- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Novel Gene Stacking Method in Plant Transformation Utilizing Split Selectable Markers
(*contributed equally to this work) Published: Vol 15, Iss 4, Feb 20, 2025 DOI: 10.21769/BioProtoc.5214 Views: 1968
Reviewed by: Xiaofei LiangHao ChenAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2023
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
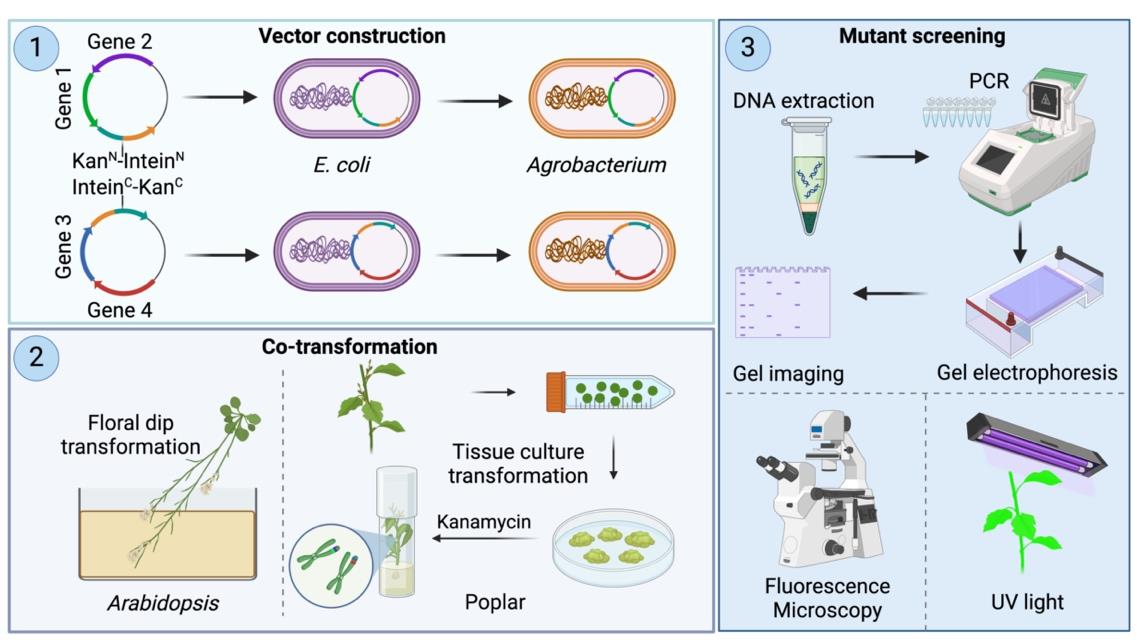
Gene stacking, the process of introducing multiple genes into a single plant to enhance desired traits, is essential for plant genetic improvement through both conventional breeding and genetic transformation. In general, transformation-based gene stacking can be achieved through either co-transformation to simultaneously introduce multiple genes or sequential multi-round transformation. While co-transformation is generally faster and more efficient than sequential multi-round transformation, it often requires two selectable marker genes, which confer resistance to antibiotics, for selecting transgenic events. However, in most cases, there is only one best selectable marker gene for a specific plant species or genotype. Also, it is harder to optimize the concentrations of two antibiotics for co-transformation than using one antibiotic for selecting transgenic events. To overcome this challenge, we recently developed an innovative split selectable marker system for plant co-transformation, allowing the use of one selectable marker gene to select transgenic events. This method involves constructing two binary vectors, each carrying a subset of genes of interest and a partial fragment of the selectable marker gene, which is connected to a partial intein fragment. Following Agrobacterium-mediated co-transformation, plants harboring both binary vectors are selected using a single antibiotic, such as kanamycin. This split-marker system can be used to co-transform multiple genes into both herbaceous and woody plants, accelerating genetic improvement of polygenic traits or integrative improvement of multiple traits to simultaneously increase crop yield and quality.
Key features
• Developed an intein-mediated split selectable marker system for efficient gene stacking in plants.
• Utilizes a single antibiotic for identifying transgenic events, simplifying the selection process of co-transformation compared to traditional methods.
• Applicable to both herbaceous and woody plant species for co-transforming multiple genes.
• Enhances scalability and feasibility of gene stacking in plant genetic engineering and crop improvement initiatives.
Keywords: InteinGraphical overview
Background
Gene stacking is a pivotal strategy in modern agriculture, involving the integration of multiple beneficial genes into a single organism to simultaneously enhance traits such as yield, disease resistance, stress tolerance, and nutritional content [1]. This approach is particularly valuable in the metabolic engineering of plants due to the intricate nature of metabolic processes and biochemical pathways, which typically involve complex interactions among multiple genes [2]. Gene stacking in plants can be achieved through several methods. First, hybrid stacking involves cross-hybridizing a plant containing one or more transgenes with another plant carrying different transgenes, resulting in the development of a multi-stack hybrid through iterative hybridization. Second, co-transformation entails transforming a plant with two or more independent transgenes, each in separate DNA constructs delivered simultaneously to the plant. Third, sequential multi-round transformation involves re-transformation of a plant already harboring a transgene with additional transgenes [3].
Co-transformation has proven to be a highly promising approach for introducing multiple genes into plants [4]. Typically, co-transformation involves using two different selectable marker genes simultaneously, which must be compatible with both the plant species and the transformation method employed. Challenges arise in ensuring these markers are effective and efficient, requiring careful optimization of transformation protocols and selection conditions to maintain high transformation efficiency. Only a limited number of marker genes, such as hygromycin phosphotransferase (hpt), neomycin phosphotransferase II (nptII), and bar, are commonly used in plant research and crop development, and no single selectable marker gene has been found to be universally effective in all situations [5,6]. To address this challenge, we recently used a split selectable marker system to facilitate gene stacking in plants [7]. This system contains two independent binary vectors, each carrying distinct genes of interest. Unlike the traditional co-transformation system, in which each vector typically contains a different selectable marker, each vector in our split selectable marker system contains a partial fragment of the selectable marker gene, which is connected to a partial intein fragment. The partial intein fragment refers to a segment of an intein that facilitates protein splicing in engineered systems. After the two vectors are co-transformed into the same plant cell, intein-mediated protein splicing occurs post-translationally, resulting in the reassembly of two marker protein fragments into a full-length selectable marker protein. In this natural process, an intein, a protein segment within a precursor protein, self-excises and ligates the remaining protein fragments (exteins) to form a functional protein [8]. This innovative system simplifies the identification of transgenic events generated by co-transformation using a single selection agent (e.g., antibiotic). This approach avoids the inefficiencies and incompatibilities associated with using two different selectable markers in the traditional co-transformation. Designing and constructing intein-mediated selectable markers is highly relevant to molecular cloning. Here, we describe in great detail the methodology—used in the original publication by Yuan et al. [7]—for creating split-marker constructs, co-transforming two vectors in two plant species (Arabidopsis thaliana and Populus tremula × P. alba clone INRA 717-1B4), and confirming transgenic events. Also, we created ready-to-use binary vectors to simplify the cloning procedure for creating DNA constructs for co-transformation.
Materials and reagents
Reagents
1. Agar (PhytoTech LABS, catalog number: HWW1000003C)
2. Agarose (GenBiotech, catalog number: RU1010)
3. Acetosyringone (Sigma-Aldrich, catalog number: D134406)
4. Cefotaxime (PhytoTech LABS, catalog number: C380)
5. NEBridge® Golden Gate Assembly kit (BsaI-HF® v2) (NEB, catalog number: E1601L)
6. NEB® 5-alpha competent E. coli (NEB, catalog number: C2987H)
7. PCR Purification kit (Inbio Highway, catalog number: K1206)
8. EHA105 Agrobacterium ElectroCompetent cells
9. Plasmid DNA Purification kit (Qiagen, catalog number: 12123)
10. Restriction enzymes: Depending on the restriction sites on the inserts that the user cloned
11. Sterilizing agents: ethanol
12. Silwet L-77
13. GoTaq® G2 Master Mixes (Promega, catalog number: 0000434787)
14. Murashige & Skoog basal medium with vitamins (MS) (PhytoTech LABS, catalog number: M5531)
15. MES (Sigma, catalog number: SLCF3242)
16. NAA (Sigma, catalog number: RNBJ597610)
17. IBA (PhytoTech LABS, catalog number: I538)
18. BAP (PhytoTech LABS, catalog number: B130)
19. 2iP; 6-(γ,γ-Dimethylallylamino)purine Solution (PhytoTech LABS, catalog number: D217)
20. 0.5 M EDTA (Millipore, catalog number: 3740903)
21. 5 M NaCl (Sigma, catalog number: SLBR7232V)
22. 1 M Tris-HCl (Invitrogen, catalog number:15567-027)
23. Chloroform, EMSURE® ACS, ISO, Reag. Ph. (VWR, catalog number: 1.02445.1000)
24. Isopropanol (2-Propanol) (VWR, catalog number: 1.09634.5000)
25. L-Glutamine (PhytoTech LABS, catalog number: HWY0229022A)
26. Phyto agar (Sigma, catalog number: P8169)
27. Kanamycin (PhytoTech LABS, catalog number: HHW47510054)
28. LB broth with agar (Sigma, catalog number: L3147)
29. LB broth (Sigma, catalog number: L3022)
30. Sucrose (Cicarelli, catalog number: 841214)
31. Sterile water
32. Timentin (GoloBio, catalog number: T-104-25)
33. Liquid nitrogen
34. Gel Extraction kit (Zymoclean Gel DNA Recovery kit, catalog number: D4008)
35. Q5® High-Fidelity 2× master mix (NEB, catalog number: M0492S)
36. TE buffer (Thermo Fisher Scientific, catalog number: 12090015)
37. Thidiazuron (TDZ) solution, 1 mg/mL (PhytoTech Labs, catalog number: T7999)
38. Cetyltrimethylammonium bromide (CTAB) (Sigma-Aldrich, catalog number: 219374)
Primers (for genotyping)
eYGFPuv_F: 5'-CACGGCAACCTCAACG-3'
eYGFPuv_R: 5'-CTCGACACGTCTGTGGG-3'
Solutions
1. Dip solution (see Recipes)
2. Callus induction media (CIM) (see Recipes)
3. Shoot induction media (SIM) (see Recipes)
4. Shoot elongation media (SEM) (see Recipes)
5. Root induction media (RM) (see Recipes)
6. LB agar medium (see Recipes)
7. LB medium (see Recipes)
8. MS induction medium (see Recipes)
9. 3% CTAB extraction buffer (see Recipes)
Recipes
1. Dip solution
5% sucrose
0.03% Silwet L-77
120 mL deionized water
2. Callus induction media (CIM)
4.3 g/L MS with vitamins
30 g/L sucrose
3 g/L Phyto agar
0.2 g/L L-glutamine
0.25 g/L MES
1 mL of 10 μM/L NAA
Sterilize and add 2ip (5 μM final) 1 mL of 5 mM 2ip stock, timentin (200 mg/mL stock) 1 mL/L, cefotaxime (300 mg/mL stock) 1 mL/L, and kanamycin at 100 mg/L (final concentration).
3. Shoot induction media (SIM)
4.3 g/L MS with vitamins
30 g/L sucrose
3 g/L Phyto agar
0.2 g/L L-glutamine
0.25 g/L MES
50 μL of 1 mg/mL TDZ
Sterilize and add timentin (200 mg/mL stock) 1 mL/L, cefotaxime (300 mg/mL stock) 1 mL/L, and kanamycin at 100 mg/L (final concentration).
4. Shoot elongation media (SEM)
4.3 g/L MS with vitamins
30 g/L sucrose
3 g/L Phyto agar
0.2 g/L L-glutamine
0.25 g/L MES
100 μL of 1 mg/mL BAP
Sterilize and add timentin (200 mg/mL stock) 1 mL/L, cefotaxime (300 mg/mL stock) 1 mL/L, and kanamycin at 100 mg/L (final concentration).
5. Root induction media (RM)
2.15 g/L MS with vitamins
20 g/L sucrose
3 g/L Phyto agar
0.2 g/L L-glutamine
0.25 g/L MES
100 μL of 1 mg/mL IBA
Sterilize and add timentin (200 mg/mL stock) 1 mL/L, cefotaxime (300 mg/mL stock) 1 mL/L, and kanamycin at 100 mg/L (final concentration).
6. LB agar medium
40 g/L LB broth with agar
7. LB medium
20 g/L LB broth
8. MS induction medium
4.43 g/L MS with vitamins
20 g/L sucrose
0.5 g/L MES
20 μM (final concentration) acetosyringone
9. 3% CTAB extraction buffer (50 mL)
Dissolve 1.5 g of CTAB in 16.5 mL of distilled water on a heating plate.
Add 7.5 mL of 1 M Tris-HCl (pH 7.4), 26 mL of 5 M NaCl, and 0.2 mL of 0.5 M EDTA (pH 8).
Filter-sterilize using a 0.2 μm filter.
Laboratory supplies
1. 200 μL PCR tubes (Sigma, catalog number: Z316121)
2. 1.5 mL microcentrifuge tubes (Sigma, catalog number: SLMTBP15-EP)
3. 50 mL Falcon tubes (VWR, catalog number: 21008-940)
4. 14 mL round-bottom tubes (Sigma, catalog number: Z617806)
5. Plant culture vessel (PhytoCon, 16 oz, 473 mL)
6. Pipette and pipette tips (Eppendorf)
7. 100 mm × 15 mm Petri dishes (Sigma, catalog number: Z666246)
8. Scalpel and forceps
9. Gloves
Equipment
1. E. coli and Agrobacterium pulser transformation apparatus (Bio-Rad, model: 155103)
2. High-speed centrifuges (Thermo Fisher Scientific, SorvallTM ST8 Small Benchtop Centrifuge; catalog number: 75007200)
3. Floor model centrifuge (Thermo Fisher Scientific, SorvallTM ST8 FR Floor-Standing Refrigerated Centrifuge; catalog number: 75007208)
4. Incubator (Sigma, catalog number: Z763330)
5. Incubator shaker (Sigma, model: Innova 44 incubator shaker)
6. Biosafety cabinets (LABCONCO, catalog number: 302380011)
7. Plant growth chamber (NORLAKE Scientific)
8. Autoclave
9. Vortex (FISHER MINI VORTEXER, catalog number: 02215365)
10. Nanodrop (Denovix DS11)
11. Gel Doc (Azure Biosystems 600)
12. Cuvette (Bio-Rad, 0.2 cm electrode gap cuvette, catalog number: 1652086)
Software and datasets
1. SnapGene (GSL Biotech LLC, https://www.snapgene.com/)
Procedure
A. Design and construction of vectors for plant co-transformation
1. Construction of vectors 1 and 2 using Golden Gate Assembly
a. Vectors pSplit 1 and pSplit 2 were modified as destination vectors, ready for Golden Gate Assembly.
b. Combine the destination vector pSplit 1 with insert 1 and 2 and mix the destination vector pSplit 2 with insert 3 and 4 (Figure 1) (see General note 1).
c. Assemble all fragments according to the user manual provided by NEB (see General note 2).
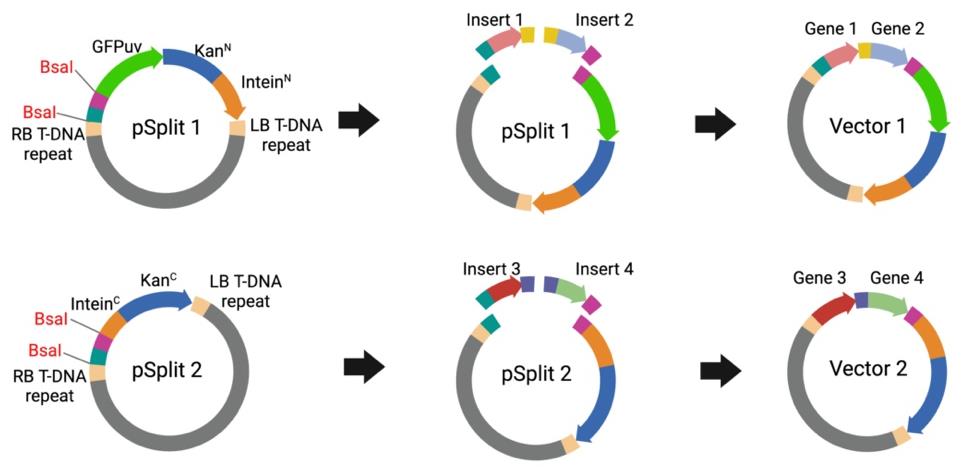
Figure 1. Vector cloning using Golden Gate Assembly. Inserts 1 and 2 contain Gene 1 and Gene 2, respectively, while Inserts 3 and 4 contain Gene 3 and Gene 4, respectively. pSplit 1 serves as the vector backbone and is mixed with Inserts 1 and 2 to assemble into Vector 1 via Golden Gate Assembly. Similarly, pSplit 2 serves as the vector backbone and is mixed with Inserts 3 and 4 to assemble into Vector 2 via Golden Gate Assembly. Although two inserts are used as examples for each vector, additional inserts can also be efficiently assembled using this method.
2. Gene sequence selection and primer design
a. Identify the gene sequences to be inserted into the vectors and their promoters and terminators (see General note 3).
b. Design primers for PCR amplification of the gene sequences, including restriction sites (BsaI) for cloning (Figure 2).
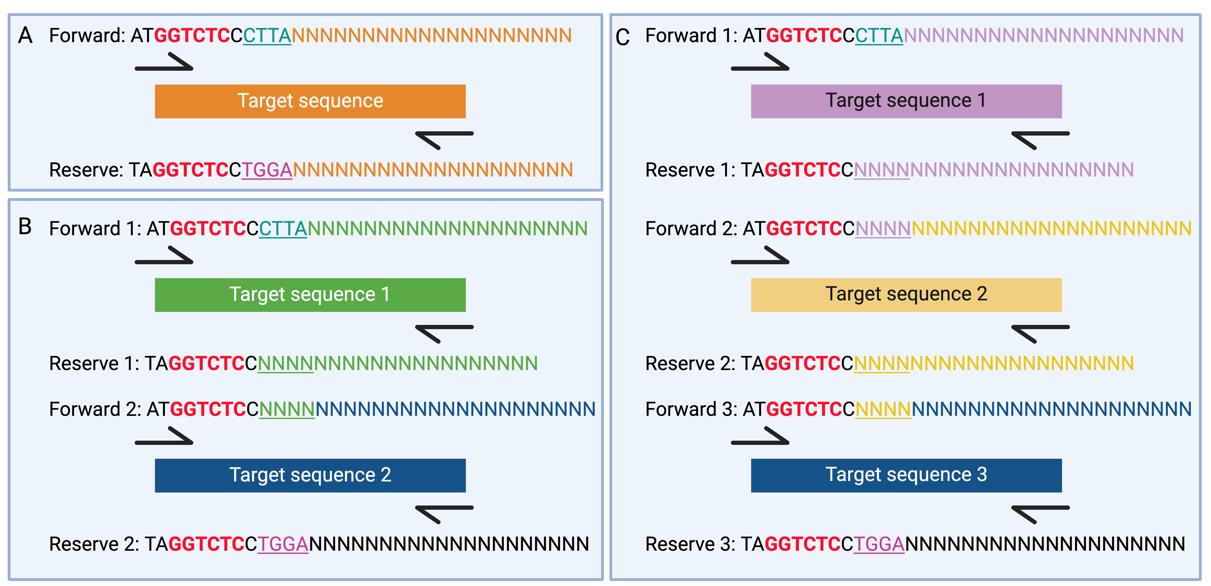
Figure 2. Primer design for vector cloning. A. Primer design of single insert. B. Primer design of double inserts. C. Primer design of three inserts. Red sequences indicate BsaI restriction sites. Underlined sequences represent the 4-bp overhangs required for Golden Gate Assembly. The sequences NNN---NNN indicate the annealing sequence with the target sequence.
3. PCR amplification
a. Perform PCR to amplify the target gene sequences using Q5 high-fidelity DNA polymerase (see General note 4).
b. Verify the PCR products by running a sample on an agarose gel.
c. Purify the PCR product using a gel extraction kit.
4. Transformation into E. coli
a. Transform the ligated products into competent E. coli cells using heat shock following the manufacturer’s protocol for C2987H.
b. Plate the transformed cells on LB agar plates containing kanamycin (50 μg/mL).
c. Incubate the plates overnight at 37 °C.
5. Colony PCR screening
a. Pick individual colonies for colony PCR using GoTaq® G2 Master Mixes.
b. Verify the PCR products by running a sample on an agarose gel.
c. Inoculate the positive colonies into liquid LB medium containing kanamycin (50 μg/mL).
d. Incubate the culture tubes overnight at 37 °C.
6. Plasmid Miniprep
a. Isolate plasmid DNA from the overnight cultures using a miniprep kit.
b. Verify the presence and orientation of the inserts by restriction digestion (see General note 5).
7. Plasmid sequencing
a. Sequence the purified plasmid DNA to confirm the correct insertion and orientation of the gene sequences.
b. Use sequencing primers that anneal to the plasmid backbone flanking the insert.
B. Agrobacterium transformation
1. Thaw the electrocompetent Agrobacterium EHA105 on ice.
2. Add 1 μL of constructed plasmid containing pSplit1 and pSplit2 into Agrobacterium separately and mix well by gently flicking.
3. Transfer the mixture immediately to the electroporation cuvette.
4. Electroporate at 2.4 kV until the apparatus beeps.
5. Add 1 mL of LB medium into the cuvette, resuspend, and transfer back to an Eppendorf tube.
6. Incubate for 1–2 h at 30 °C with shaking at 180 rpm.
7. Plate on LB agar plates with kanamycin (50 μg/mL) and rifampicin (50 μg/mL).
8. Incubate the plates for 48 h at 30 °C.
9. Pick individual colonies for colony PCR using GoTaq® G2 Master Mixes.
10. Verify the PCR products by running a sample on an agarose gel.
C. Agrobacterium-mediated co-transformation in Arabidopsis thaliana
1. Preparation of Agrobacterium tumefaciens cultures
a. Start a preculture by inoculating each of two selected Agrobacterium tumefaciens strains containing pSplit1 or pSplit2 into 5 mL of LB medium with kanamycin (50 μg/mL) and rifampicin (50 μg/mL) in a 50 mL Falcon tube. (see General note 6).
b. Incubate the culture overnight at 28 °C with shaking at 180 rpm and monitor growth. Alternatively, monitor for two nights if necessary until OD reaches 0.8–1.0 (see General note 7).
c. Inoculate 10 μL of the preculture into 100 mL of LB medium with antibiotic selection in a sterile Erlenmeyer flask.
d. Incubate overnight at 28 °C with shaking at 180 rpm.
2. Dipping of the plants
a. Verify that both cultures have grown well (see General note 7).
b. Prepare the floral dip solution (see Recipes).
c. For each cultured strain, divide the 100 mL culture into two Falcon tubes.
d. Centrifuge at 3,500× g for 7–10 min.
e. Carefully remove the supernatant.
f. Resuspend the pellet in 30 mL (total volume) of LB medium without antibiotics.
g. Pour 120 mL of floral dip solution into a plastic-covered tray and add 30 mL of bacterial suspension from each of the two strains (a total of 60 mL) to the center of the dip solution.
h. Submerge each plant completely in the liquid for a few seconds (see General note 8).
i. Place the dipped plants back in the original pots or transfer them to an appropriate container.
j. Dip all the plants of the same construct and then clean up the area.
k. Leave the plants on the bench for 48 h.
l. Transfer the plants to the growth chamber (temperature: 22–25 °C; photoperiod: 16/8 h light/dark cycle; humidity: 50%–55%).
m. Once plants are dry, you can collect the seeds.
D. Agrobacterium-mediated co-transformation in poplar (Populus tremula × P. alba clone INRA 717-1B4)
1. Preparation of Agrobacterium tumefaciens cultures
a. Streak Agrobacterium tumefaciens (EHA105) of two constructs on an LB agar plate containing kanamycin (50 μg/mL) and rifampicin (50 μg/mL). Then, incubate the plate at 28 °C for 2 days.
b. Inoculate a single A. tumefaciens colony into 5 mL of LB medium with kanamycin (50 μg/mL) and rifampicin (50 μg/mL) and grow overnight at 28 °C with shaking (180 rpm).
c. Add 100 µL of culture into 50 mL of LB medium with kanamycin (50 μg/mL) and rifampicin (50 μg/mL) and grow overnight at 28 °C with shaking (180 rpm).
d. The next day, centrifuge the cultures at 3,500× g for 15 min at room temperature to pellet the cells.
e. Discard the supernatant and proceed to the next step.
2. Resuspension of Agrobacterium pellets
a. Resuspend each Agrobacterium pellet in MS induction medium.
b. Ensure the MS induction medium contains 20 μM acetosyringone to enhance virulence.
c. Adjust the OD600nm of the suspension to 0.5 using the MS induction medium.
3. Preparation of poplar leaf disks
a. Excise approximately 150 young leaf disks (~0.5 cm diameter) from healthy poplar 717 (Populus tremula × alba clone INRA 717-1B4) leaves using a sterilized punch or scalpel (see General note 9).
b. Keep the leaf disks on sterile filter paper moistened with MS medium until ready for infection.
4. Agrobacterium infection
a. Mix an equal volume of MS induction medium containing two split constructs into a 50 mL conical flask.
b. Soak the excised leaf disks in the Agrobacterium suspension for 1 h.
c. Agitate gently during the infection process to ensure even exposure.
5. Co-culture
a. Transfer the soaked leaf disks onto solid co-culture medium (e.g., MS medium supplemented with the required hormones and 20 μM acetosyringone).
b. Incubate the plates in the dark at 22–25 °C for 2–3 days to allow co-culturing.
6. Washing
a. After co-culture, wash the leaf disks thoroughly with sterile water containing antibiotics (e.g., 300 mg/L cefotaxime and 200 mg/L timentin) for 1 h to remove excess Agrobacterium.
b. Blot the disks dry on sterile filter paper before transferring them to the callus induction medium (CIM) containing antibiotics (e.g., 300 mg/L cefotaxime, 200 mg/L timentin, and 100 mg/L kanamycin).
7. Callus induction
a. Transfer the washed leaf disks onto solid callus induction medium (e.g., MS medium supplemented with appropriate plant growth regulators (see CIM recipe).
b. Incubate the plates under controlled light conditions (16/8 h light/dark cycle) at 22–25 °C.
c. Monitor the disks for callus formation, which may take 2–4 weeks.
d. Subculture immediately if Agrobacterium re-growth is observed.
8. Shoot induction
a. Transfer the developing calli to shoot induction medium (SIM) (e.g., MS medium with cytokinin and antibiotics; see Recipes).
b. Continue incubation under the same light and temperature conditions until shoots begin to emerge.
c. Subculture every 2–3 weeks. Adventitious shoots typically emerge within 4–6 weeks. Separate transformation events as early as possible during subculturing, either at the callus or multiple shoot stages.
9. Shoot elongation
a. Transfer emerging shoots to shoot elongation medium (SEM) (e.g., MS medium with a lower concentration of cytokinin and antibiotics; see Recipes).
b. Incubate until the shoots have elongated sufficiently, which typically takes 2–3 weeks.
10. Root induction
a. Transfer elongated shoots to root induction medium (RM) (e.g., MS medium with auxin).
b. Incubate under appropriate conditions until roots develop, confirming successful transformation.
E. Confirmation of transformation
1. Sample collection: collect leaf samples (0.5–1.0 cm) from the Arabidopsis and poplar 717 lines (see General note 10).
2. Leaf grinding
a. Place the collected leaf samples in a pre-cooled mortar.
b. Grind the leaves into a fine powder using liquid nitrogen.
3. Genomic DNA isolation
a. Add 500 μL of 3% CTAB extraction buffer to 100 mg of powdered plant material in a 2.0 mL microcentrifuge tube. Vortex the mixture vigorously to ensure thorough mixing.
b. Incubate the tubes in a 65 °C water bath for 30 min. During the incubation, invert the tubes every 5–10 min to keep the contents mixed.
c. After incubation, centrifuge the samples at 13,000× g for 10 min. Carefully transfer the supernatant to a new 1.5 mL microcentrifuge tube.
d. Add 600 μL of chloroform to the supernatant and vortex thoroughly.
e. Centrifuge at 13,000× g for 10 min. Transfer the upper aqueous phase to a clean 1.5 mL microcentrifuge tube.
f. Add an equal volume of chloroform to the transferred aqueous phase and vortex to mix.
g. Centrifuge the mixture at 13,000× g for 10 min. Again, transfer the upper aqueous phase to a clean 1.5 mL tube.
h. Add an equal volume of isopropanol (2-propanol) to the aqueous phase. Mix by pipetting up and down, then precipitate the DNA by placing the tube at -20 °C for 30 min.
i. Centrifuge the precipitated mixture at 13,000× g for 20 min. Carefully discard the supernatant, avoiding disturbance of the DNA pellet, and allow the pellet to air dry briefly.
j. Add approximately 500 μL of chilled 70% ethanol to the pellet and centrifuge at 13,000× g for 5 min to wash the DNA pellet. Discard the ethanol and allow the pellet to air dry completely.
k. Finally, dissolve the DNA pellet in 100 μL of 1× TE buffer.
4. PCR setup
PCR reaction mixture (25 μL volume)
GoTaq® G2 Green master mix, 2× 12.5 μL
eYGFPuv_F (10 μM) 2.5 μL
eYGFPuv_R (10 μM) 2.5 μL
DNA template 1.0 μL
Nuclease-free water to 25 μL
a. For eYGFPuv genotyping, add the eYGFPuv_F (5'-CACGGCAACCTCAACG-3') and eYGFPuv_R (5'-CTCGACACGTCTGTGGG-3') (see General note 11).
b. Mix the reagents gently.
5. PCR cycling conditions (Table 1):
Table 1. PCR cycling conditions
| Step | Temperature | Time | Cycle |
| Initial denaturation | 95 °C | 2 min | 1 cycle |
| Denaturation | 95 °C | 10–30 s | 25–35 cycles |
| Annealing | 55–60 °C | 30 s | |
| Extension | 72 °C | 1 min/kb | |
| Final extension | 72 °C | 5 min | 1 cycle |
| Rest | 4 °C | Indefinite | 1 cycle |
6. Post-PCR analysis
Analyze the PCR products using agarose gel electrophoresis to confirm the presence of the target genes.
Validation of protocol
This protocol was used to generate the co-overexpression lines by Yuan et al. [7].
General notes and troubleshooting
General notes
1. The number of DNA fragments varies based on user needs, with successful assemblies reported ranging from 1 to 16 fragments.
2. Golden Gate Assembly protocol for using NEBridge® Golden Gate Assembly Kit (BsaI-HF®v2) (E1601) can be found at https://www.neb.com/en-us/protocols/2018/10/02/golden-gate-assembly-protocol-for-using-neb-golden-gate-assembly-mix-e1601. The manual does not mention that the destination plasmid requires pre-digestion with the BsaI enzyme followed by gel purification. However, we found that performing pre-digestion and gel purification significantly improves assembly efficiency, especially when the number of inserts exceeds two.
3. For gene overexpression, the gene sequence can either be the coding sequence of the gene of interest (without introns) or its genomic sequence (containing introns), as both are effective.
4. DNA fragments to be inserted into the binary vectors can also be commercially synthesized by vendors such as Integrated DNA Technologies (IDT, Coralville, IA) and Twist Bioscience (South San Francisco, CA). The amount of a DNA insert varies depending on the user’s purpose. Single or multiple gene cassettes can be inserted into destination plasmids through Golden Gate Assembly in a one-pot reaction.
5. Restriction digestion is optional here if positive colonies have been previously verified through colony PCR.
6. Only use a fresh colony. Do not close the Falcon tube completely and tape the lid with micropore tape.
7. The culture must be well-grown and clearly turbid.
8. Ensure the rosette is submerged during floral dipping for Arabidopsis transformation. You will observe air bubbles between the leaves and stem.
9. To achieve optimal transformation efficiency, avoid using overly mature leaves. Instead, select leaves from 1–2-month-old plants grown in vitro.
10. We detected two co-transformed vectors in both the T1 and T2 generations of Arabidopsis. Kanamycin (50 μg/mL) was used during seed germination to select for successful transformation.
11. You may design specific primers tailored to your gene of interest.
Acknowledgments
Funding: The work was supported by the Center for Bioenergy Innovation (CBI), a DOE Research Center supported by the Biological and Environmental Research (BER) program. Oak Ridge National Laboratory is managed by UT-Battelle, LLC for the U.S. DOE under Contract Number DE-AC05-00OR22725. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript or allow others to do so, for United States Government purposes. This protocol has been validated in Yuan et al. [7].
Competing interests
The authors declare no conflict of interest.
References
- Roberts, D. and Mattoo, A. (2018). Sustainable Agriculture—Enhancing Environmental Benefits, Food Nutritional Quality and Building Crop Resilience to Abiotic and Biotic Stresses. Agriculture. 8(1): 8.
- Naqvi, S., Farré, G., Sanahuja, G., Capell, T., Zhu, C. and Christou, P. (2010). When more is better: multigene engineering in plants. Trends Plant Sci. 15(1): 48–56.
- Ceccon, C. C., Caverzan, A., Margis, R., Salvadori, J. R. and Grando, M. F. (2020). Gene stacking as a strategy to confer characteristics of agronomic importance in plants by genetic engineering. Ciência Rural. 50(6): e20190207.
- Halpin, C. (2005). Gene stacking in transgenic plants – the challenge for 21st century plant biotechnology. Plant Biotechnol J. 3(2): 141–155.
- Miki, B. and McHugh, S. (2004). Selectable marker genes in transgenic plants: applications, alternatives and biosafety. J Biotechnol. 107(3): 193–232.
- Ahmed, S., Hulbert, A. K., Xin, X. and Neff, M. M. (2024). The ability of Arabidopsis to recover from Basta and its application in isolating Cas9-free mutants. Front Plant Sci. 15: e1408230.
- Yuan, G., Lu, H., De, K., Hassan, M. M., Liu, Y., Islam, M. T., Muchero, W., Tuskan, G. A. and Yang, X. (2023). Split selectable marker systems utilizing inteins facilitate gene stacking in plants. Commun Biol. 6(1): 567.
- Perler, F. B. (2002). InBase: the Intein Database. Nucleic Acids Res. 30(1): 383–384.
Article Information
Publication history
Received: Sep 3, 2024
Accepted: Jan 5, 2025
Available online: Jan 16, 2025
Published: Feb 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Yuan, G., Islam, M. T., Tuskan, G. A. and Yang, X. (2025). A Novel Gene Stacking Method in Plant Transformation Utilizing Split Selectable Markers. Bio-protocol 15(4): e5214. DOI: 10.21769/BioProtoc.5214.
Category
Plant Science > Plant transformation > Agrobacterium
Plant Science > Plant molecular biology > DNA > Gene expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.