagent | Final concentration | Quality or volume (per L) |
|---|---|---|
| Tryptone | 10 g/L | 10 g |
| Yeast extract | 5 g/L | 5 g |
| NaCl | 10 g/L | 10 g |
| Ultrapure water (e.g., Milli-Q) | 1,000 mL |
After autoclaving and cooling to approximately 50 °C, add rifampicin [final 50 μg/mL (1:1,000 dilution from 50 mg/mL stock)] and kanamycin [final 50 μg/mL (1:1,000 dilution from 50 mg/mL stock)].
3. Induction buffer
| Reagent | Final concentration | Quality or volume (per L) |
|---|---|---|
| MES (pH 5.7) | 10 mM | 1.9524 g |
| Tryptone | 10 g/L | 10 g |
| Yeast extract | 5 g/L | 5 g |
| NaCl | 10 g/L | 10 g |
| AS | 20 μM | 3.924 mg |
| Ultrapure water (e.g., Milli-Q) | 1,000 mL |
a. Prepare 500 mL of 2× LB medium, then add 400 mL of water.
b. Perform autoclave sterilization and cool to room temperature.
c. Add 100 mL of 0.1 M sterile MES (pH 5.7). Store the prepared medium at 4 °C.
d. Add AS before use to achieve a final concentration of 20 μM.
e. Add Rif and Kana (see Recipe 1) to a final concentration of 50 μg/mL (1:1,000 dilution from 50 mg/mL stock).
4. Infiltration buffer
| Reagent | Final concentration | Quality or volume (per L) |
|---|---|---|
| MES (pH 5.7) | 10 mM | 1.9524 g |
| MgCl2 6H2O | 10 mM | 2.03 g |
| AS | 150 μM | 29.4 mg |
| Ultrapure water (e.g., Milli-Q) | 1000 mL |
a. Dissolve MES and MgCl2 in 800 mL of H2O and adjust pH to 5.7 with KOH.
b. Add AS (dissolved in DMSO) and bring to 1 L. Filter-sterilize and use fresh.
5. 1× PBS
| Reagent | Final concentration | Quality (per L) |
|---|---|---|
| NaCl | 137 mM | 8 g |
| KCl | 2.7 mM | 0.2 g |
| Na2HPO4·12H2O | 10 mM | 3.58 g |
| KH2PO4 | 1.8 mM | 0.24 g |
a. Dissolve all reagents in 800 mL of ultrapure water.
b. Adjust pH to 7.4 with HCl (if needed).
c. Bring the final volume to 1 L with ultrapure water.
d. Sterilize by autoclaving (121 °C, 20 min) or filter sterilization (0.22 μm).
6. Protein extraction buffer
| Reagent | Final concentration | Quality or volume (per 100 mL) |
|---|---|---|
| SDS | 4% (w/v) | 4 g |
| 1× PBS (Recipe 5) | 1× | 100 mL (adjust final volume) |
| Triton | 1% (w/v) | 1 mL |
7. Denaturing buffer (1 M DTT)
| Reagent | Final concentration | Quantity or volume (per 1 mL) |
|---|---|---|
| DTT | 1 M | 0.15425 g |
| Ultrapure water (e.g., Milli-Q) | Up to 1 mL |
Store aliquots at -20 °C. Avoid freeze-thaw cycles.
8. 10% NP-40
| Reagent | Final concentration | Quantity or volume (per 100 mL) |
|---|---|---|
| NP-40 | 10% (v/v) | 10 mL |
| Ultrapure water (e.g., Milli-Q) | 90 mL (adjust to final volume) |
a. Use a graduated cylinder or pipette to measure 10 mL of NP-40 (wear gloves and eye protection; NP-40 is viscous and irritant).
b. Add the NP-40 to approximately 80 mL of ultrapure water in a beaker or bottle.
c. Stir gently with a magnetic stirrer or shake until fully homogeneous (NP-40 dissolves easily but is viscous).
d. Bring the final volume to 100 mL with ultrapure water.
e. Filter through a 0.22 μm filter.
f. Store protected from light.
9. 10× transfer buffer
| Reagent | Final concentration | Quality or volume (per 1,000 mL) |
|---|---|---|
| Tris base | 250 mM | 30.3 g |
| Glycine | 1.92 M | 144 g |
| dH2O | 1,000 mL |
10. Cell culture medium
| Reagent | Final concentration | Volume (per 500 mL) |
|---|---|---|
| DMEM | 500 mL | |
| FBS | 10% | 50 mL |
| Penicillin/streptomycin | 1% | 5 mL |
Store at 4 °C.
Equipment
1. Metal bath (Thermo Scientific, catalog number: 88870001)
2. Sonicator (SCIENTZ-IID, catalog number: JY92-IIN)
3. Thermal mixer (Eppendorf, catalog number: 5382000074)
4. SDS-PAGE electrophoresis system (Bio-Rad, catalog number: 1645070 and 1658001)
5. Wet transfer apparatus (Bio-Rad, catalog number: 1703935)
6. Imaging system (Tanon Chemi Dog Ultra)
Software and datasets
1. ImageLab (Bio-Rad) for gel image analysis
2. UniProt: Reference molecular weights of target proteins
3. BioRender.com online tool: Graphical overview
Procedure
Part I. Plant procedure
A. Express target protein in Nicotiana benthamiana
1. Plasmid transformation into Agrobacterium
a. Revive the strain: Streak frozen Agrobacterium glycerol stock onto an LB agar plate with Rif (50 μg/mL). Incubate at 28 °C for 48 h.
b. Liquid culture: Inoculate a single colony into 5 mL of LB liquid medium containing Rif and culture at 28 °C shaking at 200 rpm until the OD600 reaches approximately 0.6–0.8.
c. Large-scale culture: Dilute the culture at a 1:100 ratio into 50 mL of LB medium containing Rif and incubate at 28 °C shaking at 200 rpm until the OD600 reaches approximately 0.4–0.5.
d. Harvest and wash: Chill culture on ice for 15 min. Centrifuge at 5,000× g for 10 min at 4 °C. Discard supernatant. Resuspend pellet gently in 10 mL of ice-cold 10% glycerol. Repeat centrifugation. Repeat washing once more. Resuspend pellet in 10% glycerol and aliquot 50 μL into each tube. Use immediately or freeze at -80 °C.
e. Transformation via freeze-thaw method: Thaw 50 μL of competent cells on ice, add 1–5 μL of plasmid DNA (100–500 ng), mix gently, and incubate on ice for 5 min. Freeze in liquid nitrogen for 5 min, then immediately perform heat shock at 37 °C for 5 min, followed by incubation on ice for 5 min. Add 800 μL of antibiotic-free LB liquid medium and incubate at 28 °C with shaking at 200 rpm for 4–6 h. Spread 100–200 μL of the culture onto LB agar plates containing Rif (50 μg/mL) and Kana (50 μg/mL) and incubate at 28 °C for 48–72 h until colonies appear.
f. Verification of transformants: Pick colonies and amplify using plasmid-specific primers (e.g., target gene or antibiotic resistance markers).
2. Agrobacterium culture preparation
a. Day 1: Inoculate Agrobacterium harboring the target gene into 5 mL of LB medium supplemented with 50 μg/mL Rif and 50 μg/mL Kana and culture overnight at 28 °C shaking at 200 rpm.
b. Day 2: Dilute the overnight culture 1:1,000 into 100 mL of induction buffer and incubate at 28 °C shaking at 200 rpm until OD600 reaches approximately 0.6–1.0.
3. Preparation for infiltration
a. Centrifuge the culture at 3,500× g for 5 min at room temperature and discard the supernatant. Wash the pellet once with infiltration buffer to remove residual antibiotics.
b. Resuspend the pellet in infiltration buffer to an OD600 of 0.5 and incubate in the dark at room temperature for 3 h.
4. Infiltration of N. benthamiana leaves
a. Select 3–4-week-old N. benthamiana plants with fully expanded leaves that were watered on the same day.
b. Infiltrate the Agrobacterium infiltration buffer into the lower leaf surface of the N. benthamiana by pressing the tip of a needleless syringe against the leaf. The third, fourth, and fifth young leaves are used for Agrobacterium infiltration.
c. Mark the infiltrated regions on the leaf using a marker pen.
d. Incubate the infiltrated N. benthamiana seedlings for 24–48 h to allow expression of the target protein.
B. Protein extraction
1. Tissue collection: Excise the infiltrated leaf region, immediately freeze in liquid nitrogen, and grind into a fine powder.
2. Extraction: Add 1–2 mL of protein extraction buffer per 1 mL of tissue, vortex briefly, then incubate for 10 min. Centrifuge at 12,000× g for 20 min and collect the supernatant.
3. Analysis: Determine expression level by SDS-PAGE and western blot (WB) to identify the optimal tissue amount for subsequent enzymatic digestion.
Note: If a specific protein requires a specialized extraction method, follow the corresponding protocol. After extraction, precipitate the proteins using a methanol:chloroform:water mixture, and then resuspend the pellet in 4% SDS (w/v).
C. Protein denaturation
1. Add denaturing buffer to the supernatant to achieve a final concentration of DTT of 40 mM.
2. Incubate the lysate in a 95 °C metal bath for 10 min to completely denature the protein.
3. Methanol-chloroform protein precipitation:
a. Take 200 μL of denatured protein solution, add 600 μL of ice-cold methanol, 150 μL of chloroform, and 400 μL of ultrapure water.
b. Vortex thoroughly to mix and centrifuge at 10,000× g for 10 min at 22 °C.
c. Carefully discard the supernatant and wash the pellet with 600 μL of methanol. Centrifuge at 10,000× g for 10 min.
d. Repeat the methanol wash step once.
e. After discarding the supernatant, open the centrifuge tube and allow the pellet to air-dry for approximately 5 min.
f. Resuspend the pellet in 100 μL of PBS containing 1% SDS. (A brief sonication may assist in dissolving. Apply a pulsed mode with 0.1-s pulses followed by 0.1-s pauses, continuing until the protein is fully dissolved.)
Note: This step requires high-concentration SDS to prevent precipitation of heat-sensitive proteins.
D. PNGase A digestion
1. Prepare the reaction system as shown in Table 3.
Table 3. PNGase A digestion reaction system
| Group | Protein extraction | 10× NP-40 | 10× buffer | PNGase A | Ultrapure water | Total |
|---|---|---|---|---|---|---|
| Control | 10 μL | 4 μL | 4 μL | - | 22 μL | 40 μL |
| Experimental | 10 μL | 4 μL | 4 μL | 1 μL (5 U) | 21 μL | 40 μL |
2. Mixing and incubation: Vortex the reaction mixture thoroughly, avoiding excessive pipetting or bubble formation. Briefly centrifuge and then incubate at 37 °C for 4 h in a thermal mixer. Upon completion of the reaction, briefly centrifuge before opening the cap to avoid sample loss.
E. SDS-PAGE and WB
1. Add 5× loading buffer to the reaction mixture to achieve a final concentration of 1×.
2. Heat the samples at 95 °C for 5 min to fully denature the protein.
3. Briefly centrifuge (e.g., 12,000× g for 1 min).
4. Load the supernatant directly to the SDS-PAGE gel for electrophoresis at a voltage of 160 V.
5. Transfer proteins to a PVDF membrane using wet transfer apparatus at 4 °C with a current of 200 mA for 80 min.
6. Block with 5% BSA and incubate with the primary antibody (overnight at 4 °C), followed by HRP-conjugated secondary antibody incubation (1 h at room temperature).
7. Develop the signal using ECL substrate (Figure 1).
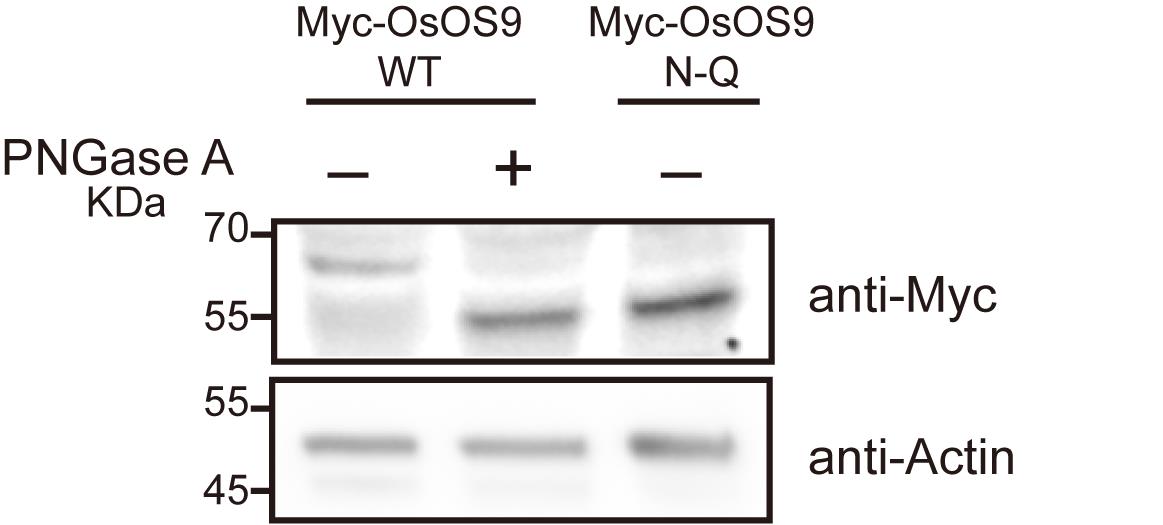
Figure 1. Validation of N-glycoproteins in Nicotiana benthamiana. Immunoblots showing Myc-OsOS9, which was expressed in N. benthamiana leaves, followed by treatment with or without PNGase A. The Myc-OsOS9 mutant, in which the N-glycosylation site was mutated to glutamine (Q), was used as a control. The lysed samples were separated using a 4%–20% gradient SDS-PAGE gel and analyzed by immunoblotting with Myc antibodies. Actin serves as the loading control.
Part II. Mammalian cell procedure
A. Cell culture
1. Culture HEK 293T cells in a 15 cm culture dish in cell culture medium.
2. Discard the culture medium, add 5 mL of ice-cold PBS to the flask/plate, swirl gently, and aspirate to remove residual serum.
3. Add 1 mL of trypsin-EDTA (0.25%), incubate at 37 °C for 3–5 min, neutralize the trypsin with 5 mL of complete medium, and transfer the cell suspension to a 15 mL centrifuge tube.
4. Centrifuge at 300× g for 5 min at 4 °C and discard the supernatant completely.
B. Protein extraction
1. Add 1 mL of ice-cold PBS to the cell pellet, gently resuspend, and centrifuge at 300× g for 5 min at 4 °C (optional: repeat washing step).
2. Add 100–200 μL of protein extraction buffer per 1 × 106 cells and briefly vortex to resuspend the pellet.
3. Sonicate to disrupt genomic DNA with the following parameters: 3-s pulses followed by 3 s pauses; repeat for 10 cycles.
4. Centrifuge at 12,000× g for 15 min at 22 °C to pellet insoluble debris.
5. Transfer the supernatant (protein lysate) to a fresh tube.
C. Protein denaturation
1. Add denaturing buffer to the supernatant to achieve a final concentration of DTT of 40 mM.
2. Incubate the lysate in a 95 °C metal bath for 10 min to completely denature the protein.
3. Methanol-chloroform protein precipitation:
a. Take 200 μL of denatured protein solution and add 600 μL of ice-cold methanol, 150 μL of chloroform, and 400 μL of ultrapure water.
b. Vortex thoroughly to mix and centrifuge at 10,000× g for 10 min at 22 °C.
c. Carefully discard the supernatant and wash the pellet with 600 μL of methanol. Centrifuge at 10,000× g for 10 min.
d. Repeat the methanol wash step once.
e. After discarding the supernatant, open the centrifuge tube and allow the pellet to air-dry for approximately 5 min.
f. Resuspend the pellet in 100 μL of PBS containing 1% SDS. (A brief sonication may assist in dissolving. Apply a pulsed mode with 0.1-s pulses followed by 0.1-s pauses, continuing until the protein is fully dissolved.)
Note: This step requires high-concentration SDS to prevent precipitation of heat-sensitive proteins.
D. PNGase F digestion
1. Prepare the reaction system as shown in Table 4.
Table 4. PNGase F digestion reaction system
| Group | Protein extraction | 10× NP-40 | 10× buffer | PNGase F | Ultrapure water | Total |
|---|---|---|---|---|---|---|
| Control | 10 μL | 4 μL | 4 μL | - | 22 μL | 40 μL |
| Experimental | 10 μL | 4 μL | 4 μL | 1 μL (500 U) | 21 μL | 40 μL |
2. Mixing and incubation: Thoroughly vortex the prepared reaction mixture, avoiding excessive pipetting or bubble formation. Briefly centrifuge and incubate at 37 °C in a thermal mixer for 4 h. Upon completion of the reaction, briefly centrifuge before opening the cap to avoid sample loss.
E. SDS-PAGE and WB
1. Add 5× loading buffer to the reaction mixture to achieve a final concentration of 1×.
2. Heat the samples at 95 °C for 5 min to fully denature the protein.
3. Briefly centrifuge (e.g., 12,000× g for 1 min).
4. Load the supernatant directly to the SDS-PAGE gel for electrophoresis at a voltage of 160 V.
5. Transfer proteins to a PVDF membrane using wet transfer apparatus at 4 °C with a current of 200 mA for 80 min.
6. Block with 5% BSA and incubate with the primary antibody (overnight at 4 °C), followed by HRP-conjugated secondary antibody incubation (1 h at room temperature).
7. Develop the signal using ECL substrate (Figure 2).
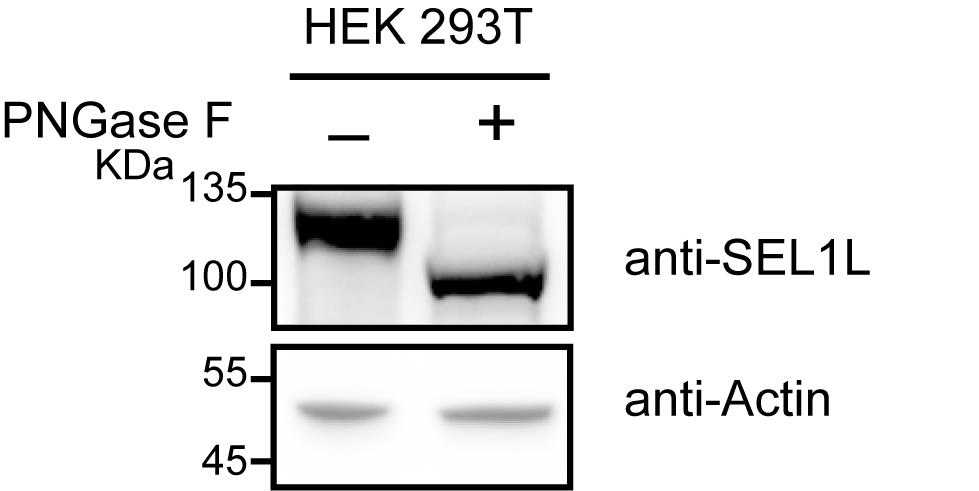
Figure 2. Validation of N-glycoproteins in HEK 293T cells. Lysed HEK 293T cells were subjected to experiments with or without PNGase F treatment, followed by separation using a 4%–20% gradient SDS-PAGE gel and immunoblotting analysis with an endogenous SEL1L antibody. Actin serves as the loading control.
Validation of protocol
This protocol has been used and validated in the following research article:
Lei et al. [11]. Chemical glycoproteomic profiling in rice seedlings reveals N-glycosylation in the ERAD-L machinery. Mol Cell Proteomics (Figures 6D–F, 7B, and 8B).
General notes and troubleshooting
General notes
1. The final SDS concentration in the reaction should not exceed 0.25%.
2. Add NP-40 to a final concentration of 1% to neutralize SDS.
3. Evaporation control: Seal the tube caps tightly during incubation. For long-term incubation, add a mineral oil overlay if necessary. For direct downstream gel electrophoresis, use PCR tubes (to reduce evaporation). For larger volume operations, use 1.5 mL Eppendorf tubes and perform intermittent centrifugation to collect condensate.
4. Enzyme-to-protein ratio: The general rule is to use 1 U of enzyme per 1 μg of protein, as recommended by NEB. For highly glycosylated proteins, such as IgG, the enzyme amount should be increased to 2–3 U/μg. In cases of uncertain glycosylation levels, it is advisable to start with 1 μL of enzyme per reaction and adjust based on post-digestion gel analysis.
5. Gel concentration selection: Choose the appropriate acrylamide gel concentration based on the MW of the target protein. For high MW proteins (>100 kDa), use a low-concentration gel (e.g., 8%), and for low MW proteins (<50 kDa), use a high-concentration gel (e.g., 12%–15%).
6. Electrophoresis time adjustment: Extend the electrophoresis time to improve resolution, especially when separating proteins with similar molecular weights. Low voltage (e.g., 80–100 V) and prolonged electrophoresis time can optimize band separation.
7. Band sharpness: Longer electrophoresis time may reduce band tailing, but overly prolonged electrophoresis can lead to band diffusion.
8. Temperature control: For long-duration electrophoresis, ensure adequate cooling (e.g., ice packs or cooled buffer) to prevent gel deformation.
9. Enzyme selection: For mammalian systems, PNGase F is preferred, but PNGase A can also be used (though it is more costly and exhibits slightly lower digestion efficiency compared to PNGase F).
10. pH requirements: PNGase F exhibits optimal activity under alkaline conditions (pH 7.5), whereas PNGase A prefers an acidic environment (pH 4.5–5.5). It is essential to strictly adhere to the enzyme buffer formulations provided by NEB to ensure compatibility.
11. Denaturation efficiency: Incomplete denaturation leaves glycosylation sites inaccessible to PNGase F and A, leading to false negatives. Boiling in SDS/DTT ensures full protein unfolding.
12. Enzyme-to-substrate ratio: Excess PNGase F (≥50 U/µg protein) is critical for complete digestion, particularly for heavily glycosylated proteins like antibodies or viral glycoproteins.
13. Glycan complexity: Proteins with multiple glycosylation sites (e.g., HIV gp120) may show smeared bands post-digestion due to residual O-glycans or microheterogeneity, necessitating complementary techniques like lectin blotting.
Troubleshooting
Problem 1: Protein precipitation during thermal denaturation.
Possible cause: Incomplete protein denaturation or insufficient SDS concentration.
Solution: Add SDS or reducing agents (e.g., DTT) to maintain solubility.
Problem 2: Electrophoretic band smearing.
Possible cause: 1) Protein degradation. 2) Inappropriate buffer pH or ionic strength.
Solution: 1) Use fresh sample for the experiment. 2) Do not load the sample immediately after the reaction. Precipitate proteins as follows: Add 60 μL of water to the reaction mixture, then add 10 volumes (600 μL) of ice-cold methanol to precipitate the proteins. Mix gently, then centrifuge at 12,000× g for 10 min at 4 °C to pellet the proteins. Air-dry the pellet at room temperature for 5–10 min (avoid over-drying). Resuspend the pellet directly in 1× SDS loading buffer. Heat the sample at 95 °C for 5 min, then load onto the SDS-PAGE gel.
Problem 3: No MW shift in electrophoresis.
Possible cause: 1) Incomplete enzyme digestion. 2) Protein bands are too dense (saturation). 3) Small MW differences, resulting in poor band separation.
Solution: 1) Increase enzyme digestion time: Extend the incubation time from 4 h to overnight (16–18 h) at 37 °C. Increase the enzyme amount: For highly glycosylated targets (e.g., IgG), use 2–3 U/μg protein. Verify enzyme activity and buffer compatibility (e.g., pH, SDS concentration). 2) Reduce the loading sample volume or dilute the sample. 3) Optimize gel electrophoresis conditions: Choose gel concentration based on the MW of your target protein. For high MW proteins (>100 kDa), use 6%–8% gels. For medium MW proteins (50–100 kDa), use 8%–12% gels. For low MW proteins (<50 kDa), use 12%–15% gels. Precast gels can be used for this step. We recommend using Invitrogen NuPAGETM gels for optimal protein separation. Run at a lower voltage and extend the running time.
Problem 4: High background in WB.
Possible cause: Not well-blotted.
Solution: Optimize the blocking solution (e.g., 5% BSA and non-fat milk).
Problem 5: Fuzzy or no bands observed in electrophoresis.
Possible cause: Incomplete transfer to the membrane.
Solution: High MW proteins are prone to precipitation in the gel. Adding no more than 0.1% SDS to the transfer buffer can effectively prevent this issue. Check the pore size of the membrane. Extend the transfer time (e.g., perform overnight wet transfer at 4 °C with 30–50 V) to ensure complete transfer of large proteins onto the membrane.
Acknowledgments
We thank all authors from the corresponding original research paper in which this protocol was used: Molecular & cellular proteomics [11]. This work was supported by the Science and Technology Innovation 2030 Major Project (2024ZD040800404), the Hainan Excellent Talent Team, the specific research fund of The Innovation Platform for Academicians of Hainan Province (YSPTZX202201), the Basic Research Project in 2023 of Yazhouwan National Laboratory, and the National Natural Science Foundation of China (92153301 and 22321005). X.C. is a recipient of Xplorer Prize. The Graphical overview was created in BioRender. Lei, C. (2025) https://BioRender.com/nxf7e3g.
Competing interests
The authors declare that they have no competing interests.
References
- Welply, J. K., Shenbagamurthi, P., Lennarz, W. J. and Naider, F. (1983). Substrate recognition by oligosaccharyltransferase. Studies on glycosylation of modified Asn-X-Thr/Ser tripeptides. J Biol Chem. 258(19): 11856–11863. https://doi.org/10.1016/s0021-9258(17)44311-0
- Strasser, R. (2016). Plant protein glycosylation. Glycobiology. 26(9): 926–939. https://doi.org/10.1093/glycob/cww023
- Zielinska, D. F., Gnad, F., Schropp, K., Wiśniewski, J. R. and Mann, M. (2012). Mapping N-Glycosylation Sites across Seven Evolutionarily Distant Species Reveals a Divergent Substrate Proteome Despite a Common Core Machinery. Mol Cell. 46(4): 542–548. https://doi.org/10.1016/j.molcel.2012.04.031
- Rips, S., Bentley, N., Jeong, I. S., Welch, J. L., von Schaewen, A. and Koiwa, H. (2014). Multiple N-Glycans Cooperate in the Subcellular Targeting and Functioning of Arabidopsis KORRIGAN1. Plant Cell. 26(9): 3792–3808. https://doi.org/10.1105/tpc.114.129718
- Lu, J., Li, N., Li, G., Tian, Z., Shi, L., Wang, Y., Cai, Y., Zhang, K., Sun, W., Wang, D., et al. (2024). N-glycosylation of SnRK2s affects NADPH maintenance in peroxisomes during prolonged ABA signalling. Nat Commun. 15(1): 6630. https://doi.org/10.1038/s41467-024-50720-3
- Chen, T., Zhang, H., Niu, G., Zhang, S. and Hong, Z. (2020). Multiple N-glycans cooperate in balancing misfolded BRI1 secretion and ER retention. Plant Mol Biol. 103: 581–596. https://doi.org/10.1007/s11103-020-01012-z
- Shen, J., Ding, Y., Gao, C., Rojo, E. and Jiang, L. (2014). N‐linked glycosylation of AtVSR1 is important for vacuolar protein sorting in Arabidopsis. Plant J. 80(6): 977–992. https://doi.org/10.1111/tpj.12696
- Häweker, H., Rips, S., Koiwa, H., Salomon, S., Saijo, Y., Chinchilla, D., Robatzek, S. and von Schaewen, A. (2010). Pattern Recognition Receptors Require N-Glycosylation to Mediate Plant Immunity. J Biol Chem. 285(7): 4629–4636. https://doi.org/10.1074/jbc.m109.063073
- Wu, H., Wan, L., Liu, Z., Jian, Y., Zhang, C., Mao, X., Wang, Z., Wang, Q., Hu, Y., Xiong, L., et al. (2024). Mechanistic study of SCOOPs recognition by MIK2–BAK1 complex reveals the role of N-glycans in plant ligand–receptor–coreceptor complex formation. Nat Plants. 10(12): 1984–1998. https://doi.org/10.1038/s41477-024-01836-3
- Jia, F., Xiao, Y., Feng, Y., Yan, J., Fan, M., Sun, Y., Huang, S., Li, W., Zhao, T., Han, Z., et al. (2024). N-glycosylation facilitates the activation of a plant cell-surface receptor. Nat Plants. 10(12): 2014–2026. https://doi.org/10.1038/s41477-024-01841-6
- Lei, C., Li, X., Li, W., Chen, Z., Liu, S., Cheng, B., Hu, Y., Song, Q., Qiu, Y., Zhou, Y., et al. (2025). Chemical Glycoproteomic Profiling in Rice Seedlings Reveals N-glycosylation in the ERAD-L Machinery. Mol Cell Proteomics. 24(2): 100883. https://doi.org/10.1016/j.mcpro.2024.100883
- Yang, J., Zhao, Y., Wang, X., Yang, J., Tang, K. and Liu, J. (2023). N-linked glycoproteome analysis reveals central glycosylated proteins involved in response to wheat yellow mosaic virus in wheat. Int J Biol Macromol. 253: 126818. https://doi.org/10.1016/j.ijbiomac.2023.126818
- Chung, C. Y., Majewska, N. I., Wang, Q., Paul, J. T. and Betenbaugh, M. J. (2017). SnapShot: N-Glycosylation Processing Pathways across Kingdoms. Cell. 171(1): 258–258.e1. https://doi.org/10.1016/j.cell.2017.09.014
- Tarentino, A. L., Gomez, C. M. and Plummer, T. H. (1985). Deglycosylation of asparagine-linked glycans by peptide:N-glycosidase F. Biochemistry. 24(17): 4665–4671. https://doi.org/10.1021/bi00338a028
- Maley, F., Trimble, R. B., Tarentino, A. L. and Plummer, T. H. (1989). Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal Biochem. 180(2): 195–204. https://doi.org/10.1016/0003-2697(89)90115-2
- Bodnar, J., Szekrenyes, A., Szigeti, M., Jarvas, G., Krenkova, J., Foret, F. and Guttman, A. (2016). Enzymatic removal of N‐glycans by PNGase F coated magnetic microparticles. Electrophoresis. 37(10): 1264–1269. https://doi.org/10.1002/elps.201500575
- Trette, V., Altmann, F. and März, L. (1991). Peptide‐N4‐(N‐acetyl‐β‐glucosaminyl)asparagine amidase F cannot release glycans with fucose attached α1 → 3 to the asparagine‐linked N‐acetylglucosamine residue. Eur J Biochem. 199(3): 647–652. https://doi.org/10.1111/j.1432-1033.1991.tb16166.x
- Freeze, H. H. and Kranz, C. (2010). Endoglycosidase and Glycoamidase Release of N‐Linked Glycans. Curr Protoc Mol Biol. 89(1): emb1713as89. https://doi.org/10.1002/0471142727.mb1713as89
Article Information
Publication history
Received: Jun 29, 2025
Accepted: Aug 11, 2025
Available online: Aug 22, 2025
Published: Sep 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Hong, W., Lei, C., Qiu, Y., Zhou, Y., Hu, Y., Chen, X., Li, X. and Li, J. (2025). Verification of N-Linked Glycosylation of Proteins Isolated from Plant or Mammalian Cell Lines Using PNGase Enzyme. Bio-protocol 15(18): e5444. DOI: 10.21769/BioProtoc.5444.
Category
Biochemistry > Protein > Modification
Biochemistry > Protein > Immunodetection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.