- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Leveraging Circular Polymerization and Extension Cloning (CPEC) Method for Construction of CRISPR Screening Libraries
Published: Vol 15, Iss 4, Feb 20, 2025 DOI: 10.21769/BioProtoc.5183 Views: 2777
Reviewed by: Kristin L. ShinglerEmmanuel Orta-ZavalzaAnonymous reviewer(s)
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Recent advancements in high-throughput functional genomics have substantially enhanced our comprehension of the genetic and molecular dimensions of cancer, facilitating the identification of novel therapeutic targets. One of the key methodological innovations in this field is the CRISPR screening strategy, which has proven efficacy in elucidating essential gene functions and pathway alterations critical to cancer cell survival and fitness. The construction of custom CRISPR libraries permits the integration of tailored single-guide RNAs (gRNAs), offering greater flexibility as well as specificity in comparison to the commercially available libraries, and enables more refined secondary screening strategies to attenuate the selection of false positive potential gene candidates. Among various molecular cloning techniques, circular polymerase extension cloning (CPEC) has emerged as a highly efficient and cost-effective approach. CPEC utilizes polymerase overlap extension to assemble overlapping DNA fragments into circular plasmids, eliminating the need for restriction digestion and ligation and thus streamlining the creation of both single and multi-fragment constructs. In this protocol, we present the application of the CPEC method to construct the EpiTransNuc knockout gRNA library, specifically designed to target epigenetic regulators, transcription factors, and nuclear proteins. The custom library, assembled using the lentiGuide-Puro backbone, comprises 40,820 gRNAs, with 10 gRNAs per gene, along with 100 non-targeting control gRNAs. Importantly, the CPEC method can be tailored to meet the specific requirements of other custom gRNA libraries, offering flexibility for diverse research applications.
Key features
• Involves PCR-based linearization of the backbone with designed primer sets.
• Facilitates flexibility in gRNA composition and number in library construction.
• Skips conventional cloning techniques such as restriction digestion and ligation.
Keywords: Circular polymerase extension cloning (CPEC)Graphical overview
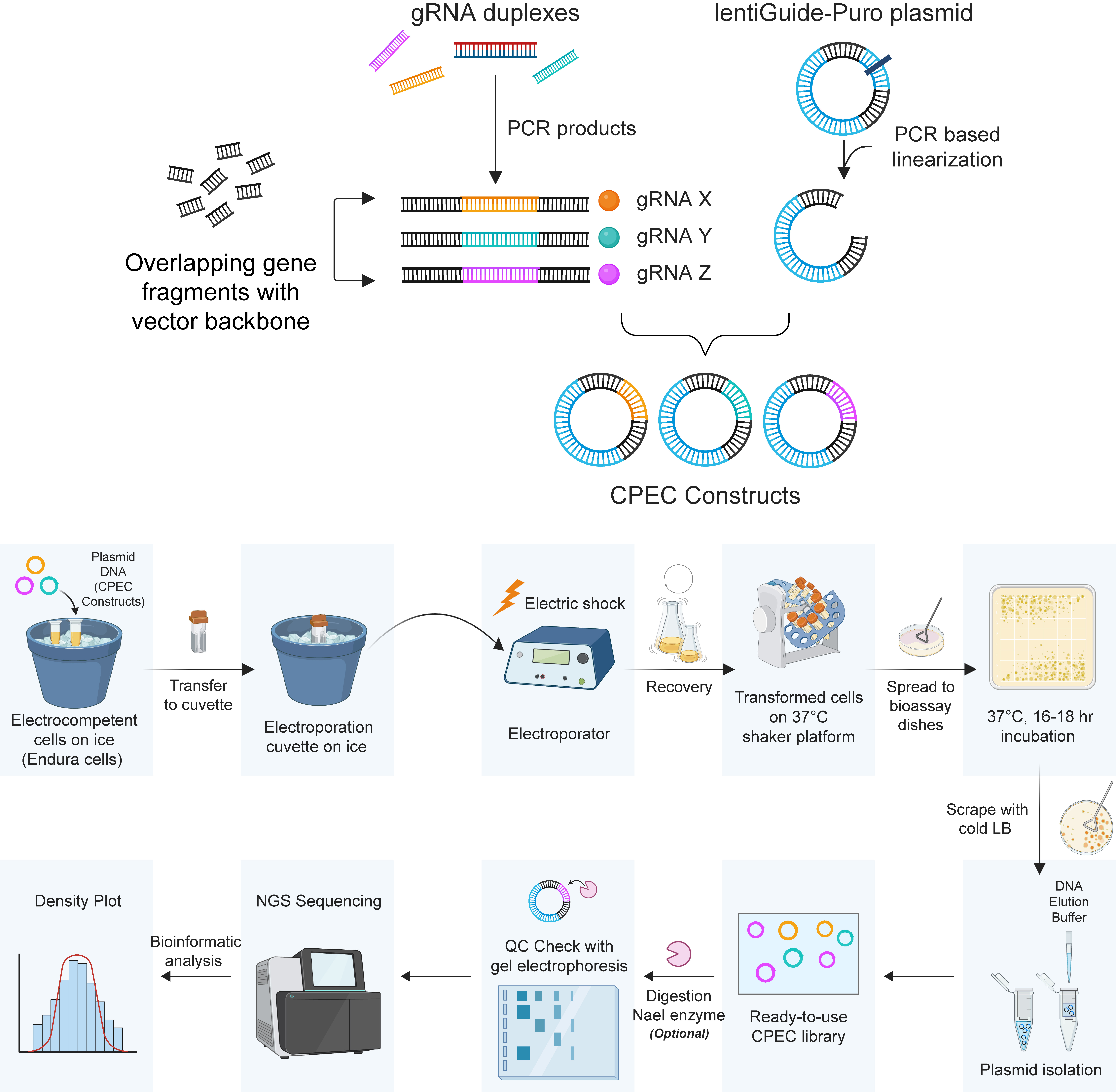
Schematic representation of the circular polymerase extension cloning (CPEC) procedure
Background
Our knowledge of the genetic and molecular aspects of cancer has advanced considerably owing to high-throughput functional genomics, which facilitates the identification of potential treatment targets or therapeutic vulnerabilities for a range of cancer types [1]. Genome-wide or focused CRISPR screening strategy has proven to be a versatile tool to unravel genes and even alterations in pathways having a major role in the survival and fitness of cancer cells, as well as therapy resistance [2,3]. Researchers can uncover details about gene functions, interactions, and regulatory networks by creating libraries containing different CRISPR constructs [4]. This understanding is crucial for deciphering complex biological systems and diseases.
Despite the availability of genome-wide or focused CRISPR libraries from commercial or repository sources, the creation of custom-built CRISPR libraries holds paramount importance for several reasons. First and foremost, de novo library construction enables the incorporation of a specific number of gRNAs, rather than being constrained by a predetermined or limited quantity. In contrast to genome-wide commercial libraries, which typically include a limited number of gRNAs per gene, custom-built libraries allow for a higher number of gRNAs per gene, thereby improving the reliability of the observed phenotypes. Moreover, to refine results and eliminate false negative candidates, secondary screens that focus on potential hits identified in primary screens are often conducted. To accomplish this, constructing a de novo library using gene sets that have already been pre-screened would become increasingly important. Second, the composition of the library can be adjusted with respect to the focus of the research. Although this study explores the CRISPR library within the context of cancer research, its application extends beyond this field. To exemplify, engineering gRNAs targeting transcription factors, metabolic genes, and immune regulation genes can be prioritized based on the specific research focus. Last but not least, a vector backbone with a Cas9 expression cassette or one that lacks Cas9 can be chosen.
In this context, researchers exploit cloning methodologies, which are classified into two primary categories: those that are sequence-dependent (Gateway cloning) and those that are sequence-independent [5], typically based on homologous recombination. Sequence-dependent strategies utilize restriction digestion-ligation methods or site-specific recombination and necessitate unique and specific sites within the insert, vector, or both. This dependence on unique sites makes these approaches less ideal for multi-fragment cloning. On the other hand, sequence-independent approaches comprise of ligation independent cloning (LIC) [6], sequence and ligation-independent cloning (SLIC) [7], Gibson assembly [8], and covalently-closed-circular synthesized (3Cs) [9]. All of these methods have pros and cons, including circular polymerization extension cloning (CPEC); however, CPEC could be exploited for the construction of CRISPR libraries whenever someone opts for a more cost-effective and streamlined approach.
Circular polymerase extension cloning (CPEC) represents a relatively cost-effective, high-efficiency cloning technique compared to the Gibson assembly and other protocols used in molecular cloning. It operates on the principle of polymerase overlap extension and is considered a robust alternative due to its omission of restriction digestion, ligation, and other procedural steps [10]. CPEC enables the transformation of overlapping DNA fragments into a double-stranded circular form via the polymerase extension mechanism, thereby facilitating the integration of the insert into the target plasmid. During the CPEC reaction, linear double-stranded inserts and vectors are first separated through increasing temperature (denaturation). Subsequently, the resulting single-stranded products anneal through their overlapping regions and use each other as templates to construct the circular plasmid. Through the CPEC method, not only a single gene but also multi-fragment assembly could be obtained. From this point of view, CPEC could have a pivotal role in the construction of CRISPR libraries (de novo). In this method, it is crucial to uniquely select the overlapping regions of the insert and vector, and the melting temperature (Tm) should be as high as possible to minimize the likelihood of vector self-ligation and concatenation.
Here, we present a robust, cost-effective, and straightforward CPEC-based protocol to engineer a custom-built gRNA library for CRISPR screening research. The knockout gRNA library, enriched for transcription factors and epigenetic regulators as well as nuclear factors, hence named EpiTransNuc, was constructed by exploiting the PCR linearized lentiGuide-Puro backbone and comprises 40820 gRNAs, comprising 10 gRNAs per gene and 100 non-targeting controls.
Materials and reagents
Biological materials
1. Endura electrocompetent E. coli bacteria (Lucigen, catalog number: 60242-1)
Reagents
1. lentiGuide-Puro backbone (Addgene, catalog number: 52963)
2. Designed primer sets for lentiGuide-Puro backbone linearization:
a. Forward primer: GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACC
b. Reverse primer: CGGTGTTTCGTCCTTTCCACAAGATATATAAAGCCAAGAAATCGAAATACTTTCAAGTTACGG
3. Q5 reaction buffer (New England Biolabs, catalog number: B9027S)
4. Q5 high GC enhancer (New England Biolabs, catalog number: B9028A)
5. 10 mM dNTPs (New England Biolabs, catalog number: N0447S)
6. Q5 high-fidelity DNA polymerase enzyme (New England Biolabs, catalog number: M0491S)
7. Ultra-pure DNase/RNase-free distilled water (Invitrogen, catalog number: 10-977-015)
8. NucleoSpin gel and PCR clean-up (Macherey-Nagel, catalog number: 740609.50)
9. EpiTransNuc library synthesized by CustomArray (Genscript) (the gRNA pool was improved upon the Addgene #51047 nuclear proteins gRNA sub-pool library through the inclusion of gRNAs from Sabatini, Brunello, GecKO, and Toronto v3)
10. Absolute ethanol (Isolab, catalog number: 9.200.262.500)
11. 10× FastDigest Esp3I (BsmBI, IIs class) enzyme (Thermo Fisher Scientific, catalog number: FD0454)
12. 10× FastDigest buffer (Thermo Fisher Scientific, catalog number: FD0454)
13. DTT (Neofroxx, catalog number: 1114GR005)
14. NEBNext high-fidelity PCR master mix (New England Biolabs, catalog number: M0541S)
15. Agarose Biomax (Prona, catalog number: 000320PR)
16. Gel loading dye, purple 6× (New England Biolabs, catalog number: B7024SVIAL)
17. 1 kb plus DNA ladder (New England Biolabs, catalog number: N3200L)
18. Isopropanol (Isolab, catalog number: 961.023.2500)
19. Electroporation cuvettes (Bio-Rad, catalog number:1652089)
20. Tryptone (AppliChem, catalog number: 1553.03)
21. Yeast extract (Nzytech, catalog number: MB16401)
22. NaCl (Merck, catalog number: M106404.1000)
23. LB broth with agar (Sigma-Aldrich, catalog number: L2897)
24. Ampicillin (Sigma-Aldrich, catalog number: A0166)
25. Tris base (Sigma-Aldrich, catalog number: T1503-1KG)
26. EDTA (Sigma-Aldrich, catalog number: E5134-500G)
27. Glacial acetic acid (Sigma-Aldrich, catalog number: 27225-2.5L-R)
28. SafeView Classic (ABM, catalog number: G108)
29. Endotoxin-free HiSpeed Plasmid DNA Maxi kit (Qiagen, catalog number: 12362)
Solutions
1. 10× Tris-acetate-EDTA buffer stock solution (10× TAE) (see Recipes)
2. Luria Bertani (LB) agar stock solution (see Recipes)
3. Luria Bertani (LB) liquid medium stock solution (see Recipes)
Recipes
Note 1: For the preparation of recipes, in-house ddH2O was used.
Note 2: A 10× TAE solution is used as stock and is stable at room temperature. Prepare LB agar stock and LB liquid medium stock solutions right before usage and keep them at 4 °C.
1. 10× Tris-acetate-EDTA buffer stock solution (10× TAE)
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Tris base | 400 mM | 48.4 g |
| EDTA | 9.9 mM | 3.7 g |
| Glacial acetic acid | 1.14% (v/v) | 11.4 mL |
| ddH2O | - | 800 mL |
| Total | n/a | Complete with ddH2O to 1,000 mL |
2. LB agar stock solution
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| LB broth with agar | 3.5% (w/v) | 35 g |
| ddH2O | - | 1,000 mL |
| Total | n/a | 1,000 mL |
3. LB liquid medium stock solution
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Tryptone | 1% (w/v) | 10 g |
| Yeast extract | 0.5% (w/v) | 5 g |
| NaCl | 1% (w/v) | 10 g |
| ddH2O | 1,000 mL | |
| Total | n/a | 1,000 mL |
Laboratory supplies
1. Nunc square bioassay dishes (Thermo Fisher Scientific, catalog number: 240835)
2. 0.2 mL PCR tubes (Labselect, catalog number: PT-02-C)
3. 1.5 mL Eppendorf (Golden Gate, catalog number: KG2211)
4. Petri dish Ø90 × 17 mm (Isolab, catalog number: 081.02.191)
5. Drigalski glass spreader (Superior, catalog number: C180024)
Equipment
1. SimpliAmp thermal cycler (Applied Biosystems, model: A24811)
2. NanoDrop2000 spectrophotometer (Thermo Fisher Scientific, model: ND-2000)
3. Microcentrifuge (Thermo Fisher Scientific, model: MicroCL 17R 230 V)
4. Balance (Sartorious, model: ENTRIS 822-1S)
5. Micropulser electroporator (Bio-Rad, catalog number: 1652100)
6. Gel electrophoresis system (Thermo Fisher Scientific, model: OWL B1A)
7. Gel Doc XR+ system with Image Lab software (Bio-Rad, model: 1708195)
8. 37 °C bacterial orbital shaker (Thermo Fisher Scientific, model: 4329)
9. 37 °C incubator (Heraeus, model: Heracell)
10. Heat block (Thermo Fisher Scientific, model: Digital Shaking Drybath)
11. Centrifuge (Eppendorf, model: 5810R)
Software and datasets
1. Python v2.7.17
2. Biopython v1.79
3. Count_spacers.py code (code is deposited at https://github.com/fengzhanglab/Screening_Protocols_manuscript)
4. BioRender (https://www.biorender.com/). The following figures were created using BioRender: Graphical overview, Senturk, S. (2025) https://BioRender.com/o79n165
Procedure
Circular polymerization extension cloning (CPEC) protocol comprises four major steps:
A. Preparation of lentiGuide-Puro backbone (Addgene #52963) via PCR-based linearization.
B. Preparation of gRNA library (in our research, every gene is targeted by 10 gRNAs).
C. Bulk cloning of amplified gRNA library into lentiGuide-Puro backbone by pursuing CPEC methodology.
D. Transformation of the gRNA cloned lentiGuide-Puro backbone into electrocompetent Endura bacteria.
A. Preparation of the lentiGuide-Puro backbone via PCR-based linearization
1. The primary objective of this methodology is to eliminate all stages that might compromise product purity and, by extension, the cloning process.
Critical: Based on our experimental assessments, gel extraction is identified as the critical initial step that needs to be addressed. From this regard, the major technical aim is to minimize or circumvent gel purification steps wherever technically feasible. To facilitate this, the lentiGuide-Puro backbone was initially linearized through PCR-based linearization.
Note: Linearize 1 ng of lentiGuide-Puro backbone with the primer pairs shared below (Table 1).
Table 1. Primer pair for backbone linearization
| Primer pair | Sequence |
|---|---|
| Forward primer | GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACC |
| Reverse primer | CGGTGTTTCGTCCTTTCCACAAGATATATAAAGCCAAGAAATCGAAATACTTTCAAGTTACGG |
2. Mix gently the reaction components as well as primer pairs designed specifically for the linearization of lentiGuide-Puro and incubate for 24 cycles following the conditions detailed below. Perform the linearization step with Q5 polymerase enzyme on ice by using 0.2 mL PCR tubes.
Note: Prepare a master mix using a 1.5 mL Eppendorf tube and then divide the reaction into 0.2 mL PCR tubes.
3. Reaction components comprise 10 μL of reaction buffer (Q5), 10 μL of GC enhancer buffer, 1 μL of dNTP (10 mM stock), 3 μL of forward primer, 3 μL of reverse primer (10 μM stock), 0.5 μL of Q5 polymerase enzyme, a maximum amount of 1 ng of lentiGuide-Puro plasmid, and ddH2O adding up to the total volume of 50 μL (Table 2).
Table 2. PCR-based backbone linearization conditions
| Reagents | Final concentration | Volume required (μL) |
|---|---|---|
| 5× Q5 reaction buffer | 1× | 10 |
| 5× Q5 HighGC enhancer | 1× | 10 |
| 10 mM dNTP | 200 μM | 1 |
| 10 μM forward primer | 0.6 μM | 3 |
| 10 μM reverse primer | 0.6 μM | 3 |
| Q5 high-fidelity DNA polymerase | 0.02 U/μL | 0.5 |
| Lenti-guidePuro backbone | Maximum 1 ng | |
| ddH2O | Varies | |
| Total | 50 |
4. Follow the thermocycling conditions shown in Table 3.
Table 3. Thermocycling conditions for the PCR reaction
| Step | Temp. (°C) | Duration | No. of cycles |
| Initial denaturation | 98 | 3 min | 1 |
| Denaturation | 98 | 10 s | 24 |
| Annealing | 60.9 | 30 s | |
| Extension | 72 | 8 min | |
| Final extension | 72 | 10 min | 1 |
| Hold | 4 | ∞ | - |
5. Perform in total 10 different PCR linearization reactions.
6. Pool those 10 reactions and column purify samples using NucleoSpin Gel and PCR clean-up kit.
7. To detail this section, first add ethanol into concentrated buffer NT3. Then, mix previously pooled and purified samples and buffer NT1 supplied by the kit in a 1:2 ratio. Insert NucleoSpin Gel and PCR clean-up column into the collection tubes (2 mL) supplied with the kit and then load a 700 μL sample. Perform all steps and centrifugation at room temperature. Centrifuge samples at 11,000× g for 30 s. This step is followed by the removal of the flowthrough. Wash silica membrane with 700 μL of buffer NT3 twice and subsequently centrifuge samples at 11,000× g for 30 s. After removal of the flowthrough, dry silica membranes by centrifugation again at 11,000× g for 1 min.
Note 1: Since residual ethanol may interfere with downstream enzymatic reactions, columns were incubated at 70 °C. In the elution step, transfer columns into new 1.5 mL Eppendorf tubes and keep samples initially with 15–30 μL of buffer NE (composition of elution buffer NE: 5 mM Tris/HCl, pH 8.5) for at least 1 min, followed by centrifugation at 11,000× g for 1 min).
Note 2: The product resulting from the linearization PCR measures 8,303 bp, and its termini align with the Esp3I (also referred to as BsmBI) cleavage sites present in the original vector.
8. After the elution step, the linearized PCR product is ready for upcoming experimental steps.
Critical: To minimize the occurrence of gRNA-free colonies on negative control LB agar plates, utilize a maximum of 1 ng of lentiGuide-Puro per PCR reaction (see Troubleshooting 1). Additionally, to compensate for potential losses during purification, conduct a total of 10 PCR reactions.
Note: Scale up the number of reactions whenever there is a loss during consecutive purification steps.
9. Subsequently, for product size validation, 1) pool the eluted PCR-linearized lentiGuide-Puro into a single tube, and 2) mix the PCR linearization product with gel loading dye purple 6× to a final concentration of 1×. Then, run 5 μL of the validation reaction alongside uncut lentiGuide-Puro on a 0.8% (w/v) agarose gel cast in 1× TAE buffer with SafeView Classic dye (Figure 1, gel image on the left).
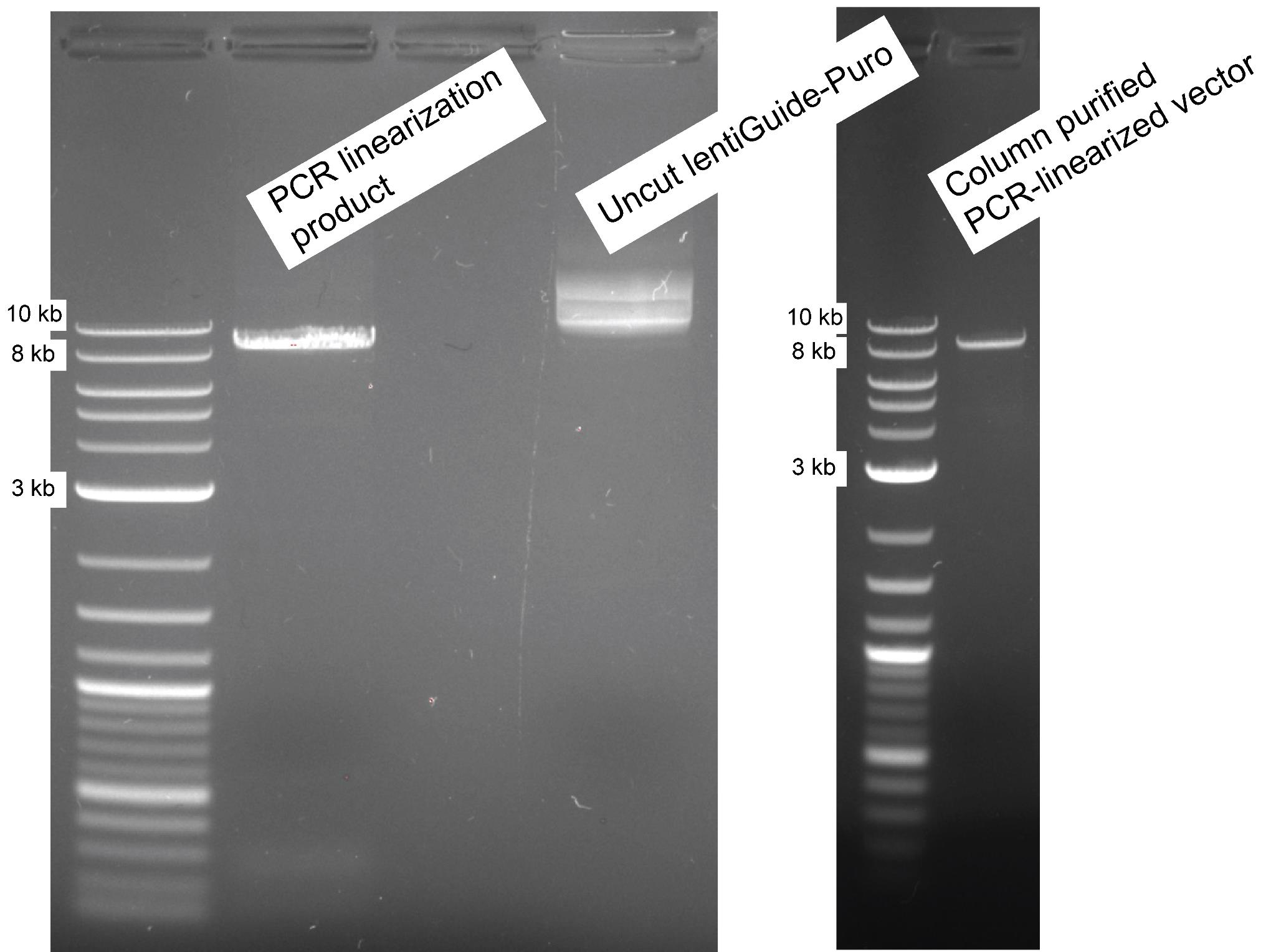
Figure 1. lentiGuide-Puro PCR linearization. The gel image on the left displays the result of the PCR linearization product on a 0.8% (w/v) agarose gel, with the last lane demonstrating 100 ng of uncut lentiGuide-Puro backbone as a control. As a DNA ladder, 1 kb plus DNA ladder was used. The gel image on the right shows the column-purified vector for pre-cloning after Esp3I (BsmBI) restriction digestion.
10. Following the pooling of PCR products and column purification of the lentiGuide-Puro backbone, which serves as the template for linearization, incubate with the Esp3I (BsmBI) restriction enzyme at 37 °C for 4 h.
Note 1: Initial optimization experiments employed a 1 h digestion, with 4 h ultimately identified as the optimal condition in our experimental setup.
Note 2: This procedure was intended to diminish the colony-forming potential of the intact lentiGuide-Puro vector used in the PCR-based linearization.
Critical: FastDigest Esp3I (BsmBI) enzyme requires dithiothreitol (DTT) and should be prepared freshly (see Troubleshooting 1). Reaction components for restriction digestion comprise 14 μL of nuclease-free water, 2 μL of 10× FastDigest Buffer, 1 μL of DTT (20 mM), 1 μL of FastDigest enzyme Esp3I (BsmBI), and 1 μg of the PCR product. Mix all the reaction components on ice, followed by incubation at 37 °C for 4 h.
11. Inactivate the restriction enzyme by incubating the reaction at 65 °C for 15 min.
12. Subsequently, repurify the linearized Esp3I (BsmBI) digested PCR product using NucleoSpin Gel and PCR clean-up Kit and verify for purity and integrity on 0.8% (w/v) agarose (as described in step A9) (Figure 1, gel image on the right). All modifications made at this stage should successfully eliminate factors affecting impurity.
B. PCR amplification and purification of the gRNA library
Note 1: The gRNA pool was improved upon the nuclear proteins gRNA sub-pool library (Addgene #51047 Nuclear Proteins gRNA sub-pool Library): an expanded list of genes targeting epigenetic regulators, transcription factors, and nuclear factors (labeled as EpiTransNuc) was assembled by adding validated gRNA sequences from various libraries, including Sabatini, Brunello, GecKO, and Toronto v3, all available from Addgene. For each gRNA, forward (F) and reverse (R) sequences were appended to both ends of the gRNA sequences. Oligonucleotide sequences consist of a 5' universal flanking sequence (TATCTTGTGGAAAGGACGAAACACCG) and a 3' universal flanking sequence (GTTTTAGAGCTAGAAATAGCAAGTTAAAAT). Oligonucleotides featuring a 5' and 3' extension compatible with the Esp3I (BsmBI) enzyme were synthesized in array format by CustomArray (Genscript).
Note 2: The EpiTransNuc library was designed so that each gene is targeted by 10 distinct gRNAs, covering a total of 4,072 genes. Additionally, 100 non-targeting control gRNAs were included in the library composition [retrieved from Addgene #51048 Control targets (most diverse) gRNA sub-pool library].
1. To transition from the gRNA oligonucleotide library to the PCR library, amplification was performed using NEBNext high-fidelity PCR master mix. The reaction was setup using the following primer pair: GTAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACC (forward primer) and ACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC (reverse primer).
2. Set up a 6× gRNA oligonucleotide amplification reaction (Table 4) by following the thermocycling conditions shown in Table 5. Prepare the master mix in a 1.5 mL Eppendorf tube, then transfer it into 0.2 mL PCR tubes.
Table 4. Oligonucleotide library amplification
| Reagents | Final concentration | Volume required (μL) |
|---|---|---|
| NebNext, HF, PCR MM, 2× | 1× | 12.5 |
| Pooled oligonucleotide library template | 0.04 ng/μL | 1 |
| Oligonucleotide forward primer | 0.5 μM | 1.25 |
| Oligonucleotide reverse primer | 0.5 μM | 1.25 |
| Ultra-pure water | 9 | |
| Total | 25 |
Table 5. Thermocycling conditions for the PCR reaction
| Step | Temp. (°C) | Duration | No. of cycles |
| Initial denaturation | 98 | 30 s | 1 |
| Denaturation | 98 | 10 s | 20 |
| Annealing | 63 | 10 s | |
| Extension | 72 | 15 s | |
| Final extension | 72 | 2 min | 1 |
| Hold | 4 | ∞ | - |
3. After the amplification, 5 μL of the PCR product was electrophoresed on a 2.5% (w/v) agarose gel for validation (as described in step A9) (Figure 2).
Note: The PCR amplicon is observed at approximately 140 bp. Notably, the 120 bp primer dimer reaction, highlighted as a concern in the study by Joung et al. [11], was not detected in our samples. To mitigate potential skewing in gRNA representation, PCR amplification was restricted to 20 cycles (see Troubleshooting 5) in accordance with recommendations by Joung et al. [11].
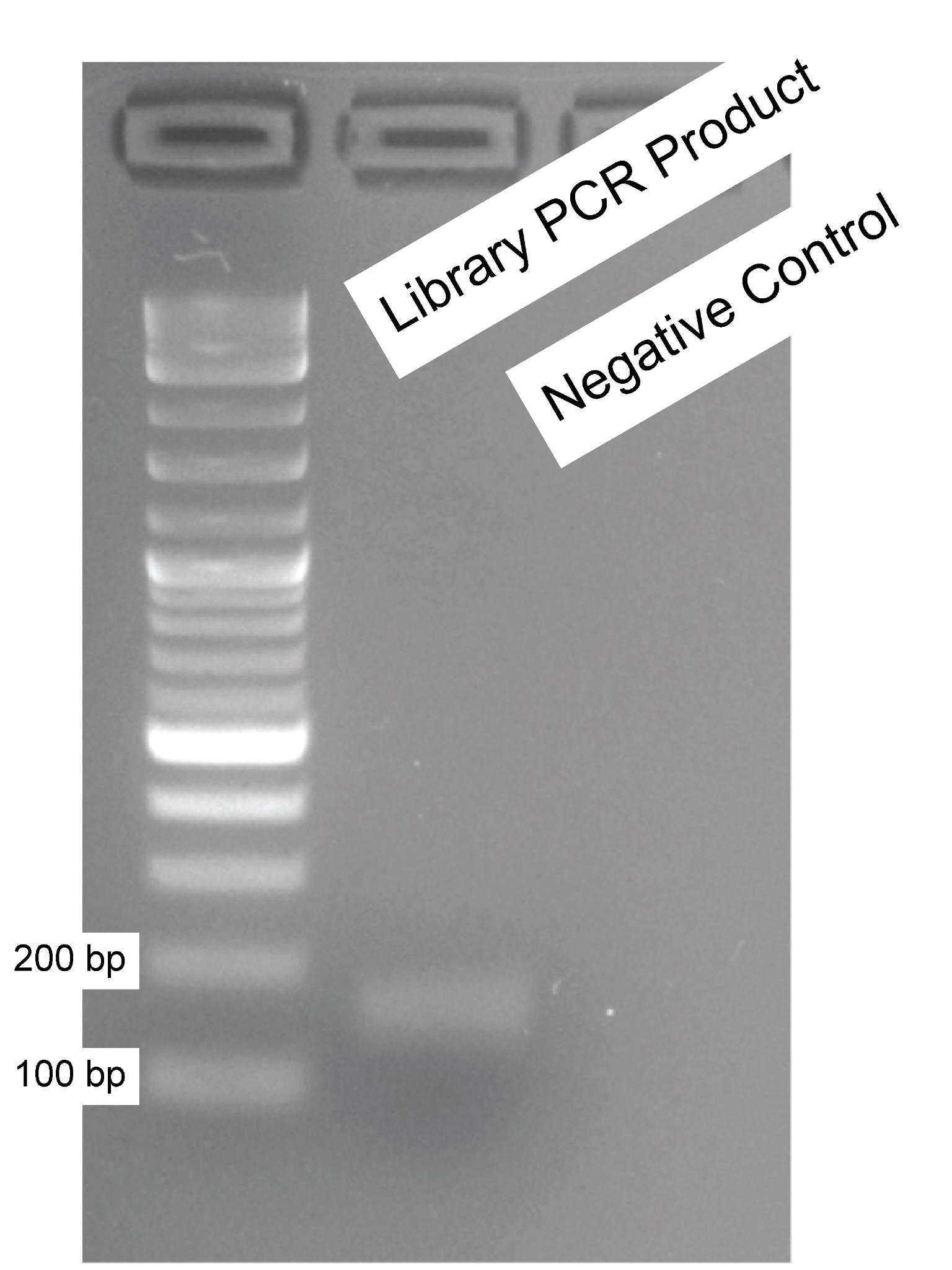
Figure 2. PCR amplification of gRNA oligonucleotides. The oligonucleotides synthesized through a service provider were amplified using long primer pairs suggested by Joung et al. [11], and a small amount (5 μL) was analyzed on a 2.5% (w/v) agarose gel for validation purposes. A 1 kb plus DNA ladder was used. The first well represents the library PCR product, while the second well serves as the negative control (water as input).
4. gRNA PCR library was purified with the NucleoSpin Gel and PCR clean-up kit, following the protocol provided by the manufacturer.
Note: In line with the approach used for the linear vector preparation, the gel extraction step was omitted during the preparation of the gRNA PCR library to ensure purity.
C. Circular polymerization extension cloning (CPEC)
Note: Components of CPEC protocol comprise NEBNext HF PCR master mix, previously PCR-linearized lentiGuide-Puro backbone (A), PCR-amplified gRNA library (B), and distilled water.
1. Use 100 ng of lentiGuide-Puro backbone while utilizing only 16.9 ng of PCR amplified and purified gRNA library (Table 6). Limit the CPEC reaction to four cycles and perform at least 10 separate reactions following the thermocycling conditions shared in Table 7. Include negative controls involving the same setup without gRNA library addition.
Table 6. Circular polymerization and extension cloning reaction
| Reagents | Final concentration | Volume required (μL) |
|---|---|---|
| NebNext, HF, PCR MM, 2× | 1× | 12.5 |
| lentiGuide-Puro (Esp3I (BsmBI)) | 1 M | 100 ng (varies) |
| PCR product (gRNA library) | 10 M | 16.9 ng (varies) |
| ddH2O | varies | |
| Total | 25 |
Table 7. Thermocycling conditions for the CPEC reaction
| Step | Temp. (°C) | Duration | No. of cycles |
| Initial denaturation | 98 | 30 s | 1 |
| Denaturation | 98 | 10 s | 4 |
| Primer annealing | 72 | 5 min | |
| Extension | 72 | 5 min | |
| Final extension | 72 | 5 min | 1 |
| Hold | 4 | ∞ | - |
2. Following this, purify the CPEC products using isopropanol, as recommended by Joung et al. [11]. In this procedure, mix the CPEC product with isopropanol at a 1:1 ratio. Subsequently, add NaCl solution to achieve a final concentration of 50 mM.
Note: This precipitation step is intended to concentrate the library prior to bacterial transformation and to reduce the salt concentration from prior reactions, which could adversely impact electroporation efficiency (see Troubleshooting 4).
3. Vortex the reaction and incubate at room temperature for 15 min. Centrifuge the CPEC product at 15,000× g for 15 min at room temperature.
Critical: At this stage, the pellet may be difficult to visualize. Carefully aspirate the supernatant from the side opposite to where the pellet is located to avoid accidentally aspirating the CPEC product (see Troubleshooting 2). Use the vacuum aspiration system at the lowest setting.
4. Add ice-cold 80% ethanol (150 μL) without disturbing the pellet and centrifuge samples at 11,000× g for 5 min at 4 °C. Aspirate the supernatant carefully and repeat this step one more time. Remove any residual ethanol and air-dry samples for 1 min.
5. Incubate the reaction at 55 °C for 10 min with a TE buffer.
6. Determine the concentration using a NanoDrop2000 spectrophotometer.
D. Electroporation of the CPEC product
1. Transfer the CPEC reaction (600 ng of total gRNA plasmid library) to Endura electrocompetent bacteria. At this step, carry out three separate electroporations, each one with 25 μL of electrocompetent bacteria.
2. Perform electroporation by following the EC1 program on Bio-Rad MicroPulser.
Note 1: In this setting, the convenient cuvette size is 0.1 cm, the voltage is 1.8 kV, and the number of pulses is 1 (6 ms). Electroporation conditions are based on 10 microfarads and 600 ohm resistance.
Note 2: Use 2 mL of recovery medium.
3. Following this, pool bacteria and incubate for 75 min at 225 rpm and 37 °C conditions.
4. Spread the bacteria carrying the target library onto two prewarmed LB agar bioassay dishes (500 cm2) and incubate for 18 h at 37 °C culture conditions (Figure 3).
Note 1: Simultaneously, in order to determine the library coverage, spread the bacteria in different dilutions (1/10,000, 1/1,000, and 1/10) onto ampicillin-containing (1:1,000) agar plates.
Note 2: In Figure 3, there were 1,761 CPEC (+) colonies in 1/1,000 dilution, whereas there were only 22 colonies in CPEC (-) plate (see Troubleshooting 3).
Note 3: Our library coverage was calculated as ~43× using the following formula:
Coverage = (CPEC (+) colony number - CPEC (-) colony number) * Dilution factor/Library size
According to our results, the formula applies as (1761 - 22 = 1739) * 1,000 (dilution factor) = 1,739,000. Divide the result by the library size; in our case, this is 40,820 gRNAs.
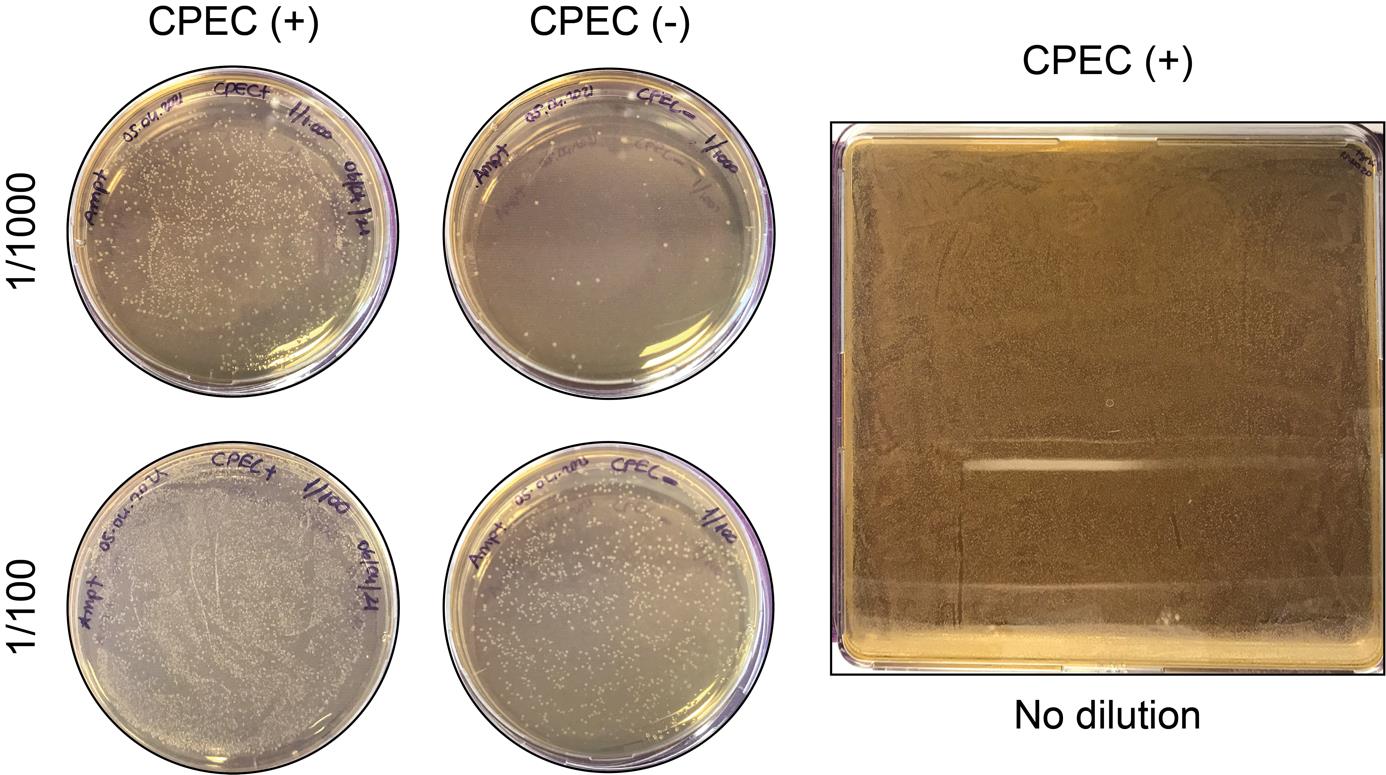
Figure 3. Electroporation application of commercial Endura bacteria. Circular polymerase extension cloning (CPEC) (+) and CPEC (-) products in varying dilutions on 10 cm Petri dishes (left). CPEC (+) product spread onto two LB agar bioassay dishes (representative image on the right).
5. After 18 h, gently scrape the electrocompetent bacteria from LB agar bioassay dishes by using a bio-spreader with 20 mL of cold LB liquid medium per dish. Centrifuge (previously set to 4 °C) Falcon tubes with bacteria for 30 min at 3,214× g and, after removal of supernatants, weigh pellets.
6. Purify the products with an endotoxin-free HiSpeed Plasmid DNA Maxi kit.
Data analysis
The gRNA distribution in the library was confirmed by Next-generation sequencing (NGS) using HiSeq X with 2× 150 bp paired-end reads, providing a sequencing depth of 6 Gb per sample. NGS library was constructed following the protocols and reagents described by Joung et al. [11]. The raw sequencing data was retrieved and processed using the count_spacers.py script in Python v2.7.17 [11].
Validation of protocol
We validated the process using the count_spacers.py code along with additional analyses. To run count_spacers.py, three essential inputs are required: 1) the .fastq file containing sequencing data (Figure 4), 2) a .csv file with gRNA sequence information, and 3) a key sequence (CGAAACACC) that facilitates the identification of gRNAs within the sequencing reads. The key sequence may be tailored based on the specific sequence composition of the library.
Figure 4. Subset of the NGS sequencing .fastq file used in the analysis
When necessary, the .fastq files were trimmed to remove adapter sequences using the trimming algorithm like Cutadapt. The trimmed .fastq files were analyzed using the count_spacers.py code (https://github.com/fengzhanglab/Screening_Protocols_manuscript/blob/master/count_spacers.py), which generated two output files: 1) a text file containing analysis statistics (Figure 5) and 2) a .csv file listing the read counts for each gRNA (Table 8).
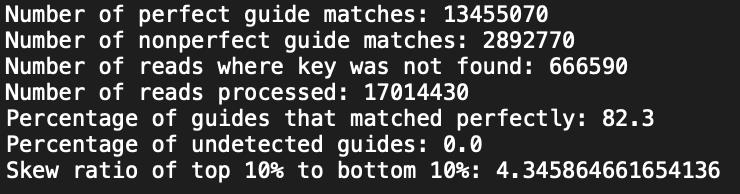
Figure 5. Statistics.txt output file from count_spacers.py code
Table 8. Read counts for the top 10 gRNAs from .csv file
| gRNA Sequence | Read count |
|---|---|
| TCCGCGCCTTCGCCTACACC | 1,818 |
| TGCTGTCCACCGCTCCTCCC | 1,553 |
| TGCCTGTCCTGTGTCAAGTC | 1,533 |
| TGAACTCGTCCAGCACCGCC | 1,505 |
| AGCCGCGCCTCACCGGGTGC | 1,447 |
| TCACTCACCTGCATCTGCCC | 1,447 |
| TCTGCCTTGTTCCCTGCCTG | 1,446 |
| TGTCTGCTGCTCCTGCCTTT | 1,416 |
| GTCCCAGTTCTCCGCCCTCC | 1,407 |
| TTCCTCCTCGCTCTCCTCTC | 1,402 |
To visualize and assess the distribution of gRNAs, a density plot was generated using the log2-transformed counts of the gRNAs (Figure 6). The density plot was created using R (v4.2.2) and the ggplot2 package (v3.5.1). The analysis revealed that over 90% of the gRNAs were represented within a 10-fold range of read counts, indicating a highly uniform representation across the library. This suggests minimal over- or under-representation of individual gRNAs, ensuring the integrity of the library for downstream applications.
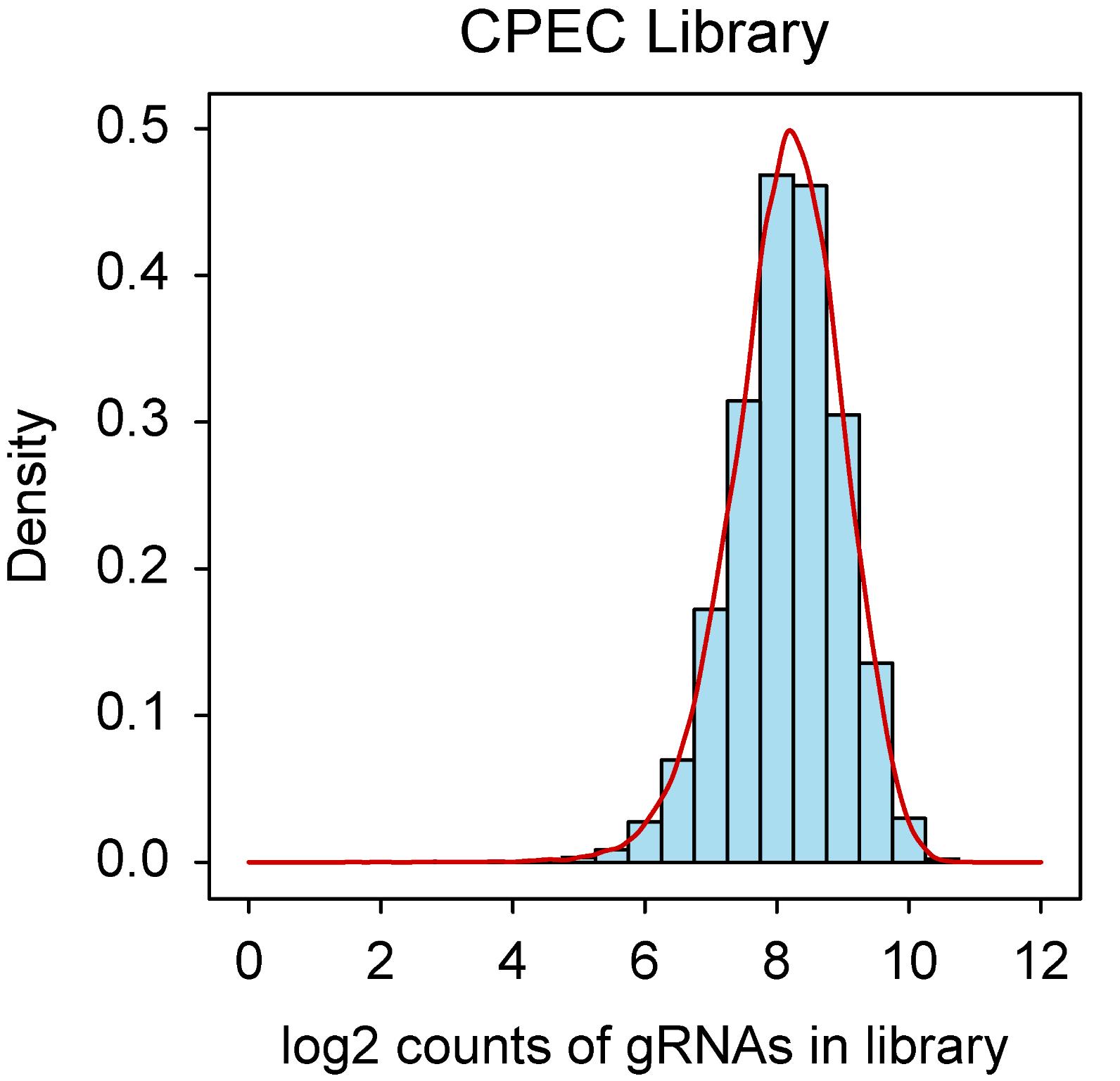
Figure 6. Density plot depicting the gRNA representation in EpiTransNuc library. The distribution of gRNAs in the library was verified using NGS. The bars in the graph illustrate the log2-transformed read counts for each gRNA.
General notes and troubleshooting
General notes
1. To prevent inter-colony competition that may result in skewing of gRNA library distribution, a solid culture amplification rather than a liquid one is recommended [11].
2. Multi-fragment assembly can be accomplished via CPEC methodology so that at least two gRNAs can be cloned tandemly in one vector backbone in order to target different genes.
Troubleshooting
Problem 1: High number of colonies on the negative control agar plates.
Possible cause: Recovery of backbone, incomplete restriction of uncut plasmid.
Solution 1: In the PCR-based linearization, usage of 1 ng of backbone per reaction is strictly recommended.
Solution 2: For 1 μg of PCR-linearized product, usage of 1 μL of Esp3I (BsmBI) is quite significant; do not reduce the enzyme amount.
Solution 3: FastDigest Esp3I (BsmBI) enzyme requires DTT in the reaction. DTT should be prepared and added freshly.
Problem 2: Aspiration of CPEC products after isopropanol precipitation step.
Possible cause: Pellet is loose or difficult to see.
Solution 1: Perform a second round of centrifugation.
Solution 2: Use GlycoBlue, a reagent that comprises a blue dye covalently conjugated to glycogen. It serves as a nucleic acid co-precipitant, with the dye facilitating enhanced visualization of the nucleic acid pellet.
Problem 3: Insufficient number of colonies on the positive agar plates, low transformation efficiency.
Possible cause: Gel extraction step interferes with the CPEC reaction and mitigates positive clones. Moreover, check your electrocompetent cells with an intact, commercial plasmid.
Solution: Skip gel extraction steps wherever it is feasible and scale up the number of electroporation reactions.
Problem 4: Problems or arcing during electroporation.
Possible cause: Salts coming from buffers, higher ionic strength of samples.
Solution: Perform isopropanol precipitation, desalt as much as possible before electroporation, and ensure that there are no air bubbles in the electroporation cuvette.
Problem 5: Insufficient representation of the gRNA library.
Possible cause: PCR-based bias
Solution: Minimize the number of PCR cycles during oligonucleotide amplification to prevent potential biases; limit PCR cycles to 20.
Acknowledgments
The authors acknowledge Izmir Biomedicine and Genome Center (IBG) for providing financial and administrative support. This research was funded by The Scientific and Technological Research Council of Türkiye (TÜBİTAK) with grant number 119Z540. Bengisu Dayanc was supported by YÖK 100/2000 PhD Scholarship and TÜBİTAK-BİDEB 2211/C National PhD Scholarship programs. Sude Eris was supported by TÜBİTAK-BİDEB 2210/A National MSc/MA scholarship program. Serif Senturk acknowledges support from the Turkish Academy of Sciences (TUBA GEBIP 2017) and the Science Academy (BAGEP 2019). We acknowledge Dr. Minoo Karimi (University of Montreal) and Dr. Ece Cakiroglu (Dokuz Eylul University) for technical help throughout the construction of the gRNA library.
Competing interests
The authors declare no competing interests.
References
- Shalem, O., Sanjana, N. E. and Zhang, F. (2015). High-throughput functional genomics using CRISPR–Cas9. Nat Rev Genet. 16(5): 299–311.
- Zhang, Z., Wang, H., Yan, Q., Cui, J., Chen, Y., Ruan, S., Yang, J., Wu, Z., Han, M., Huang, S., et al. (2023). Genome-wide CRISPR/Cas9 screening for drug resistance in tumors. Front Pharmacol. 14: e1284610.
- Chan, Y. T., Lu, Y., Wu, J., Zhang, C., Tan, H. Y., Bian, Z. x., Wang, N. and Feng, Y. (2022). CRISPR-Cas9 library screening approach for anti-cancer drug discovery: overview and perspectives. Theranostics. 12(7): 3329–3344.
- Sanson, K. R., Hanna, R. E., Hegde, M., Donovan, K. F., Strand, C., Sullender, M. E., Vaimberg, E. W., Goodale, A., Root, D. E., Piccioni, F., et al. (2018). Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat Commun. 9(1): 5416.
- Quan, J. and Tian, J. (2009). Circular Polymerase Extension Cloning of Complex Gene Libraries and Pathways. PLoS One. 4(7): e6441.
- Aslanidis, C. and de Jong, P. J. (1990). Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18(20): 6069–6074.
- Li, M. Z. and Elledge, S. J. (2007). Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods. 4(3): 251–256.
- Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A. and Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 6(5): 343–345.
- Wegner, M., Husnjak, K. and Kaulich, M. (2020). Unbiased and Tailored CRISPR/Cas gRNA Libraries by Synthesizing Covalently-closed-circular (3Cs) DNA. Bio Protoc. 10(1): e3472.
- Quan J, Tian J. (2014). Circular Polymerase Extension Cloning. In: Valla S, Lale R (Eds.). DNA Cloning and Assembly Methods. Humana Press, Totowa, NJ, pp 103–117.
- Joung, J., Konermann, S., Gootenberg, J. S., Abudayyeh, O. O., Platt, R. J., Brigham, M. D., Sanjana, N. E. and Zhang, F. (2017). Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc. 12(4): 828–863.
Article Information
Publication history
Received: Sep 25, 2024
Accepted: Dec 8, 2024
Available online: Dec 31, 2024
Published: Feb 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Dayanc, B., Eris, S. and Senturk, S. (2025). Leveraging Circular Polymerization and Extension Cloning (CPEC) Method for Construction of CRISPR Screening Libraries. Bio-protocol 15(4): e5183. DOI: 10.21769/BioProtoc.5183.
Category
Molecular Biology > DNA > DNA cloning
Cancer Biology > General technique > Molecular biology technique
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.