- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Multiplex Genome Editing of Human Pluripotent Stem Cells Using Cpf1
Published: Vol 14, Iss 22, Nov 20, 2024 DOI: 10.21769/BioProtoc.5108 Views: 2682
Reviewed by: Thirupugal GovindarajanVishal NehruAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2020
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Targeted genome editing of human pluripotent stem cells (hPSCs) is critical for basic and translational research and can be achieved with site-specific endonucleases. Cpf1 (CRISPR from Prevotella and Francisella) is a programmable DNA endonuclease with AT-rich PAM sequences. In this protocol, we describe procedures for using a single vector system to deliver Cpf1 and CRISPR RNA (crRNA) for genome editing in hPSCs. This protocol enables indel formation and homologous recombination–mediated precise editing at multiple loci. With the delivery of Cpf1 and a single U6 promoter-driven guide RNA array composed of an AAVS1-targeting and a MAFB-targeting crRNA array, efficient multiplex genome editing at the AAVS1 (knockin) and MAFB (knockout) loci in hPSCs could be achieved in a single experiment. The edited hPSCs expressed pluripotency markers and could differentiate into neurons in vitro. This system also generated INS reporter hPSCs with a 6 kb cassette knockin at the INS locus. The INS reporter cells can differentiate into β-cells that express tdTomato and luciferase, permitting fluorescence-activated cell sorting of hPSC-β-cells. By targeted screening of potential off-target sequences that are most homologous to crRNA sequences, no off-target mutations were detected in any of the tested sequences. This work provides an efficient and flexible system for precise genome editing in mammalian cells including hPSCs with the benefits of less off-target effects.
Key features
• A single-vector system to deliver Cpf1 and crRNA enables the sorting of transfected cells
• Efficient and simultaneous multi-modular genome editing exemplified by mutation of MAFB and knockin of AAVS1 loci in a single experiment
• Edited PSCs showed minimal off-target effects and can be differentiated into multiple cell types
Keywords: Human pluripotent stem cells (hPSCs)Graphical overview
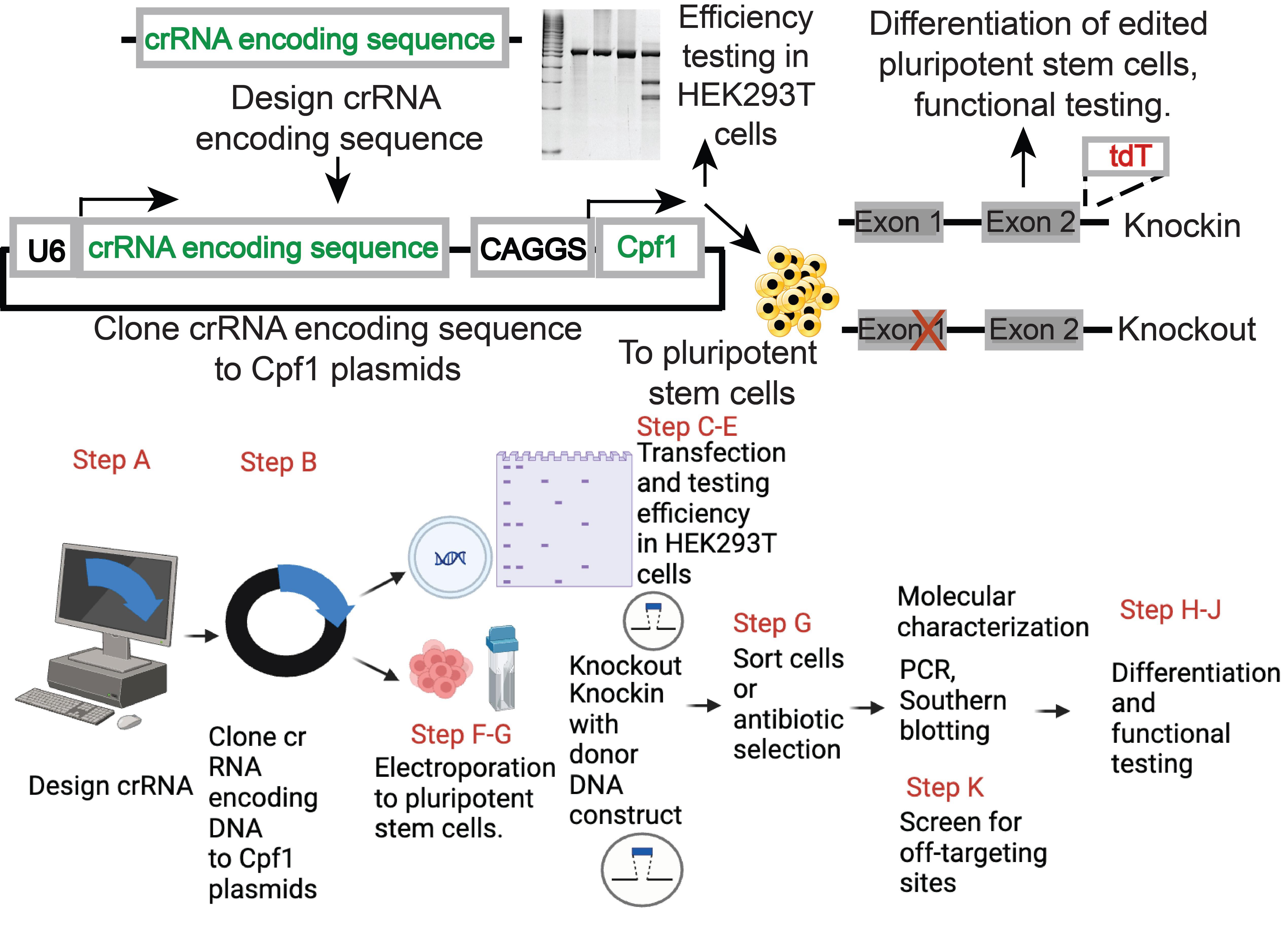
Genome editing of human pluripotent stem cells (hPSCs) using Cpf1. The top panel provides a brief overview of the approach; the bottom panel indicates additional details corresponding to the procedures of the protocol. The bottom panel was generated with biorender.com.
Background
Human pluripotent stem cells (hPSC) [1,2] and their differentiated cell types, such as pancreatic β-cells [3], enable cell replacement therapies as a potential treatment for multiple diseases [4]. Genetic modifications of hPSCs can correct genetic lesions and provide functions including increased immune tolerance by overexpressing PDL1 [5]. Multiple programmable nucleases have been developed for efficient genome editing in hPSCs: zinc finger nucleases [6], TALE nucleases [7], and CRISPR-Cas9 [8,9]. Complementary utilization of these nucleases is a critical component for hPSC-based regenerative medicine [10,11].
The CRISPR-Cas9 system enables efficient genome editing applicable to diverse cell types [12]. However, wild-type SpCas9 can generate double-strand DNA breaks in off-target sites partially complementary to guide RNA. The off-target effects of SpCas9 can be reduced by modifying the basic amino acid residues of the DNA binding domain of SpCas9, but the modified SpCas9 could also show reduced activity compared to wild-type SpCas9 [13]. Base-editing [14] and prime-editing [15] technologies enable genome editing without generating double-stranded DNA breaks. However, introducing large DNA fragments to specific loci of cells remains challenging as recent studies showed insertions in the range of hundreds of nucleotides [16]. Furthermore, to achieve effective multiplex editing at the single-cell level with a DNA vector, each guide RNA requires its promoter, complicating multiplex applications [17].
CRISPR-Cpf1 family endonucleases confer efficient genome editing in transformed mammalian cell lines [18] and mouse embryos [19,20]. Additionally, Francisella novicida Cpf1 (FnCpf1) and Acidaminococcus Cpf1 (AsCpf1) have RNase activity to process their cognate CRISPR RNA (crRNA) [18,21], making it possible to deliver multiple crRNAs from a pre-crRNA array driven by a single U6 promoter [22]. Furthermore, Cpf1 has less off-target effects in genome editing experiments with transformed human cell lines [23,24]. These unique features make Cpf1 a system that complements Cas9.
This protocol describes a detailed genome-editing procedure that uses a single vector system that expresses AsCpf1 and its crRNA. With donor constructs, locus-specific knockin alleles of hPSC were generated and could be differentiated into neurons and β-cells. Furthermore, by introducing a pre-crRNA array composed of AAVS1-targeting guide and MAFB-targeting guide, we observed 100% editing of MAFB locus in AAVS1 targeted clones, providing an example for genome editing in multiple loci in hPSCs. This protocol provides a flexible system for genome editing in hPSCs.
Materials and reagents
Biological materials
H1-OCT4-GFP pluripotent stem cells (WiCell Research Institute, catalog number: H1 OCT4-EGFP, NIH Human Embryonic Stem Cell Registry ID: NIHhESC-10-0043)
WIBR3 pluripotent stem cells (Whitehead Institute, NIH Human Embryonic Stem Cell Registry ID: NIHhESC-10-0079)
HEK293T cells (ATCC, catalog number: CRL-3216)
Stbl3 Chemically Competent E. coli (Invitrogen, catalog number: C737303)
DR4 mice [Dnmt1tm3Jae Hprt1b-m3 Tg(pPWL512hyg)1Ems/J] (Jackson Laboratory, catalog number: 003208)
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratory, catalog number: 005557)
DMEM-F12 medium (Life Technologies, catalog number: 11330-057)
KSR (Life Technologies, catalog number: 10828-028)
FGF2 (Life Technologies, catalog number: PHG0261)
2-Mercaptoethanol (2-ME) (Life Technologies, catalog number: 21985-023)
100× L-glutamine (Life Technologies, catalog number: 25030-081)
100× GlutaMAX (Life Technologies, catalog number: 35050-061)
100× MEM-NEAA (Life Technologies, catalog number: 11140-050)
100× penicillin/streptomycin (Life Technologies, catalog number: 15140-122)
100× Insulin-transferrin-selenium-ethanolamine (Life Technologies, catalog number: 51500-056)
Collagenase IV (Life Technologies, catalog number: 17104019)
mTeSR1 medium (STEMCELL Technologies, catalog number: mTeSR1)
Accutase (STEMCELL Technologies, catalog number: 7920)
MCDB131 medium (Life Technologies, catalog number: 10372019)
Y-27632 (Thermo Fisher Scientific, catalog number: 688000-100MG)
Puromycin (Sigma-Aldrich, catalog number: P7255-100MG)
CHIR99021 (Cayman Chemical, catalog number: 252917-06-9)
Matrigel (Thermo Fisher Scientific, catalog number: CB40234)
Activin A (R&D Systems, catalog number: 338-AC-050)
KGF (PeproTech, catalog number: 100-19-250UG)
Vitamin C (Sigma-Aldrich, catalog number: A4544-25G)
Retinoic acid (Sigma-Aldrich, catalog number: R2625-100MG)
SANT-1 (Sigma-Aldrich, catalog number: S4572-5MG)
LDN193189 (Sigma-Aldrich, catalog number: SML0559-5MG)
Heparin (Sigma-Aldrich, catalog number: H3149)
TPB (Tocris, catalog number: 5343)
EGF (PeproTech, catalog number: AF-100-15)
T3 (EMD, catalog number: 64245)
ALK5 inhibitor II (Axxora, catalog number: ALX-270-445-M005)
Gamma secretase inhibitor XX (VWR, catalog number: 82602-300)
Trace Elements A (Corning, catalog number: 25-021-CI)
Trace Elements B (Corning, catalog number: 25-022-CI)
Fatty acid–free BSA (Thermo Fisher Scientific, catalog number: 50412870)
D-Luciferin potassium salt (Perkin Elmer, catalog number: 122799)
RPMI 1640 (Gibco, catalog number: 31800-089)
DMEM high glucose medium (Cytiva, catalog number: SH30022)
FBS (Cytiva, catalog number: SH30396.03HI)
BbsI-HF (New England Biolabs, catalog number: R3539S)
SphI-HF (New England Biolabs, catalog number: R3182L)
CutSmart Buffer (New England Biolabs, catalog number: B6004)
T7 Endonuclease I (New England Biolabs, catalog number: M0302S)
Lipofectamine 2000 transfection reagent (Invitrogen, catalog number: 11668019)
Plasmid Mini Kit (Omega Bio-Tek, catalog number: D6943-02)
DNA Clean & Concentrator (Zymo Research, catalog number: D4033)
T4 PNK (New England Biolabs, catalog number: M0201S)
T4 DNA ligase (New England Biolabs, catalog number: M0202M)
DNeasy Blood & Tissue Kit (Qiagen, catalog number: 69504)
Q5 HiFi 2× Master Mix (New England Biolabs, catalog number: M0492L)
10% TBE PAGE gel (Bio-Rad, catalog number: 3450053)
Mouse anti-OCT3/4 antibody, 1:100 (BD Transduction Laboratories, catalog number: 611203)
Rabbit anti-NANOG antibody, 1:100 (Thermo Fisher Scientific, catalog number: PA1-097)
Mouse anti-TRA-1-60, 1: 100 (Life Technologies, catalog number: 411000)
Mouse anti-SSEA4 antibody, clone MC-813-70, 1:100 (Thermo Fisher Scientific, catalog number: MA1-021)
Mouse anti-Nestin antibody, 1:100 (Santa Cruz, catalog number: SC-23927)
Mouse anti-TUBB3 antibody, 1:1000 (BioLegend, catalog number: 801201)
Alexa 488 conjugated anti-mouse IgG antibodies, used at 1:400 (Life Technologies, catalog number: A21202)
Alexa 488 conjugated anti-rabbit IgG antibodies, used at 1:400 (Life Technologies, catalog number: A21206)
ZymoPURE II Plasmid Midiprep Kit (Zymo Research, catalog number: D4200)
AmpliTaq Gold 360 Master Mix (Thermo Fisher Scientific, catalog number: 4398881)
E.Z.N.A.® Gel Extraction kit (Omega Bio-Tek, catalog number: D2500-02)
HindIII-digested Lamda DNA (New England Biolabs, catalog number: N3012S)
Pyruvate (100 mM) (Corning, catalog number: 25-000-CIR)
Insulin solution (Sigma, catalog number: I9278)
Neurobasal medium (Life Technologies, catalog number: 21103049)
AlbuMAX I Lipid-Rich BSA (Life Technologies, catalog number: 11020021)
N2 NeuroPlex (GeminiBio, catalog number: 400163)
Lactic syrup (Sigma, catalog number: L4263-100ML)
Biotin (Sigma, catalog number: B4639-500MG)
Gem21 without vitamin A (GeminiBio, catalog number: 400-161-010)
Proteinase K (Promega, catalog number: C3021)
Sodium citrate tribasic dihydrate (Sigma, catalog number: C8532)
Primers
F NGS seq primer for MAFB of WIBR3 602-1 ATATGGTCAAGTGCGAGAAACTCG
F NGS seq primer for MAFB of WIBR3 602-2 AGAGGGTCAAGTGCGAGAAACTCG
F NGS seq primer for MAFB of WIBR3 602-3 ACACGGTCAAGTGCGAGAAACTCG
F NGS seq primer for MAFB of WIBR3 602-4 GCGCGGTCAAGTGCGAGAAACTCG
F NGS seq primer for MAFB of WIBR3 602-5 GTGTGGTCAAGTGCGAGAAACTCG
F NGS seq primer for MAFB of WIBR3 602-6 CTCTGGTCAAGTGCGAGAAACTCG
F NGS seq primer for MAFB of WIBR3 602-7 TTAAGGTCAAGTGCGAGAAACTCG
F NGS seq primer for MAFB of WIBR3 602-8 TTGGGGTCAAGTGCGAGAAACTCG
F NGS seq primer for MAFB of WIBR3 602-9 TTCCGGTCAAGTGCGAGAAACTCG
F NGS seq primer for MAFB of WIBR3 602-10 CCTTGGTCAAGTGCGAGAAACTCG
R NGS seq primer for MAFB of WIBR3 GCAGGGACAGGGTCCGGGGTAG
AAVS1 off 1 F CTGCTGAACACTCAGCATCTGCC
AAVS1 off 1 R CAGAGGAGCGAGTGGAGCAGACAG
AAVS1 off 2 F ATTGCAATATCCTCCTATTAGCC
AAVS1 off 2 R CACTAGAGTCACCCTATGGCTCCC
AAVS1 off 3 F CATGGGACAAGTTGATAGCTAAG
AAVS1 off 3 R GAAGTTACTCTGAAACGTATAGCAC
AAVS1 off 4 F GCTGCTCCTGGATTTAGCAAAC
AAVS1 off 4 R GCCCAGCCCAACACTTTTGGTC
AAVS1 off 5 F GTGTGCTAGTATCATTGCAAAAG
AAVS1 off 5 R CACTATTGCGTTCTCTCATTTCTC
MAFB off 1 F TCTGAGGTCCGTCTCACACACTG
MAFB off 1 R CACTGTTCAAAGAGTTTGAACATTCC
MAFB off 1 F CTCCTGACTATTGCAGTTGCTGGTCACC
MAFB off 1 R AACAGAGGAGCGAGTGGAGC
INS off 1 F CGGAGTCTCACTTTGTTGCCATG
INS off 1 R TGGTTAGAACTTCCTGCCCACAG
Gelatin (Sigma, catalog number: G2500)
Mitomycin C (Sigma, catalog number: M4287)
CMV-AsCpf1-2A-GFP-U6-AAVS1-crRNA plasmid (Addgene, #194716)
AVS1-tdT targeting plasmid (Addgene, #194728)
CAGGS-AsCpf1-2A-GFP-U6-crRNA-cloning vector (Addgene, #159281)
CMV:AsCpf1-2A-GFP-U6-crRNA-cloning vector (Addgene, #194715)
CAGGS-AsCpf1-2A-GFP-U6-tdT-crRNA plasmid (Addgene, #194724)
CAGGS-AsCpf1-2A-GFP-U6-AAVS1-crRNA plasmid (Addgene, #194723)
CAGGS-AsCpf1-2A-GFP-U6-AAVS1-MAFB-crRNA (Addgene, #194725)
CAGGS-AsCpf1-2A-GFP-U6-INS-crRNA plasmid (Addgene, #159283)
INS-2A-luciferase-2A-tdT donor plasmid (Addgene, #159348)
LB broth (Sigma, catalog number: L3522-1KG)
Agar (Fisher Scientific, catalog number: 1423-500)
Na2HPO4·7H2O (Sigma, catalog number: S9390)
Na2HPO4 (Fisher Scientific, catalog number: BP329-500)
NaCl (Sigma, catalog number: S271-3)
KCl (Fisher Scientific, catalog number: BP366-1)
KH2PO4 (Sigma, catalog number: P0662)
CaCl2 (Sigma, catalog number: C8106-500G)
MgCl2·6H2O (Sigma, catalog number: M2670-500G)
Tris (Millipore, catalog number: 648311-1KG)
EDTA (Sigma, catalog number: E6758-500g)
SDS (Fisher Scientific, catalog number: BP166-500)
H3PO4 (85%) (Sigma, catalog number: 345245-500ML)
Ascorbic acid (Sigma, catalog number: A4544-25G)
REDTaq PCR Reaction Mix (Sigma, catalog number: R2523)
Prime-It II Random Primer Labeling Kit (Agilent, catalog number: 300385)
CHROMA SPIN + TE-30 (Takarabio, catalog number: 636093)
Hybond-XL membrane (Cytiva, catalog number: RPN2020S)
BioMax MS film (Carestream, catalog number: 8294958)
NaOH (Sigma, catalog number: S5881-500G)
HCl (Sigma, catalog number: H1758)
Dorsomorphin (R & D Systems, catalog number: 3093/10)
Acetic acid (Sigma, catalog number: A6283)
Ethidium bromide (Sigma, catalog number: E1510)
[a-32P]dCTP (Revvity, catalog number: BLU513H)
LB agar (see Recipes)
Gelatin solution (see Recipes)
10× PBS (see Recipes)
Ca2+ and Mg2+ solution (see Recipes)
PBS+ (see Recipes)
MEF medium/HEK293T cells medium (see Recipes)
MEF inactivation medium (see Recipes)
DNA-extraction buffer (see Recipes)
2× phosphate buffer (pH 7.2) (see Recipes)
Hybridization buffer (see Recipes)
20× SSC buffer (pH 7.0) (see Recipes)
hPSC medium (see Recipes)
NGD medium (see Recipes)
50× TAE buffer (see Recipes)
LB agar
Reagent Final concentration Quantity or Volume LB broth 2.5% w/vol 10 g Agar 1.5% w/vol 6 g MilliQ water n/a Add to 400 mL Total (optional) n/a 400 mL Gelatin solution
Reagent Final concentration Quantity or Volume Gelatin 0.2% (w/v) 0.8 g H2O n/a 400 mL Total n/a 400 mL 10× PBS
Reagent Final concentration Quantity or Volume Na2HPO4·7H2O 25.6 g Na2HPO4 13.56 g NaCl 80 g KCl 2 g KH2PO4 2 g H2O To 1,000 mL Total 1,000 mL Adjust pH to 7.4 and autoclave for 15 min at 121 °C.
Ca2+ and Mg2+ solution
Reagent Final concentration Quantity or Volume CaCl2 5 g MgCl2·6H2O 5 g H2O To 500 mL Total 500 mL Filter the solution through a 0.22 μm filter and autoclave for a 45 min liquid cycle. After autoclaving, keep the solution at 4 °C.
PBS+
Reagent Final concentration Quantity or Volume 1× PBS (diluted from 10× PBS) 990 mL Ca2+ and Mg2+ solution (Recipe 4) 10 mL Total 1000 mL Stir the solution for approximately 30 min until it clears up. Then, filter through a 0.22 μm filter and keep refrigerated at 4 °C.
MEF medium/HEK293T cells medium
Reagent Final concentration Quantity or Volume DMEM high glucose 435 mL FBS 10% 50 mL Penicillin/streptomycin 1% 5 mL NEAA 1% 5 mL Pyruvate (100 mM) 1 mM 5 mL Total 500 mL MEF inactivation medium
Reagent Final concentration Quantity or Volume DMEM high glucose 87 mL FBS 10% 10 mL Penicillin/streptomycin 1% 1 mL NEAA 1% 1 mL Pyruvate (100 mM) 1 mM 1 mL Mitomycin C 5 µg/mL 0.5 mg Total 100 mL DNA-extraction buffer
Reagent Final concentration Quantity or Volume 1 M Tris (pH 8.0) 50 mM 25 mL 0.5 M EDTA (pH 8.0) 10 mM 10 mL 5 M NaCl 100 mM 10 mL 10% SDS 0.5% 25 mL MilliQ H2O 430 mL 20 mg/mL Proteinase K 0.5 mg/mL 25 μL for 1 mL, add just before use 2× phosphate buffer (pH 7.2)
Reagent Final concentration Quantity or Volume Na2HPO4·7H2O 0.5 M 67 g H3PO4 (85%) 4 mL MilliQ H2O Add to 1 L Hybridization buffer
Reagent Final concentration Quantity or Volume 2× phosphate buffer (Recipe 9) 1× 50 mL 0.5 M EDTA (pH 8.0) 1 mM 0.2 mL 5 M NaCl 100 mM 5 mL 20% SDS 7 % 35 mL MilliQ H2O 15 mL BSA 1% 1 g, add before use in 100 mL buffer warmed to 60 °C 20× SSC buffer (pH 7.0)
Reagent Final concentration Quantity or Volume NaCl 175.3 g Sodium citrate tribasic dihydrate 88.2 g MilliQ H2O Add to 1 L, adjust pH to 7.0 with a few drops of HCl hPSC medium
Reagent Final concentration Quantity or Volume DMEM-F12 385 mL KSR 5% 25 mL FBS 15% 75 mL Penicillin/streptomycin 1% 5 mL NEAA 1% 5 mL L-glutamine 1% 5 mL FGF2 4 ng/mL 2-ME 1 μM Total (optional) n/a 500 mL NGD medium
Reagent Final concentration Quantity or Volume Neurobasal medium 500 mL Gem21 without vitamin A 1% 5 mL AlbuMAX I 0.2% w/v 1 g NeuroPlex N2 0.5% 2.5 mL NaCl (5 M) 5 mL Pyruvate (100 mM) 1 mM 5 mL Penicillin/streptomycin (100×) 1× 5 mL GlutaMAX (100×) 1× 5 mL Biotin (5 mg/mL in 1 M NaOH) 0.35 μL Ascorbic acid (100 mM) 50 μL Lactic syrup (85%) 100 μL 50× TAE
Reagent Final concentration Quantity or Volume Tris 2 M 121 g EDTA (0.5 M, pH 8) 50 mM 50 mL Acetic acid 1 M 28.55 mL H2O To 500 mL Total 500 mL
Laboratory supplies
6-well ultra-low attachment plates (Corning, catalog number: CLS3471-24EA)
AggreWell 400 (StemCell Technologies, catalog number: 34425)
12-well plates (Corning, catalog number: 3512)
6-well plates (Corning, catalog number: 3506)
Cell lifters (Corning, catalog number: 3008)
Electroporation cuvettes, 0.2 cm (Bio-Rad, catalog number: 1652086)
PCR tubes and caps (USA Scientific, catalog number: 1402-2500)
Equipment
SMZ1270 microscope (Nikon, model: SMZ1270)
Eclipse Ti microscope (Nikon, model: Eclipse Ti)
Thermal Cycler (Bio-Rad, model: T100)
Accuspin micro desktop centrifuge (Fisher Scientific, catalog number: 75002461)
ST1 Plus desktop centrifuge (Sorvall ST1 Plus, with =TX-400 rotor and adaptors, catalog number: 75016030)
Heracell 150i CO2 incubator (Thermo Scientific, catalog number: 13998034)
Incushaker (Benchmark Scientific, catalog number: 31-206)
Gene Pulser Xcell Eukaryotic System (Bio-Rad, catalog number: 1652661)
Nanodrop 2000 spectrometer (Thermo Scientific, catalog number: ND2000)
AlphaImager gel documentation system (Alpha Innotech, model: AlphaImager 2200)
Xenogen IVIS Spectrum imager (Xenogen, model: IVIS Spectrum)
FACSAria cell sorter (BD Biosciences)
Software and datasets
Cpf1 crRNA design was performed at https://portals.broadinstitute.org/gppx/crispick/public
Prediction of off-targets was obtained using Cas-OFFinder, http://www.rgenome.net/cas-offinder
Procedure
The following are step-by-step procedures for the process of designing crRNA, validation of crRNA, genome editing of hPSCs (knockout, knockin, or multiplex genome editing), characterizing genome-edited hPSCs, testing potential off-targeting effects, and differentiation of hPSC. Some steps can be skipped based on the experimental design.
Design DNA oligos for Cpf1 crRNAs
Design crRNA. Sequences encoding Cpf1 crRNAs are designed using 23nt crRNA from the website https://portals.broadinstitute.org/gppx/crispick/public. An example of designing Cpf1 crRNA targeting the AAVS1 locus is shown in Figure 1A with the PAM sequence in magenta. The target sequence is in yellow and matches the guide sequence in the Cpf1 crRNA.
Design the DNA oligonucleotide by adding the auxiliary bases (highlighted in cyan in Figure 1B) to the crRNA guide sequence design. The addition of the auxiliary bases generates annealed products with adhesive ends (Figure 1C) to facilitate cloning the annealed products to the Cpf1 crRNA backbone plasmids.
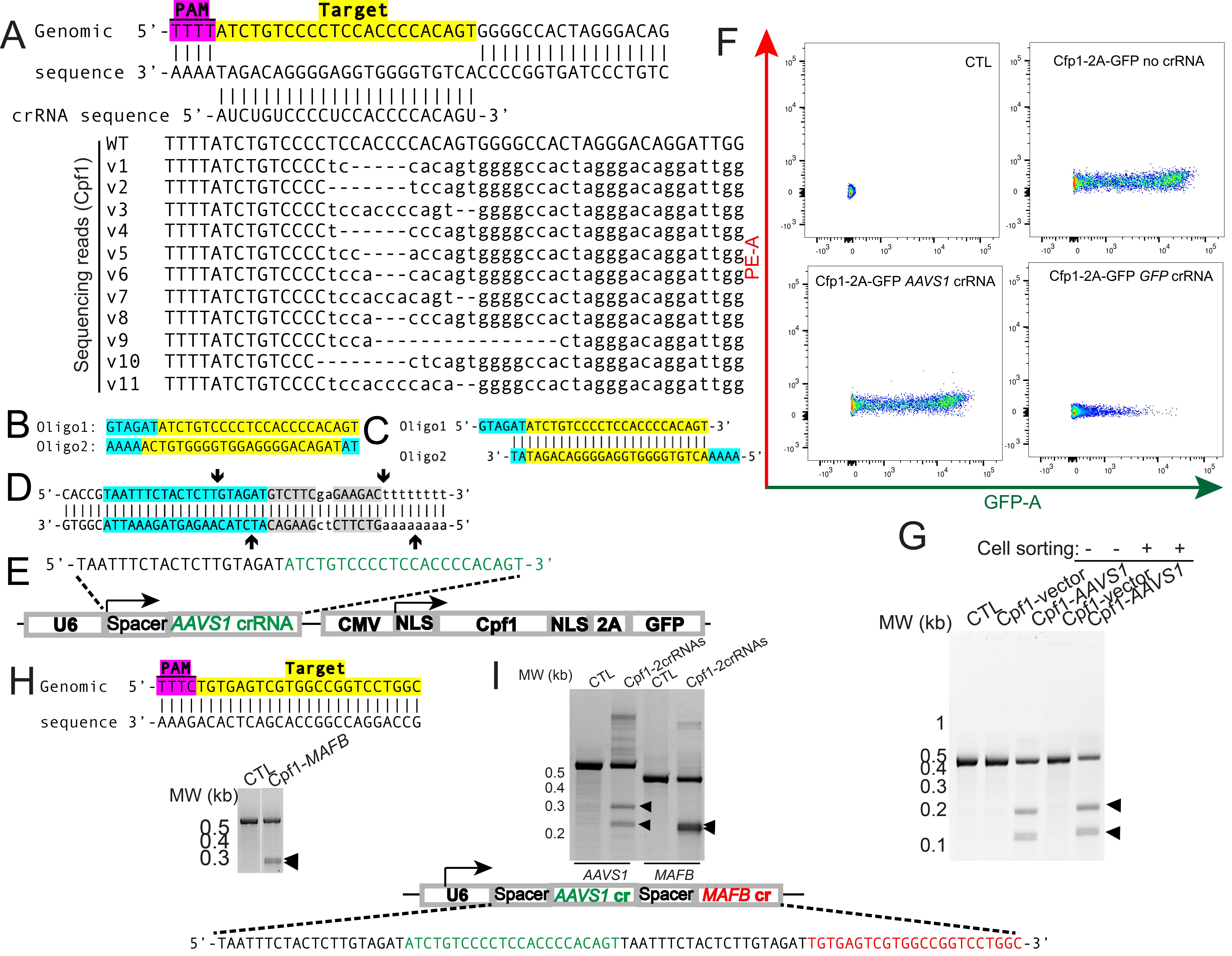
Figure 1. Design gene-specific crRNAs to clone to the bicistronic AsCpf1 plasmids and validation in HEK293T cells.A. Example of Cpf1 crRNA design for the genomic sequence at the AAVS1 locus. Magenta denotes the TTTN PAM sequence and yellow denotes gene-specific target sequence. B. Example of oligo design for the targeted genomic sequence at the AAVS1 locus. C. Annealed products from the oligos in B. Cyan denotes the sequence for adhesive ends to facilitate cloning and yellow denotes a target-specific sequence. D. BbsI cloning sites of Cpf1 plasmids. Cyan denotes spacer sequence, gray denotes BbsI cognitive sites, and arrows denote cleavage sites upon BbsI digestion. E. Design of a bicistronic vector delivering both guide RNA (AAVS1 sg RNA shown as nucleotides in green) and AsCpf1 followed by 2A-GFP. F. Representative FACS scatter plots of control HEK293T cells, AsCpf1-2A-GFP expressing, AsCpf1-2A-GFP-AAVS1 expressing HEK293T cells. G. T7 endonuclease I (T7EI) assay to detect mutations in the AAVS1 locus from unsorted and sorted cells. H. Diagram showing the AsCpf1 MAFB crRNA in yellow. The PAM sequence for AsCpf1 was highlighted in magenta. I. T7EI assay of mutations in the MAFB locus from transfected cells. J. Design of an AAVS1-targeting (green) and MAFB-targeting (red) crRNA array driven by a U6 promoter. K. T7EI assay to detect mutations in the AAVS1 and MAFB locus from transfected cells.
Clone crRNA encoding oligos to AsCpf1-2A-GFP-U6-crRNA-cloning vector
The CAGGS-AsCpf1-2A-GFP-U6-crRNA-cloning vector and the CMV:AsCpf1-2A-GFP-U6-crRNA-cloning vector can be obtained from Addgene (159281 and 194715, respectively). Use the samples from Addgene to streak LB agar plates supplemented with 100 μg/mL ampicillin and incubate the agar plates at 30 °C overnight. Pick a single colony to inoculate 5 mL of LB medium with 100 μg/mL ampicillin and shake at 220 rpm at 30 °C overnight.
Purify plasmids with the Omega Miniprep kit and measure concentration with a Nanodrop spectrometer.
Set up the following restriction digestion reaction (Table 1).
Table 1. Components for the restriction endonuclease reaction
Reagent Amount CutSmart Buffer 3 μL BbsI-HF 0.5 μL (10 units) Plasmid 3–5 μg Water Fill up to 30 μL Incubate the enzymatic reaction at 37 °C for 3 h.
Purify the DNA from the enzymatic digestion using the Zymo DNA Clean & Concentrator and elute in about 25 μL of elution buffer. Measure the concentration using a Nanodrop spectrometer. The DNA sequences between the arrows of Figure 1D will be excised, leaving adhesive ends compatible with the annealed DNA oligo in Figure 1C.
Dilute the DNA oligos to 100 μM and anneal the oligos by setting up the following reaction (Table 2).
Table 2. Components for the oligo annealing reaction
Reagent Amount Forward oligo (100 μM) 1 μL Reverse oligo (100 μM) 1 μL 10× T4 DNA ligase buffer 1 μL T4 PNK 0.5 μL Water 4.5 μL Incubate the reaction on a thermocycler at 37 °C for 1 h. Then, anneal the reaction by using the following conditions:
1. 95 °C, 10 min
2. 95–85 °C, −2 °C/s
3. 85 °C, 1 min
4. 85–75 °C, −0.3 °C/s
5. 75 °C, 1 min
6. 75–65 °C, −0.3 °C/s
7. 65 °C, 1 min
8. 65–55 °C, −0.3 °C/s
9. 55 °C, 1 min
10. 55–45 °C, −0.3 °C/s
11. 45 °C, 1 min
12. 45–35 °C, −0.3 °C/s
13. 35 °C, 1 min
14. 35–25 °C, −0.3 °C/s
15. 25 °C, 1 min
16. 25–4 °C, −0.3 °C/s
17. 4 °C, hold
Perform a 10× dilution of the annealed oligos and ligate the annealed oligo with BbsI-digested plasmid by setting up the following reaction (Table 3).
Table 3. Components for the ligation reaction
Reagent Amount Purified, BbsI-linearized plasmid (30 ng/μL) 2 μL Annealed oligo (1:10 diluted) 1 μL 10× T4 DNA ligase buffer 0.5 μL T4 DNA ligase 0.5 μL Water 1 μL Ligate at room temperature or 16 °C overnight. Use 1 μL of ligation product to transform Stbl3 chemically competent E. coli based on the manufacturer’s protocol and incubate the LB plates at 30 °C overnight.
Pick 3–6 individual colonies, inoculate 5 mL of LB medium with 100 μg/mL ampicillin, and shake at 220 rpm at 30 °C overnight. Make miniprep from the samples and sequence with U6 primer (5'-GAGGGCCTATTTCCCATGATTCC-3') to determine clones with the correct crRNA sequence insertion.
Make midi-prep of the correct plasmids using ZymoPURE II Plasmid Midiprep kit.
Validation of crRNA by transfecting Cpf1 plasmids to HEK293T cells for genome editing
Culture HEK293T cells in DMEM high glucose medium supplemented with 10% FBS, 1× L-glutamine, 1× MEM-NEAA, and 1× penicillin/streptomycin. Passage the cells 1:5 with Trypsin-EDTA upon reaching near confluency.
Plate HEK293T cells in 12-well plates. Upon 60%–80% confluence, transfect the cells with 0.5 μg of CMV-AsCpf1-2A-GFP-U6-AAVS1-crRNA plasmid (Addgene #194716, Figure 1E) with Lipofectamine 2000 transfection reagent (Invitrogen) according to manufacturer’s guideline.
Three days after transfection, dissociate cells with trypsin, followed by DNA purification with DNeasy Blood & Tissue Kit. Optionally, GFP-expressing cells could be sorted on a FACSAria (Figure 1F) as starting materials for DNA purification with the DNeasy Blood & Tissue Kit.
Set up PCR reactions as below (Table 4).
Table 4. Components for the genotyping PCR
Reagent Amount Genomic DNA 0.5 μg AmpliTaq Gold 360 Master Mix 10 μL Forward primer (5'-CCCCTTACCTCTCTAGTCTGTGC-3') 2 μL 2 μM solution (0.1 μM final concentration) Reverse primer (5'-CTCAGGTTCTGGGAGAGGGTAG-3') 2 μL 2 μM solution (0.1 μM final concentration) Water Fill up to 20 μL The PCR condition is:
One cycle of 95 °C 5 min
35 cycles of 95 °C 30 s
56 °C 30 s
72 °C50 s
One cycle of 72 °C 5 min
Purify the PCR products using a PCR purification kit. Anneal a total of 200 ng PCR products in NEB buffer 2 using the annealing conditions stated above and digest with 5 units of T7EI (NEB) for 1 h at 37 °C before resolving on 10% TBE PAGE gels (Bio-Rad) run at 100 V for 2 h. Observe DNA fragments with staining with ethidium bromide and take images with an AlphaImager gel documentation system (Figure 1G).
Similarly, MAFB-targeting crRNA containing Cpf1 plasmids can be generated with the procedures described above by annealing the following primers: MAFB-crRNA F: 5'-GTAGATTGTGAGTCGTGGCCGGTCCTGGC-3', MAFB-crRNA R: 5'- AAAAGCCAGGACCGGCCACGACTCACAAT-3'. The validation results are in Figure 1H–I using the following T7EI primers: MAFB-T7EI F: 5'-GGGGCTACGCCCAGTCTTGCAGGTATAAACG-3', MAFB-T7EI R: 5'- TCTCTCTCTCCGGCTCTGCTCGAGTCTAGGAGG-3'.
Multiple crRNAs could be generated from a single U6 promoter-driven guide RNA array separated by a spacer. In the example in Figure 1J, the crRNA sequence of the AAVS1 locus (green) and the MAFB locus (red) are separated by a space sequence (black). T7EI assay showed that transfection of the plasmids with the 2crRNAs showed similar indel efficiency compared to each crRNA individually (Figure 1K).
Knockin of the AAVS1 locus HEK293T cells by using the validated crRNA and a targeting construct
An AAVS1-tdT targeting plasmid can be obtained from Addgene (#194728) and purified following the previous procedures.
Transfect HEK293T cells from one well of a 12-well plate with 0.8 μg of targeting vector AAVS1-CAGGS-tdTomato (Figure 2A) and 0.4 μg of AsCpf1-AAVS1 plasmid using Lipofectamine 2000 transfection reagent.
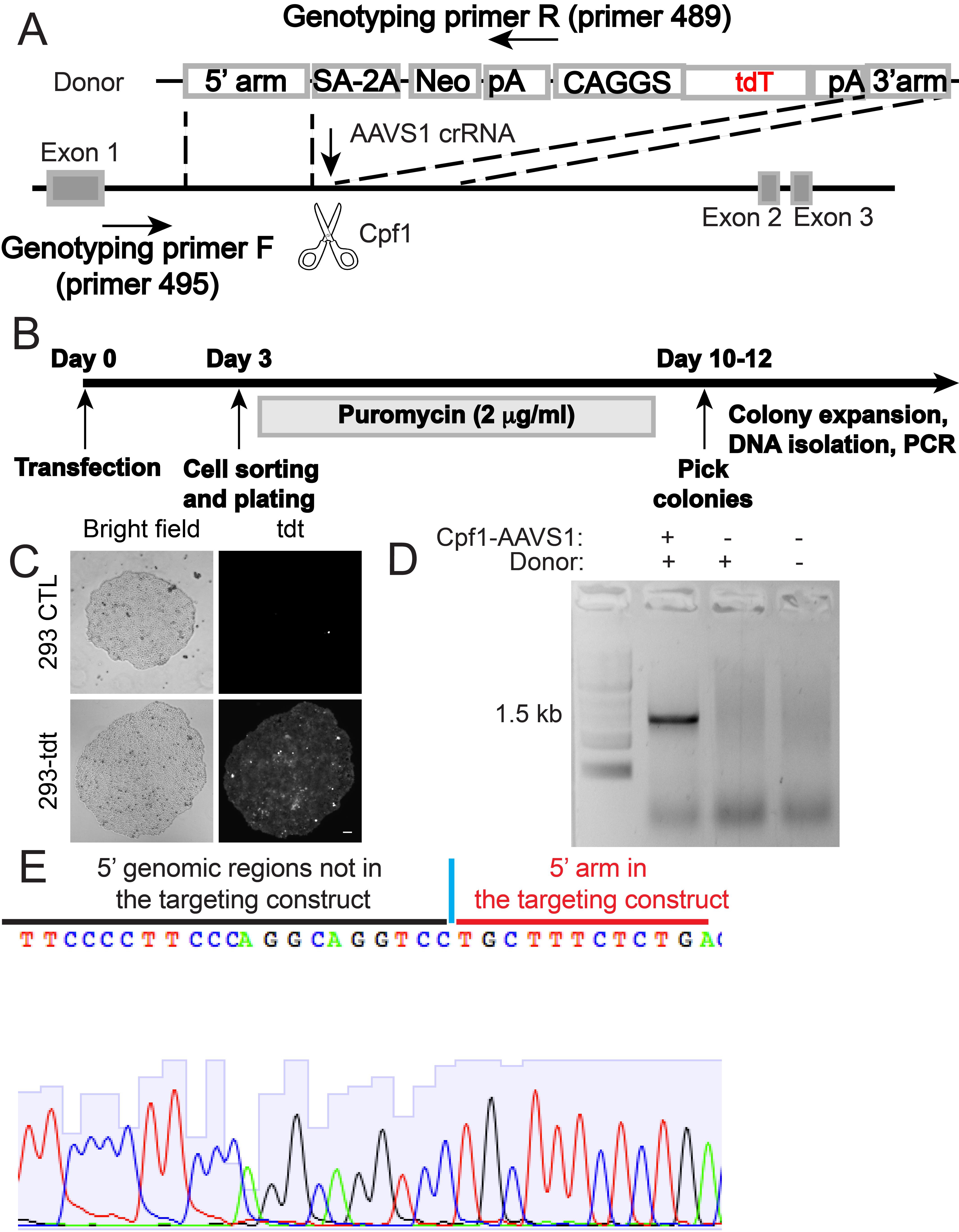
Figure 2. AsCpf1-mediated knockin of a tdTomato-expressing cassette to the AAVS1 locus in HEK293T cells. A. Schematic illustrating the targeting strategy with AsCpf1. B. Experimental timeline to generate HEK293T cells with CAGGS-tdTomato knockin at the AAVS1 locus. C. Expression of tdTomato in AsCpf1-targeted HEK293T cells (bottom panel) but not in the control cells (top panel). Scale bar: 20 µm. D. Representative genotyping PCR results with primer 495 and 489 in A to identify HEK293T cell clones with the CAGGS-tdTomato cassette integrated at the AAVS1 locus. E. Representative Sanger sequencing chromatogram by sequencing the junction region of the PCR amplicon from targeted cells. Sequences inside and outside of the homology arm are shown in red and black, respectively.Sort GFP and tdTomato (tdT) double-positive cells 3 days post-transfection on a FACSAria. Plate sorted cells at a density of approximately 2,000 cells per 10 cm culture plate and subject to selection with 2 μg/mL puromycin until colonies are formed (Figure 2B).
Mechanically passage individual tdT-expressing colonies to individual wells of a 12-well plate for expansion (Figure 2C).
To genotype HEK293T clones with correct knockin, use 0.5 μg of genomic DNA for a PCR reaction with primer 495 (5'- TCTCTCTCCTGAGTCCGGACC-3') and primer 489 (5'-ACTGAGCTCTCAGGCACCGGGCTTGCGG-3') (Figure 2A) with REDTaq PCR Reaction Mix with the following PCR condition: 95 °C 5 min, followed by 35 cycles of 95 °C 30 s, 57 °C 30 s, 72 °C 1 min 40 s, then 72 °C for 5 min. Resolve PCR products with 1% agarose gels (Figure 2D). Extract DNA from bands of the expected size with a gel purification kit (Omega Bio-tek) and subject to Sanger sequencing with primer 495 (Figure 2E).
Knockout of tdT in HEK293T-tdT cells by a tdT-crRNA
We use the CAGGS-AsCpf1 construct for introducing a crRNA targeting the tdTomato-coding sequence (Figure 3A–3B) and transfected HEK293T cells that stably express tdTomato (HEK293-tdT) (Figure 2C). As expected, we observed expression of GFP and increased tdTomato-low and negative cell proportion in the GFP-positive population compared to control cells (Figure 3C). At 4 days post-transfection, tdTomato-low cells can be observed in the HEK293T-tdTomato cells, particularly in cells with high expression of GFP (Figure 3C). The loss of expression of tdTomato becomes clearer after the propagation of sorted cells with low tdTomato expression (Figure 3D). The following are the experimental procedures.
Design a tdT crRNA to target the 5’ region of the tdT-coding sequence (Figure 3A). Design the oligos for annealing according to Section A.
Anneal the tdT-targeting crRNA oligos and ligate to CAGGS-AsCpf1-2A-GFP-U6-crRNA-cloning vector (Addgene, #159281) based on the procedures described in section B, giving rise to the CAGGS-AsCpf1-2A-GFP-U6-tdT-crRNA plasmid (Addgene, #194724).
Transfect the CAGGS-AsCpf1-2A-GFP-U6-tdT-crRNA plasmid to HEK293T-tdT cells based on the procedures described in section C. GFP-expressing cells showed reduced expression of tdT starting from 4 days post-transfection (Figure 3C).
The cells with reduced tdT expression can be isolated with FACS sorting, followed by expansion and isolation of individual clones.
Purify DNA from isolated clones and use it for PCR with the following primers. tdT-forward primer: 5’-GCAACGTGCTGGTTATTGTGCTG-3’; tdT-reverse primer: 5’-TCGCCCTCGCCCTCGATCTCGAA-3’. Purify and sequence PCR amplicons with the tdT-forward primer to identify mutations in the tdT coding sequence.
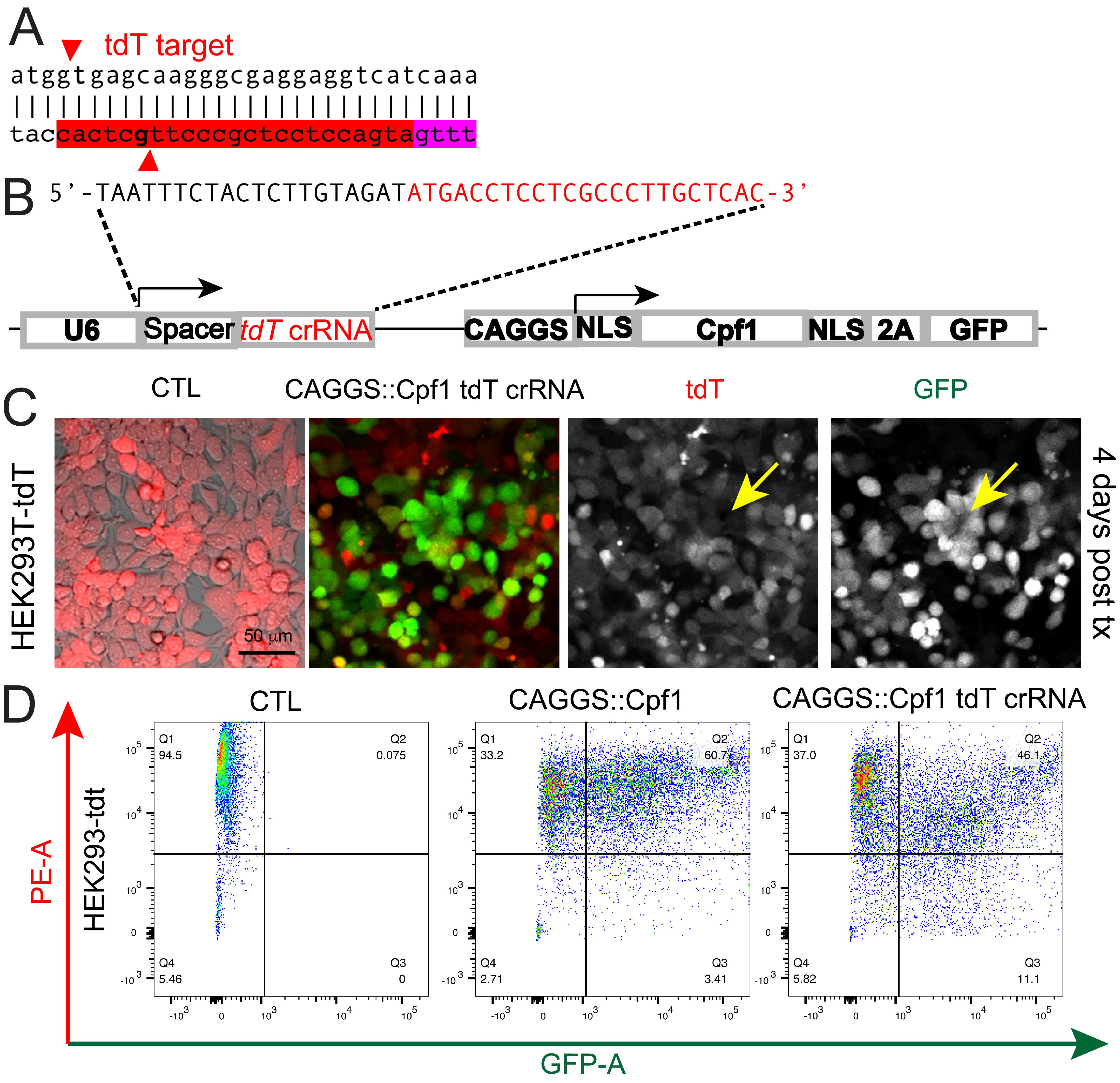
Figure 3. Disruption of tdTomato-expression in HEK293T-tdT cells with CAGGS-Cpf1 construct that expresses a tdT crRNA. A. Diagram showing the crRNA targeting the tdTomato-coding sequence. The PAM sequence is highlighted in magenta, and the guide sequence is in yellow. B. Diagram of the CAGGS-Cpf1-tdT-crRNA construct. C. Representative micrographs showing the expression of Cpf1 and the tdT crRNA in HEK293T cells reduced tdTomato fluorescence signals 4 days after transfection. Scale bar: 50 µm. D. Flow cytometry analyses showing control HEK293T-tdT cells (left panel), HEK293T-tdT cells expressing AsCpf1 driven by CAGGS promoter (middle panel), and HEK293T-tdT cells expressing AsCpf1 and the tdT crRNA.
Genome editing of hPSCs by ablating expression of tdTomato
The following procedures describe the ablation of tdTomato expression of H1-OCT4-GFP hPSCs with heterozygous integration of CAGGS-tdTomato expression cassette at the AAVS1 locus [25] by using the CAGGS-AsCpf1-2A-GFP-tdT crRNA construct.
Coat plates with gelatin. Prepare the 0.2% gelatin solution by autoclaving 0.8 g of gelatin in 400 mL of MilliQ H2O for 15 min. To coat 6-well plates, add 2 mL of cooled gelatin solution to each well of a 6-well plate and coat overnight. Coating of other tissue culture vessels could be performed by using the gelatin solution proportional to the surface area of the wells.
Generation of mouse embryonic fibroblast cells: All animal experiments were performed following the protocols approved by the Animal Research Regulation Committee at the Whitehead Institute and guidelines from the Department of Comparative Medicine at the Massachusetts Institute of Technology. Mating pairs of DR4 mice [26] were set up, and timed pregnancies were determined by detection of vaginal plugs (E0.5). At E13.5, pregnant DR4 female mice were euthanized by carbon dioxide (CO2) inhalation. After hair clipping of the abdomen and cleaning of exposed skin with 1% Wescodyne solution followed by 70% ethanol, uteri with embryos were dissected using autoclaved surgical tools and put in PBS with penicillin/streptomycin in a biosafety cabinet. Each uterus was transferred to a 10 cm tissue culture plate and the embryo was dissected with autoclaved surgical tools. After removing internal organs and extra-embryonic tissues, the remaining embryonic tissue was minced into small pieces with a scalpel. Then, 0.5 mL of 0.25% trypsin/EDTA was added to each embryo and incubated for 15 min in a 37 °C CO2 incubator. DNaseI solution (10 μL of 10 mg/mL dissolved in PBS) was added, and the tissue was mechanically dissociated by pipetting with a 1 mL pipette until no sticky components remained. Then, cells were transferred to a 15 cm tissue culture plate with 40 mL of MEF medium and incubated in a CO2 incubator as P0 cells. Upon confluency in approximately 5 days, the cells were passaged by 1:5 as P1 and then 1:5 as P2.
Remove the medium from confluent P2 MEF plates and add MEF medium with 5 μg/mL mitomycin C (13 mL for a 15 cm dish). After incubation in a CO2 incubator for 2 h, dispose of the mitomycin-containing medium in a mitomycin C waste container, rinse the cells with PBS+ 3 times, and dispose of the wash buffer in the mitomycin C container. Then, wash the cells with PBS once and harvest cells after trypsinization.
Plate mitomycin C-inactivated MEF cells at a density of 0.4 million cells per well of a gelatin-coated 6-well plate after removing the gelatin solution and culture overnight.
Maintain human pluripotent stem cell line H1-OCT4-GFP with a heterozygous knockin of a CAGGS-tdTomato expression cassette H1-tdT [25] in DMEM-F12 supplemented with 20% KSR, 4 ng/mL FGF2, 1× 2-mercaptoethanol, 1× L-glutamine, 1× MEM-NEAA, and 1× penicillin/streptomycin. Passage cells with 1 mg/mL collagenase IV every 4–6 days on mitomycin C-inactivated MEF feeders cultured in DMEM medium supplemented with 10% FBS, MEM-NEAA, 1× penicillin/streptomycin, and 1× L-glutamine.
Electroporation of plasmids to hPSC: Electroporate the CAGGS-AsCpf1-2A-GFP-U6-tdT-crRNA plasmid (Addgene, #194724) to H1-tdT cells to test the editing of the tdTomato-encoding sequence. Culture hPSCs in the medium supplemented with 10 μM Rho kinase (ROCK) inhibitor Y-27632 overnight before dissociation and electroporation. Also, prepare MEF plates during the day before electroporation. Dissociate approximately 10 million hPSCs into single cells with Accutase, filter with a 40 μm cell strainer, wash with medium containing Y-27632, centrifuge, and resuspend in 800 μL of PBS with 100 μg of Cpf1 plasmid. Load the mixture to a 0.4 cm gap Gene Pulser electroporation cuvette and incubate on ice for 5 min before electroporation with a Gene Pulser Xcell System with 1 pulse of 250 V, 500 μF. Then, incubate the cuvette on ice for 5 min before plating cells on two 6-well plates of MEF. GFP-expressing cells showed reduced expression of tdT starting from 4 days post-transfection (Figure 4A).
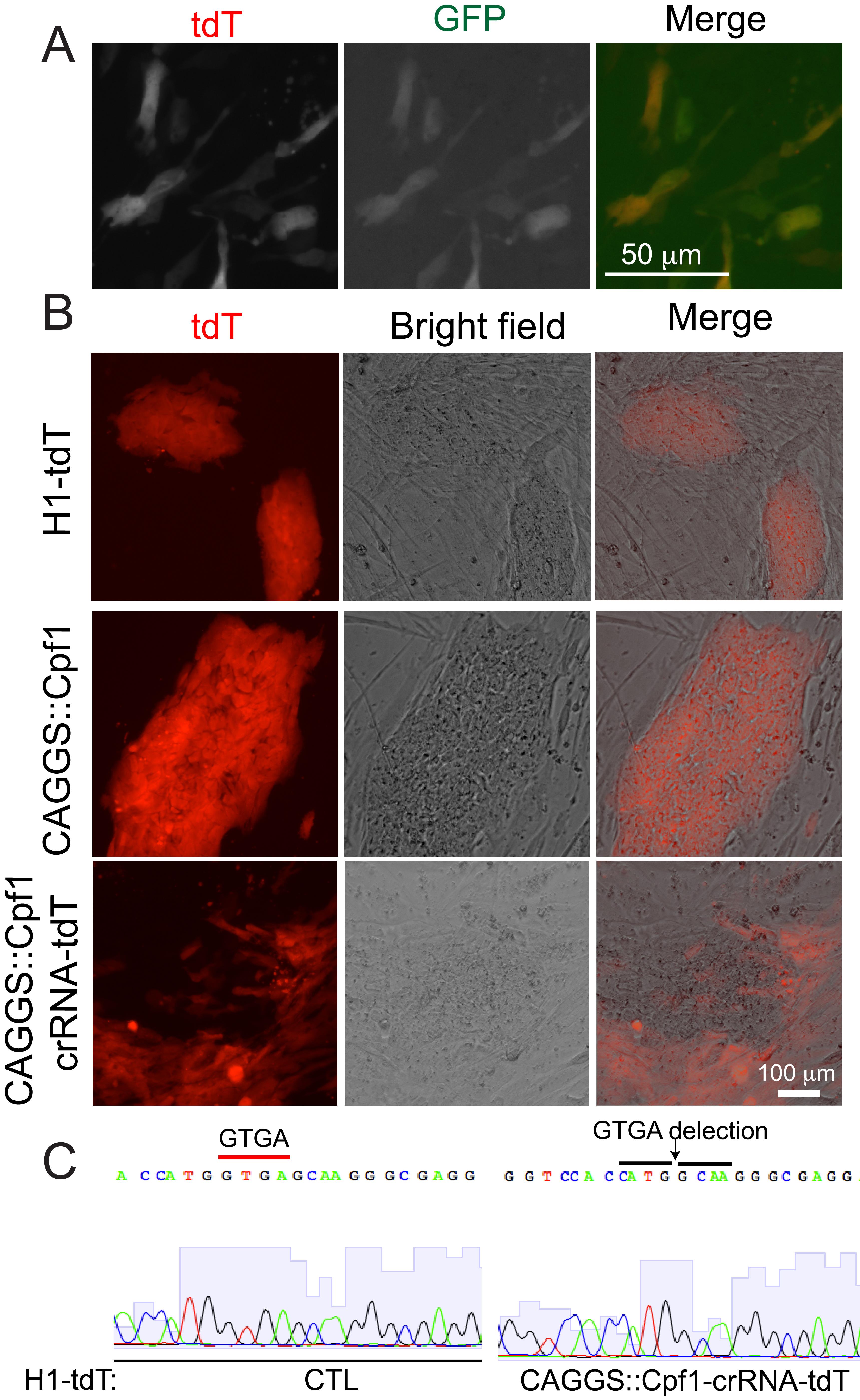
Figure 4. AsCpf1-mediated mutation formation in hPSCs. A. Representative fluorescent micrographs of hPSCs electroporated with the corresponding constructs on the left. B. Disruption of tdTomato expression in H1-tdT cells by CAGGS promoter driven AsCpf1 with a tdT crRNA. A 4-nucleotide deletion was identified in cells that lost tdTomato expression but not in control cells. Scale bar: 100 μm.Manually passage individual colonies onto MEF-coated 12-well plate wells.
To test if the loss of tdTomato expression in cells was caused by mutations of the tdTomato-coding sequence, tdT_Fwd (5'- GCAACGTGCTGGTTATTGTGCTG-3') and tdT_Rev (5'- TCGCCCTCGCCCTCGATCTCGAA-3') primers were used to amplify the potentially edited regions of tdT with PCR. Sanger sequencing showed out-of-frame mutation in electroporated cells but not in control (Figure 4B).
Knockin of hPSC and multiplex genome editing
Maintain WIBR3 hPSCs [27] in DMEM-F12 supplemented with 15% FBS, 5% KSR, 4 ng/mL FGF2, 1× 2-mercaptoethanol, 1× L-glutamine, 1× MEM-NEAA, and 1× penicillin/streptomycin. Passage cells with 1 mg/mL collagenase IV every 4–6 days on mitomycin C-inactivated MEF feeders cultured in DMEM medium supplemented with 10% FBS, MEM-NEAA, 1× penicillin/streptomycin, and 1× L-glutamine.
Electroporation of plasmids to WIBR3 hPSCs: One day before electroporation, culture cells in the medium supplemented with 10 μM Rho kinase (ROCK) inhibitor Y-27632. Dissociate approximately 10 million hPSCs into single cells with Accutase, filter with a 40 μm cell strainer, wash with medium containing 10 μM Y-27632, centrifuge, and resuspend in 800 μL of PBS with 100 μg of CAGGS-AsCpf1-2A-GFP-U6-AAVS1-crRNA plasmid (Addgene, #194723) and 40 μg of AAVS1-tdTomato donor construct (Figure 5A) (Addgene, #194728). Load the mixture to a 0.4 cm gap Gene Pulser electroporation cuvette and incubate on ice for 5 min before electroporation with a Gene Pulser Xcell System with 1 pulse of 250 V, 500 μF. Then, incubate the cuvette on ice for 5 min before plating cells on two 6-well plates of DR4 MEF. Add puromycin-containing medium (0.5 μg/mL) 3–4 days after electroporation (Figure 5B). Manually passage individual colonies onto MEF-coated 12-well plate wells (Figure 5C), expand, and subject to DNA purification.
Purify genomic DNA by isopropanol precipitation. Lyse cells from puromycin-resistant clones in DNA-extraction buffer with freshly added 0.5 mg/mL proteinase K and incubate on a 55 °C rocking platform overnight. Then, add an equivalent volume of isopropanol. Precipitate DNA samples by centrifugation at 4 °C at 16,000× g for 10 min, followed by 70% ethanol wash and centrifugation at the same condition. Dry DNA samples and dissolve in 10 mM Tris (pH 8.5) overnight on a 55 °C rocking platform.
Test the purified genomic DNA with PCR using the primers described in step D5.
Perform Southern blots by digesting 15 μg of purified DNA with 50 units of SphI-HF for 8–10 h at 37 °C in 50 μL. Resolve digested samples in a 0.8% agarose 1× TAE gel with ethidium bromide (EtBr) using HindIII-digested Lamda DNA for approximately 10 h at 75 V. Image the gel with AlphaImager and mark ladder bands with Indian ink. Soak the gel in 0.25 M HCl for 15 min at room temperature on an orbital shaking platform, followed by incubation with 0.4 M NaOH for 15 min at room temperature. Then, transfer the DNA of the gel to the Hybond-XL membrane presoaked in 0.4 M NaOH for 3–4 h or overnight at room temperature by botting with tissue stacks with weight on top.
After the transfer, wash the membrane with 0.2 M Tris buffer (pH 7) at room temperature for 5 min, followed by 2× SSC buffer at room temperature for 5 min. Dry the membrane before prehybridization in hybridization buffer in a hybridization tube placed in a hybridization oven set to 60 °C with constant rotation of the tubes.
Generate AAVS1 internal probe by PCR using AAVS1-tdTomato plasmid as a template and the following primers: 5'- GAATTCGCCCTTTGCTTTCTCTGAC-3', and 5'-TGAGCTCTCGGACCCCTGGAAGAT-3'. Generate AAVS1 external probe by PCR with H1 genomic DNA and the following primer: 5'-ACAGGTACCATGTGGGGTTC-3', and 5'-CCCTTGCCTCACCTGGC GAT-3'. The PCR products were gel purified with E.Z.N.A. Gel Extraction Kit after separation on an agarose gel.
Use Prime-It II Random Primer Labeling Kit to generate radioactive probes and add 100 ng of purified probe DNA to 10 μL of random 9-mer primer, 5 μL of OLB buffer from the kit, and DNase- and RNase-free water to 34 μL. Denature the mixture at a 98 °C heating block for 5 min before chilling on ice for 2 min. Then, add 10 μL of Klenow buffer containing dATP, dGTP, dTTP, but no dCTP, 1 μL of Klenow-Exo- from the kit, and 5 μL (about 50–100 μCi) fresh [a-32P]dCTP (Revvity), mix well, and spin down before incubation at 37 °C for 10–20 min. Purify the labeled probe using CHROMA SPIN + TE-30 Columns based on the product manual.
Denature the purified probe at 95–100 °C for 3 min before putting it on ice for 2 min. Then, add the probe to the pre-hybridized membrane to hybridize with internal or external probes overnight at 60 °C.
Wash the hybridized membrane in 60 °C 2× SSC with 0.2% SDS for 5 min in a water bath shaker.
Then, wash the membrane in 60 °C 0.2× SSC with 0.2% SDS for 15 min in a water bath shaker.
Lastly, wash the membrane in 60 °C 0.2× SSC with 0.5% SDS for 45 min in a water bath shaker.
Dry and wrap the membrane with Saran wrap and expose to autoradiography films such as BioMax MS film or compatible films in an autoradiography cassette in a -80 °C freezer overnight before developing the film (Figure 5D). The first 4 clones are correctly targeted with heterozygous knockin of CAGGS-tdT in the AAVS1 locus.
Immunofluorescence analyses of pluripotency markers in targeted WIBR3-tdT cells are performed according to previous methods [28] with the following antibodies: mouse anti-OCT3/4, 1:100 (BD Transduction Laboratories, 611203); rabbit anti-NANOG, 1:100 (Thermo Fisher Scientific); mouse anti-TRA-1–60, 1: 100, Life Technologies); mouse anti-SSEA4 antibody, 1:100 (Thermo Fisher Scientific, clone MC-813-70). Alexa 488 conjugated secondary antibodies (Life Technologies) were used at 1:400. Targeted hPSC clones expressed the pluripotency markers as control WIBR3 cells (WIBR3 CTL, Figure 5E).
Multiplex genome editing experiments targeting AAVS1 and MAFB are performed in the same way, except using the CAGGS-AsCpf1-2A-GFP-U6-AAVS1-MAFB-crRNA (Addgene, #194725) instead of #194723. The correctly targeted clones are identified with PCR and sequencing validation of the correct junction sequences (Figure 5F-5G). Then, correct clones are further verified with Southern blotting, and the correctly targeted clones are labeled in green (Figure 5H).
Next-generation sequencing is used to examine gene editing of MAFB in AAVS1 and MAFB multiplex experiments by using the same reverse primer (R NGS seq primer for MAFB of WIBR3: 5’- GCAGGGACAGGGTCCGGGGTAG-3’) and differentially barcoded forward primers (primers in the reagent lists). Set up PCR reactions based on the conditions described in step C4 and 30 s of amplification time. Resolve amplicons on a 1.5% agarose gel followed by gel purification. Dissolve purified PCR products in 35 µL of 10 mM Tris buffer (pH 8.0) at a concentration of 10–20 ng/µL. Samples were pooled and submitted to CCIB DNA Core Facility at Massachusetts General Hospital (Cambridge, MA) for CRISPR-sequencing. Briefly, partial Illumina adaptor sequences and unique identifiers (barcodes) were attached by ligation to both the 5’ and 3’ ends of the PCR amplicons. A subsequent low-cycle PCR amplification step was used to complete the addition of full-length adaptor sequences. Paired-end sequencing (2 × 150 bp) of the resulting TruSeq-compatible paired-end Illumina libraries was performed on an Illumina MiSeq platform, using V2 chemistry. The NGS data are in Figure 5I–5J.
Teratoma assays are performed to test if targeted hPSCs could form teratoma with three germ layers. Single-plex and multiplex targeted hPSCs, each from two near-confluent six-well plates, are dissociated with collagenase and resuspended in DMEM without serum. The cells are injected intramuscularly into NSG mice between 8 and 12 weeks of age using 1 mL syringes with 23-gauge needles. The formation of teratomas is monitored twice weekly for about 2 months. Palpable tumors reaching between 7 and 10 mm in diameter are dissected and fixed in buffer-neutralized formalin solution. Paraffin embedding and histological analysis using hematoxylin and eosin (H&E) staining are performed with standard protocols. Targeted hPSCs from both methods generated teratomas with three germ layers including pigmented ectodermal cells, cartilage-like mesodermal cells, and glandular endodermal cells (Figure 5K). All animal experiments were performed following the protocols approved by the Animal Research Regulation Committee.
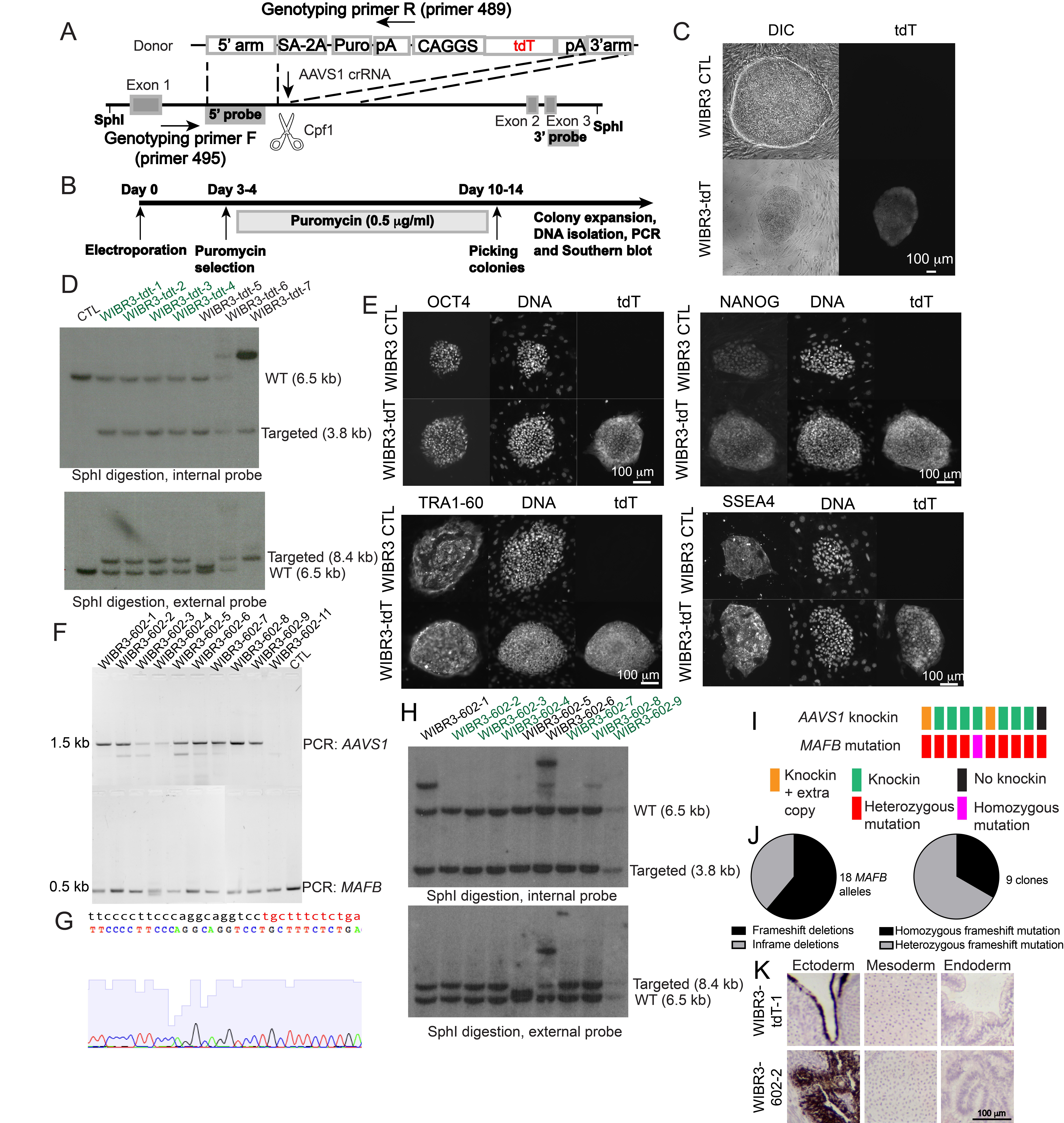
Figure 5. Multiplex genome editing in hPSCs with AsCpf1. A. Schematic illustrating the strategy for knockin at the AAVS1 locus in hPSCs with Cpf1. B. Timeline for the experiments. C. Expression of tdTomato in a representative knockin clone but not in control. D. Verification of correct knockin with Southern blot with internal probe (top panel) and external probe (bottom panel). Clones showing correct knockin are labeled in green. E. Expression of pluripotency markers OCT4, NANOG, TRA-1-60, and SSEA4 in targeted hPSC clones. F. PCR-based characterization of knockin at the AAVS1 locus in WIRB3 with the guide RNA array shown in Figure 1J. G. Example of Sanger sequencing chromatogram showing the correct junction of the sequence not in the homologous arm of the targeting construct (black letters) and sequence in the homologous arm (red letters). H. Southern blotting analyses of knocking of the AAVS1 locus with the guide RNA array shown in Figure 1J. I. Diagram showing the distribution of knockin and MAFB mutations in isolated hPSC clones. Each vertical bar pair represents an isolated hPSC clone. J. Distribution of different mutation alleles in the MAFB gene upon AsCpf1 mediated editing with the guide array in Figure 1J. K. H&E staining of teratoma tissues formed upon injection of WIBR3-tdTomato cells (top panels) or WIBR3-602-1 cells into immunocompromised mice. Scale bars in C, E, and K: 100 μm.
Differentiation to neural precursors neurons
Neuronal differentiation is performed based on the published protocol [29].
For neural progenitor cells (NP) differentiation, plate a total of 5 million dissociated WIBR3 or WIBR3-tdT cells to one Matrigel-coated 6-well plate well in NGD medium supplemented with 10 ng/mL FGF2, 1:500 insulin, and 2.5 μM dorsomorphin (R & D Systems) for 14 days with daily medium changes to keep medium from turning very yellow. Passage NP cells every 3 days in NGD medium with 10 ng/mL FGF2, 1:500 insulin.
For neuronal differentiation, plate about 0.2 million NP cells in each 12-well plate well in 1 mL of NGD medium and change the medium every 2–3 days. Fix NP and neurons after 4–6 weeks of differentiation using the immunofluorescence protocols described above in G14. Nestin expression showed proper NP differentiation of WIBR3-tdT cells (Figure 6A), and neuronal maker TUBB3 was expressed similarly in unedited WIBR3 and Cpf1 edited WIBR3-tdT cells (Figure 6A), indicating Cpf1 editing did not affect neural differentiation of hPSCs.
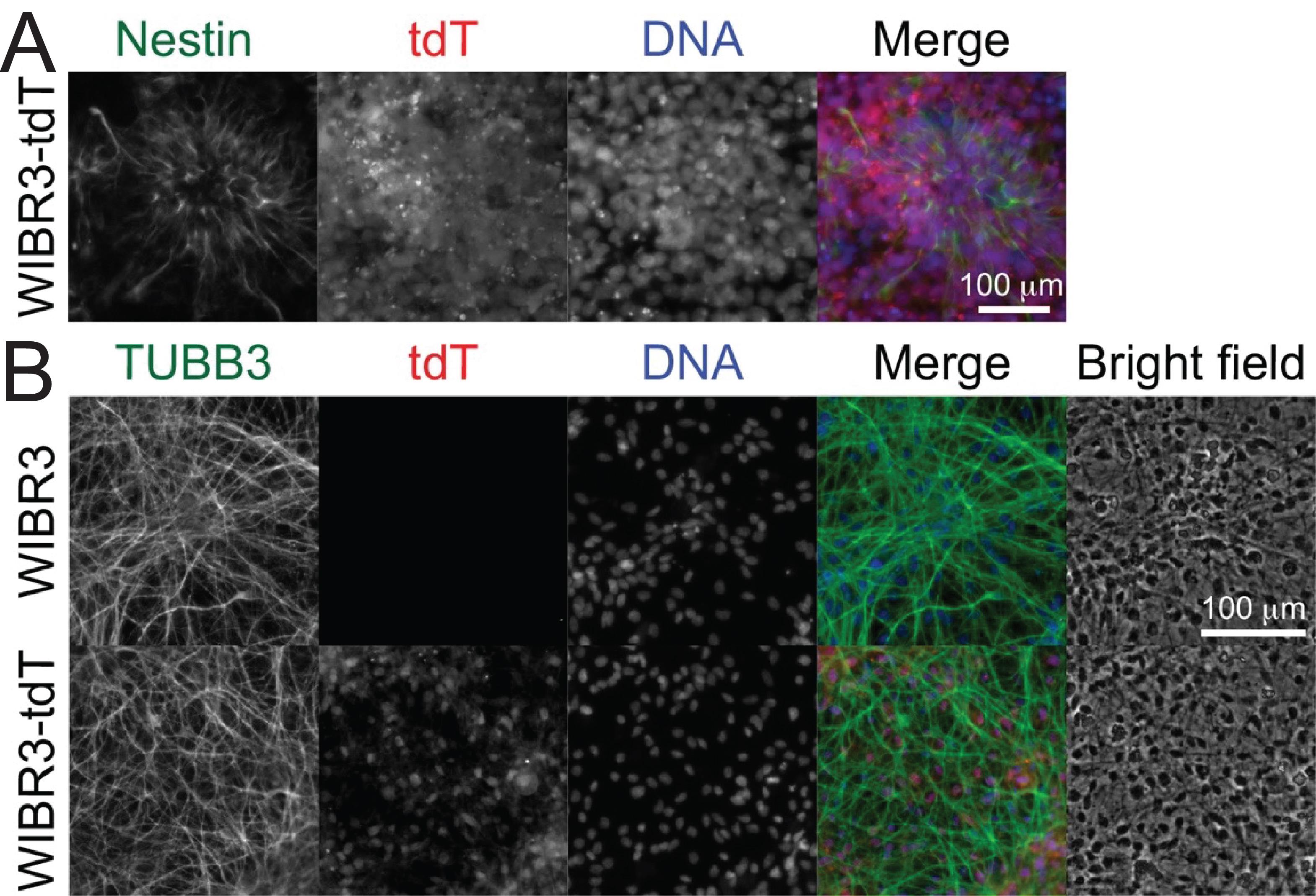
Figure 6. Differentiation of Cpf1-edited WIBR3-tdT cells to neural precursor cells and neurons. A. WIBR3-tdT cells were differentiated into neural precursor (NP) cells for 14 days. Then, NP cells were passaged, fixed, and stained with an anti-Nestin antibody (1:100, SC-23927). The formation of tdT-expressing (red) neural rosette structures (green) was shown. B. The WIBR3-tdT-differentiated NP cells were differentiated toward neurons with growth factor withdrawal for four weeks. Neurite structures were shown by anti-TUBB3 (1:1,000) staining. Scale bars in A and B: 100 μm.
Generating INS reporter hPSC cells
The INS crRNA (Figure 7A) was designed, generated, and validated in a similar way as described before. Electroporation was performed as described before. The generated CAGGS-AsCpf1-2A-GFP-U6-INS-crRNA plasmid is available from Addgene (159283). The primers 417-1 (5'- TGGGGCAGGTGGAGCTGGGCGG-3') and primer 418-1 (5'-ACCACCCCTGGCCCCTCAGAGACC-3') were used for T7EI assays.
The H1-OCT4-GFP hPSCs [30] were cultured in the same way as H1-tdT described before.
The INS-2A-luciferase-2A-tdT donor plasmid (Figure 7B) was described before [5] and is available from Addgene (159348). To target the INS locus in H1-OCT4-GFP, electroporation was performed using the procedures described before by using 100 μg of CAGGS-AsCpf1-2A-GFP-U6-INS-crRNA and 40 μg of donor construct INS-2A-luciferase-2A-tdT. Add the puromycin-containing medium (0.5 μg/mL) 3–4 days after electroporation. Manually passage individual colonies onto MEF-coated 12-well plate wells, expand, and genotype with PCR using the primer 662 (5'-GGCCTTTGGTGCAGTGACCAGAGTGTCAGG-3') and primer 663 (5'- GATTCTCCTCGACGTCACCGCATGTTAGC-3'). Subject clones with correct knockin to Southern blotting analysis.
For the INS targeted cells, amplify the internal probe with PCR with the following primers and firefly luciferase gene as a template: forward: 5'-ATGGAAGACGCCAAAAACATAAAGAAAGGCCC-3'; reverse: 5'-CACGGCGATCTTTCCGCCCTT-3'. Amplify the external probe with PCR with the following primers and genomic DNA from H1-OCT4-GFP hPSCs: forward: 5'-GACTCCCCACTTCCTGCCCATCT-3'; reverse: 5'-TCTTCTCCCAGCCCCGTCCTCAC-3'. The Southern blotting process is based on the procedures described in section G. Clones with correct heterozygous knockin were 1, 2, 7, 8, 13, and 15 in Figure 7C. Note that the control H1-OCT4-GFP has two different bands for the external probe, possibly because of the different sequences of the two alleles at the INS locus.
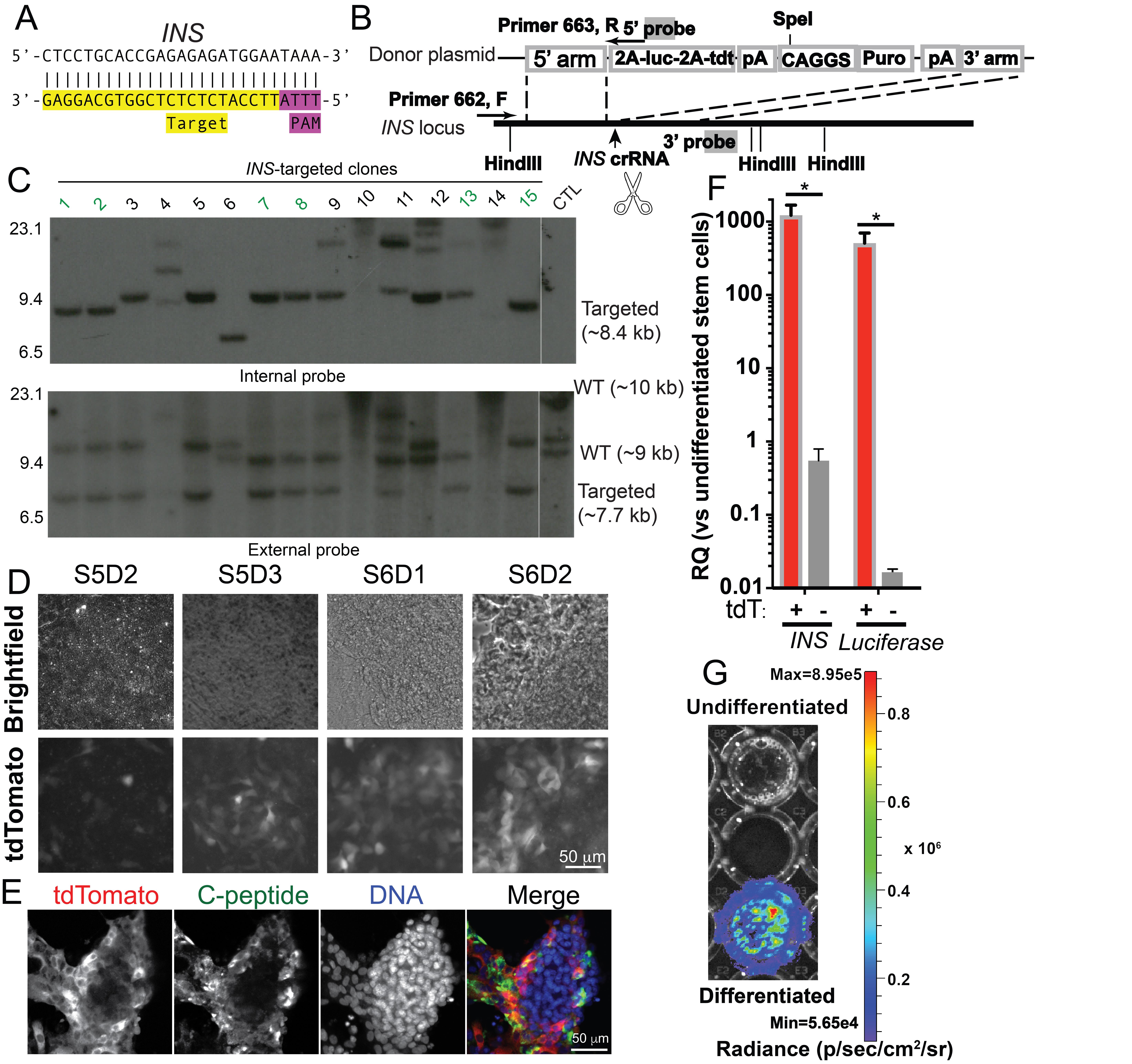
Figure 7. AsCpf1-mediated targeting in the INS locus in hPSCs enables monitoring of INS expression. A. The design of an INS targeting crRNA. The PAM sequence is highlighted in magenta, and the guide sequence is highlighted in yellow. B. The targeting strategy for generating INS luciferase and tdTomato reporter INS-luciferase-tdT allele by knocking the 2A-luciferase-2A-tdt cassette to the 3' end of the INS coding sequence. C. Southern blots with internal probe (left panel) and external probe (right panel) to identify clones with a single copy of the 2A-luciferase-2A-tdt cassette inserted into the INS locus. Correct clones were labeled in green. The double bands in the control samples for the external probe (right panels) correspond to two wild-type INS alleles of the H1-OCT4-GFP cells. D. Expression of tdTomato in cells during the differentiation of the INS-luciferase-tdT reporter hPSCs toward pancreatic β-like cells. E. Representative confocal micrographs of INS-luciferase-tdT hPSC differentiated β-like cells with anti-tdTomato staining (red) and anti-C-peptide staining (green). F. Quantitative RT-PCR analysis of sorted tdTomato positive cells (red bars) showed significantly higher expression of INS and luciferase compared to sorted tdTomato negative cells (grey bars) (* P < 0.05, t-test, n = 3 experiments). G. β-like cells specific luciferase signal at stage 6, day 7. Scale bars in D and E: 50 μm.
Differentiation of β-cells
Differentiation of β-cells was based on a published protocol [5] with the following steps.
Adapt H1-OCT4-GFP-INS-tdT hPSC cells for feeder-free conditions by culturing in Matrigel-coated tissue culture plates in mTeSR. For coating plates, dilute Matrigel 1:100 in cold DMEM-F12 medium. Then, add 2 mL of diluted Matrigel solution to one well of a 6-well plate well and coat overnight at room temperature before pre-warming in a 37 °C incubator for 1 h before using. Culture cells in mTeSR medium in 6-well plates and passage every 3–4 days with a 1:4–6 passage ratio. To prevent differentiation, passage cells before complete confluency.
Treat PSCs with mTeSR medium supplemented with 10 μM Rho kinase (ROCK) inhibitor Y-27632 overnight. Then, dissociate the cells into single cells with Accutase. Dislodge cells by tapping the sides of the plates and add RI-supplemented mTeSR to dissociate cells into single cells. After cell counting with Countess, plate about 1.8 million live cells to each well of a 6-well Matrigel-coated plate in mTeSR medium supplemented with 10 μM ROCK inhibitor Y-27632.
After overnight culture, replace the Y-27632-containing mTeSR medium with the mTeSR medium. Differentiation is initiated when cells reach around 90% confluency (usually between 1–2 days after plating cells). Wash cells with PBS- and then perform daily medium changes as follows.
S1 (3 days) base medium: MCDB131 with 0.5% FAF-BSA, 1.5 g/L NaHCO3, 10 mM glucose, 1× GlutaMAX, 1× penicillin/streptomycin. Freshly supplemented factors: 100 ng/mL Activin A and 3 μM CHIR99021 (day 1); 100 ng/mL Activin A, and 0.3 μM CHIR99021 (day 2); 100 ng/mL Activin A (day 3).
S2 (2 days) base medium: MCDB131 with 0.5% FAF-BSA, 1.5 g/L NaHCO3, 10 mM glucose, 1× penicillin/streptomycin, 1× GlutaMAX, and 0.25 mM Vc. Freshly supplemented factors: 50 ng/mL KGF.
S3 (2 days) base medium: MCDB131 with 2% FAF-BSA, 1.5 g/L NaHCO3, 10 mM glucose, 1× penicillin/streptomycin, 1× GlutaMAX, 0.25 mM Vc, and 0.5× insulin-transferrin-selenium-ethanolamine. Freshly supplemented factors: 50 ng/mL KGF, 0.25 μM SANT-1, 1 μM retinoic acid, 100 nM LDN193189, and 200 nM TPB.
S4 (4 days) base medium: MCDB131 with 2% FAF-BSA, 1.5 g/L NaHCO3, 10 mM glucose, 1× penicillin/streptomycin, 1× GlutaMAX, 0.25 mM Vc, and 0.5× insulin-transferrin-selenium-ethanolamine. Freshly supplemented factors: 50 ng/mL KGF, 50 ng/mL EGF, 0.25 μM SANT-1, 0.1 μM retinoic acid, 200 nM LDN193189, and 100 nM TPB. For the last medium change, 10 μM Y-27632 was added to the S4 medium.
S5 (3 days) starts with a transition from 2D culture to 3D culture. Cells are washed with PBS-, dissociated with Accutase, washed with H1 S5 base medium (see below), and about 6 million live cells are plated in 3 mL of S5 medium (see below) supplemented with 10 μM Y-27632 in each well of a 6-well AggreWell 400 plates. After overnight culture, move the clusters to 6-well ultra-low adherent culture plates placed on an orbital shaker set at 95–100 rpm for 2 more days in S5 medium. The rest of the differentiation is carried out in ultra-low attachment plates placed on an orbital shaker with the same setting.
S5 base medium: MCDB131 with 2% FAF-BSA, 1.5 g/L NaHCO3, 20 mM glucose, 1× penicillin/streptomycin, 1× GlutaMAX, 0.25 mM Vc, 0.5× insulin-transferrin-selenium-ethanolamine, 10 µg/mL heparin, and 10 μM ZnSO4. Freshly supplemented factors: 10 μM ALK5 inhibitor II, 0.25 μM SANT-1, 0.05 μM retinoic acid, 100 nM LDN193189, and 1 μM T3.
S6 (7 days) base medium: MCDB131 with 2% FAF-BSA, 1.5 g/L NaHCO3, 20 mM glucose, 1× penicillin/streptomycin, 1× GlutaMAX, 0.25 mM Vc, 0.5× 100× insulin-transferrin-selenium-ethanolamine, 10 µg/mL heparin, 10 μM ZnSO4. Freshly supplemented factors: 10 μM ALK5 inhibitor II, 100 nM gamma-secretase inhibitor XX, 100 nM LDN193189, and 1 μM T3.
S7 (7–14 days) medium: MCDB131 with 2% FAF-BSA, 1.5 g/L NaHCO3, 8 mM glucose, 1× penicillin/streptomycin, 1× GlutaMAX, 1× MEM-NEAA, 10 μg/mL heparin, 10 μM ZnSO4, 1× trace element A, and 1× trace element B.
The INS-tdT-targeted hPSCs are tdTomato negative at the pluripotent stem cell state. During the differentiation of the targeted INS reporter hPSCs toward β-cells, tdTomato signal emerges at day 2 of stage 5 (Figure 7D, S5D2), and more tdTomato expressing cells are detected during later stages (Figure 7D). Immunofluorescence analyses show that C-peptide expression cells also expressed tdTomato (Figure 7E). Quantitative RT-PCR analysis of cells sorted based on tdTomato expression levels show high levels of INS and luciferase expression in tdTomato-positive cells compared to tdTomato-negative or lowly expressing cells (Figure 7F). To test the expression of luciferase, 15 mg/mL D-luciferin is added to undifferentiated hPSC or INS reporter cells differentiated to β-cells. After mixing for 1 min, luciferase signals are acquired with an IVIS Spectrum imager (Xenogen) with an exposure of 1 min and mid-bin setting. Imaging quantification is performed with Living Image (Version 4.5.4). A control well is used to subtract the background signal from the measurements of samples. Consistent with the qPCR results (Figure 7F), specific luciferase signals were identified in differentiated β-like cells but not in undifferentiated pluripotent stem cells (Figure 7G).
Test potential off-targeting effects
The predicted off-target sites can be obtained from http://www.rgenome.net/cas-offinder with the following guide sequences (PAM sequences in italic font, position allowing for variable bases in bold). Sequences showing most similarities are shown with differences in lower case. No less than 4-nucleotide differences off-target sites were identified.
AAVS1: TTTTATCTGTCCCCTCCACCCCACAGT
MAFB: TTTCTGTGAGTCGTGGCCGGTCCTGGC
INS: CTCCTGCACCGAGAGAGATGGAATAAA
PCR reactions with primers (AAVS1 off 1 to INS off 1 in the primer list) were performed with 0.5 μg of genomic DNA from parental and targeted pluripotent stem cell clones. PCR products were resolved with 1% agarose gel followed by purification with a gel purification kit and Sanger sequencing with the sequencing primers listed in Table 5.
Table 5. Experimental evaluation of off-targeting effects of Cpf1-mediated genome editing in hPSCs. The nucleotides in lower case denote the differences between the potential off-target site and the corresponding crRNA.
Putative off target sequence Genomic position* (direction) Mutation/tested AAVS1 crRNA: TTTNTGTGAGTCGTGGCCGGTCCTGGC
DNA: TTTCTGTGAGTCcTGGCCctTCCTGGa
7: 131811-131837 (-) 0/5 AAVS1 crRNA: TTTNTGTGAGTCGTGGCCGGTCCTGGC
DNA: TTTTATCTcTCCCCTCCACCCaAaAcT
13: 91233988-91234014 (+) 0/5 AAVS1 crRNA: TTTNATCTGTCCCCTCCACCCCACAGT
DNA: TTTCATCTcTCCCCTaCtCCCCACtGT
2: 18590645-18590671 (+) 0/5 AAVS1 crRNA: TTTNATCTGTCCCCTCCACCCCACAGT
DNA: TTTTATCTtcCCgCTgCACCCCACAGT
2: 235878805-235878831 (-) 0/5 AAVS1 crRNA: TTTNATCTGTCCCCTCCACCCCACAGT
DNA: TTTAATtTtgCCaCTCCACCCCACAGT
6: 123920988-123921014 (-) 0/5 MAFB crRNA: TTTNTGTGAGTCGTGGCCGGTCCTGGC
DNA: TTTATGTGAaTCaTGGCCaGTCCTGtC
11: 134335239-134335265 (+) 0/10 MAFB crRNA: TTTNTGTGAGTCGTGGCCGGTCCTGGC
DNA: TTTCTGTGAGTCcTGGCCctTCCTGGa
7: 131811-131837 (-) 0/10 INScrRNA: TTTNTTCCATCTCTCTCGGTGCAGGAG
DNA: TTTATTCCATCTCTCTgctTcCAGGAG
11: 62432165-62432191 (+) 0/6
Validation of protocol
This protocol or parts of it has been used and validated in the following research article:
Ma et al. [5]. Human T-cells expressing a CD19 CAR-T receptor provide insights into mechanisms of human CD19 positive b-like cell destruction. Cell Rep Med. (Figure 1 and Figure S1).
General notes and troubleshooting
General notes
Working with hPSCs could require intuitional approval and appropriate MTA. Research needs to adhere to approved protocols that follow the ISSCR guidelines regarding hPSC research. Keeping hPSC cell density from reaching complete confluency reduces the chance of differentiation.
All work involving vertebrate animals including mice requires Institutional Animal Care and Use Committee (IACUC) approval.
Appropriate licenses for working with radioactive isotopes, comprehensive training, protective equipment, and users’ dosimeters are needed to work with radioactive Southern blotting probes. Digoxigenin-labeled probes could be an alternative approach for radioactive probes.
For efficient editing of hPSCs, the CAGGS-AsCpf1-2A-GFP-U6-crRNA-cloning vector (Addgene, #159281) is recommended. If the transfected cells do not express GFP, it is helpful to FACS sort GFP-expressing cells to enrich cells that express Cpf1.
For cell lines such as HEK293T cells, both CAGGS-AsCpf1-2A-GFP-U6-crRNA-cloning vector (Addgene, #159281) and CMV:AsCpf1-2A-GFP-U6-crRNA-cloning vector (Addgene, #194715) can be used. Lower editing efficiency could be observed compared to Cas9-mediated gene editing using T7EI assays.
The AAVS1-tdT targeting construct (Addgene, #194728) was modified based on AAVS1-tdT (Addgene, #159275) by modifying a segment with high sequence similarity to the Cpf1 AAVS1 crRNA to avoid potential digestion of the targeting construct by Cpf1.
The starting cell density for differentiation of β-cells would need optimization. For H1-OCT-GFP cells, Figure 8A shows a suitable density to start differentiation, whereas Figure 8B and Figure 8C show a lower and higher density than the optimal condition, respectively. The cellular morphologies of the first two steps were published before (Figure 1A, first three images of [31]). A representative image after pancreatic precursor 2 (PP2) is shown in Figure 8D.
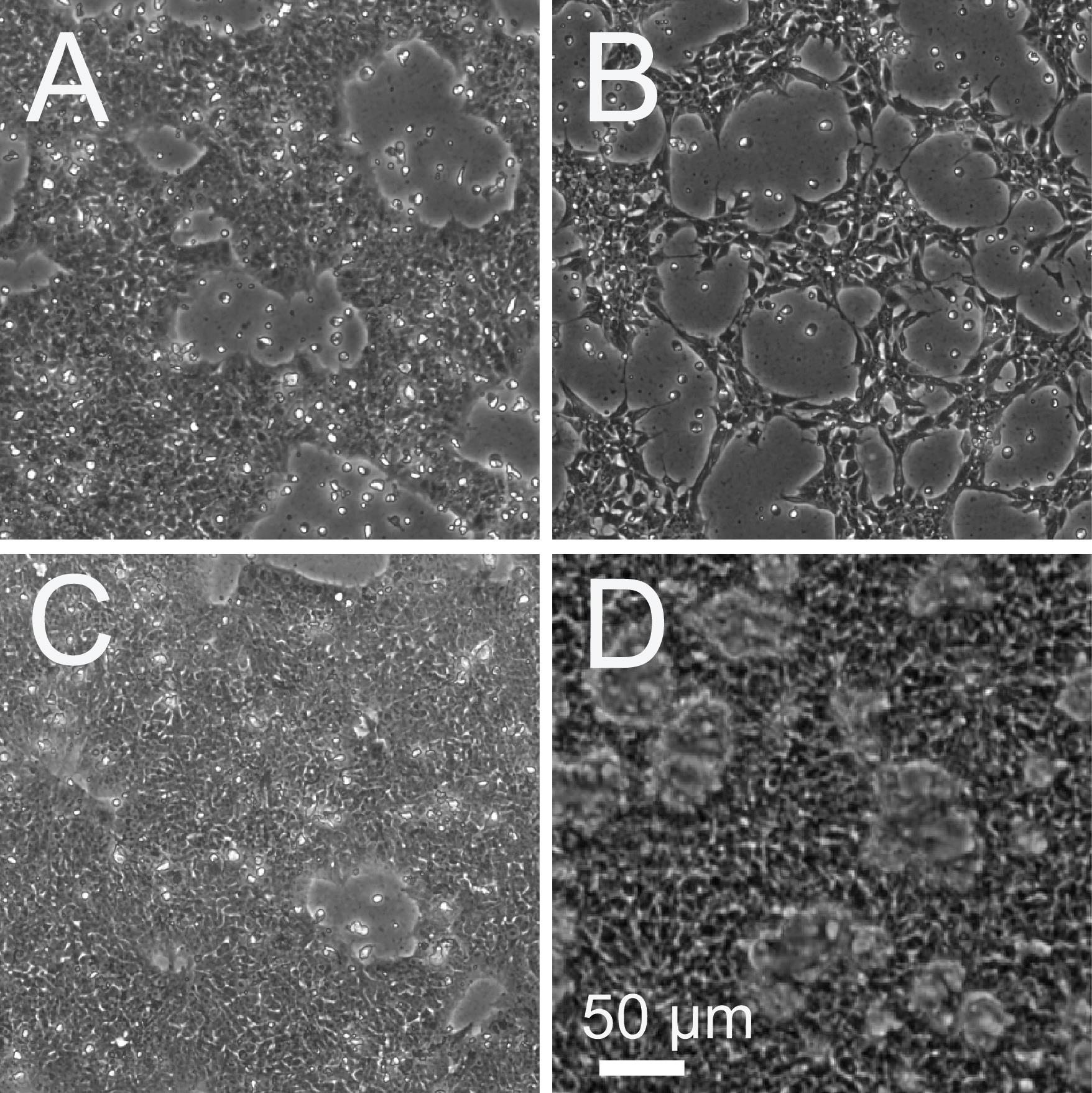
Figure 8. Cell density optimization for starting β-cell differentiation. A. Suitable cellular density for differentiation. B. Representative cellular density that is lower than optimal densities. C. Representative cell density that is higher than optimal densities. E. Representative image of PP2 cells before dissociation to form spheroids. Scale bar: 50 μm.
Troubleshooting
Problem 1: Low editing efficiency of hPSCs.
Possible cause: Insufficient expression of Cpf1.
Solution: Use the CAGGS promoter to drive Cpf1 expression by cloning crRNA to the CAGGS-AsCpf1-2A-GFP-U6-crRNA-cloning vector (Addgene, #159281). Increase the amount of plasmids used in electroporation or transfection. Furthermore, including the crRNA targeting your gene of interest in a crRNA assay together with AAVS1 crRNA (illustrated in Figure 1J) and performing simultaneous targeting of the AAVS1 locus using a donor plasmid that allows puromycin selection (Figure 5A) can assist the identification of edited hPSC clones.
Problem 2: Poor survival rate of hPSCs after electroporation.
Possible cause: Insufficient ROCK inhibitor treatment and non-optimal MEF plates.
Solution: Before dissociation and electroporation, hPSCs are treated with a final concentration of 10 µM ROCK inhibitor-supplemented hPSC medium overnight. After electroporation, the hPSCs should be plated in the medium with 10 µM ROCK inhibitor for overnight attachment to MEF feeder cells. The plates should have a confluent monolayer of MEF cells. It is important to wash off mitomycin C after MEF inactivation. MEF inactivated with irradiation (3000 rad) could also be used as feeder cells, but they tend to last shorter than mitomycin C-inactivated MEF.
Acknowledgments
The authors wish to thank Prof. Rudolf Jaenisch and members of Prof. Jaenisch’s laboratory for advice and discussion, Prof. Feng Zhang’s laboratory for sharing pX458 and pY010 plasmid through Addgene, Dr. Huan Yang of Prof. Hongkui Deng’s laboratory for sharing INS targeting construct, Patrick Autissier, Eleanor Kincaid, and Hanna Aharonov of FACS core laboratory of Whitehead Institute for sorting cells, Gary Rogers of the Whitehead Institute for assistant with medium preparation, the Center for Computational and Integrative Biology (CCIB) at Massachusetts General Hospital for the use of the CCIB DNA Core Facility (Cambridge, MA), which provided NGS and data analysis service, and Arend Vischer of the Ma’s laboratory for feedback on this work. This study was supported by a generous gift from Liliana and Hillel Bachrach and in part by the NIH (RO1-CA084198) to R.J., and the University of Illinois start-up funding to H.M.
Competing interests
H.M. declares no competing interests.
References
- Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K. and Yamanaka, S. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 131(5): 861–872. https://doi.org/10.1016/j.cell.2007.11.019.
- Wernig, M., Meissner, A., Foreman, R., Brambrink, T., Ku, M., Hochedlinger, K., Bernstein, B. E. and Jaenisch, R. (2007). In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 448(7151): 318–324. https://doi.org/10.1038/nature05944.
- Pagliuca, F. W., Millman, J. R., Gurtler, M., Segel, M., Van Dervort, A., Ryu, J. H., Peterson, Q. P., Greiner, D. and Melton, D. A. (2014). Generation of functional human pancreatic beta cells in vitro. Cell. 159(2): 428–439. https://doi.org/10.1016/j.cell.2014.09.040.
- Hochedlinger, K. and Jaenisch, R. (2015). Induced Pluripotency and Epigenetic Reprogramming. Cold Spring Harb Perspect Biol. 7(12). https://doi.org/10.1101/cshperspect.a019448.
- Ma, H., Jeppesen, J. F. and Jaenisch, R. (2020). Human T Cells Expressing a CD19 CAR-T Receptor Provide Insights into Mechanisms of Human CD19-Positive beta Cell Destruction. Cell Rep Med. 1(6): 100097. https://doi.org/10.1016/j.xcrm.2020.100097.
- Hockemeyer, D., Soldner, F., Beard, C., Gao, Q., Mitalipova, M., DeKelver, R. C., Katibah, G. E., Amora, R., Boydston, E. A., Zeitler, B., et al. (2009). Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 27(9): 851–857. https://doi.org/10.1038/nbt.1562.
- Hockemeyer, D., Wang, H., Kiani, S., Lai, C. S., Gao, Q., Cassady, J. P., Cost, G. J., Zhang, L., Santiago, Y., Miller, J. C., et al. (2011). Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol. 29(8): 731–734. https://doi.org/10.1038/nbt.1927.
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science. 339(6121): 819–823. https://doi.org/10.1126/science.1231143.
- Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., Norville, J. E. and Church, G. M. (2013). RNA-guided human genome engineering via Cas9. Science. 339(6121): 823–826. https://doi.org/10.1126/science.1232033.
- Cornu, T. I., Mussolino, C. and Cathomen, T. (2017). Refining strategies to translate genome editing to the clinic. Nat Med. 23(4): 415–423. https://doi.org/10.1038/nm.4313.
- Cox, D. B., Platt, R. J. and Zhang, F. (2015). Therapeutic genome editing: prospects and challenges. Nat Med. 21(2): 121–131. https://doi.org/10.1038/nm.3793.
- Hsu, P. D., Lander, E. S. and Zhang, F. (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell. 157(6): 1262–1278. https://doi.org/10.1016/j.cell.2014.05.010.
- Slaymaker, I. M., Gao, L., Zetsche, B., Scott, D. A., Yan, W. X. and Zhang, F. (2016). Rationally engineered Cas9 nucleases with improved specificity. Science. 351(6268): 84–88. https://doi.org/10.1126/science.aad5227.
- Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. and Liu, D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 533(7603): 420–424. https://doi.org/10.1038/nature17946.
- Anzalone, A. V., Randolph, P. B., Davis, J. R., Sousa, A. A., Koblan, L. W., Levy, J. M., Chen, P. J., Wilson, C., Newby, G. A., Raguram, A., et al. (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 576(7785): 149–157. https://doi.org/10.1038/s41586-019-1711-4.
- Liu, B., Dong, X., Zheng, C., Keener, D., Chen, Z., Cheng, H., Watts, J. K., Xue, W. and Sontheimer, E. J. (2023). Targeted genome editing with a DNA-dependent DNA polymerase and exogenous DNA-containing templates. Nat Biotechnol. https://doi.org/10.1038/s41587-023-01947-w.
- Sakuma, T., Nishikawa, A., Kume, S., Chayama, K. and Yamamoto, T. (2014). Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci Rep. 4: 5400. https://doi.org/10.1038/srep05400.
- Zetsche, B., Gootenberg, J. S., Abudayyeh, O. O., Slaymaker, I. M., Makarova, K. S., Essletzbichler, P., Volz, S. E., Joung, J., van der Oost, J., Regev, A., et al. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 163(3): 759–771. https://doi.org/10.1016/j.cell.2015.09.038.
- Hur, J. K., Kim, K., Been, K. W., Baek, G., Ye, S., Hur, J. W., Ryu, S. M., Lee, Y. S. and Kim, J. S. (2016). Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nat Biotechnol. 34(8): 807–808. https://doi.org/10.1038/nbt.3596.
- Kim, Y., Cheong, S. A., Lee, J. G., Lee, S. W., Lee, M. S., Baek, I. J. and Sung, Y. H. (2016). Generation of knockout mice by Cpf1-mediated gene targeting. Nat Biotechnol. 34(8): 808–810. https://doi.org/10.1038/nbt.3614.
- Fonfara, I., Richter, H., Bratovic, M., Le Rhun, A. and Charpentier, E. (2016). The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature. 532(7600): 517–521. https://doi.org/10.1038/nature17945.
- Zetsche, B., Heidenreich, M., Mohanraju, P., Fedorova, I., Kneppers, J., DeGennaro, E. M., Winblad, N., Choudhury, S. R., Abudayyeh, O. O., Gootenberg, J. S., et al. (2017). Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol. 35(1): 31–34. https://doi.org/10.1038/nbt.3737.
- Kim, D., Kim, J., Hur, J. K., Been, K. W., Yoon, S. H. and Kim, J. S. (2016). Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol. 34(8): 863–868. https://doi.org/10.1038/nbt.3609.
- Kleinstiver, B. P., Tsai, S. Q., Prew, M. S., Nguyen, N. T., Welch, M. M., Lopez, J. M., McCaw, Z. R., Aryee, M. J. and Joung, J. K. (2016). Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol. 34(8): 869–874. https://doi.org/10.1038/nbt.3620.
- Ma, H., Wert, K. J., Shvartsman, D., Melton, D. A. and Jaenisch, R. (2018). Establishment of human pluripotent stem cell-derived pancreatic beta-like cells in the mouse pancreas. Proc Natl Acad Sci U S A. 115(15): 3924–3929. https://doi.org/10.1073/pnas.1702059115.
- Tucker, K. L., Wang, Y., Dausman, J. and Jaenisch, R. (1997). A transgenic mouse strain expressing four drug-selectable marker genes. Nucleic Acids Res. 25(18): 3745–3746. https://doi.org/10.1093/nar/25.18.3745.
- Lengner, C. J., Gimelbrant, A. A., Erwin, J. A., Cheng, A. W., Guenther, M. G., Welstead, G. G., Alagappan, R., Frampton, G. M., Xu, P., Muffat, J., et al. (2010). Derivation of pre-X inactivation human embryonic stem cells under physiological oxygen concentrations. Cell. 141(5): 872–883. https://doi.org/10.1016/j.cell.2010.04.010.
- Li, C., Ma, H., Wang, Y., Cao, Z., Graves-Deal, R., Powell, A. E., Starchenko, A., Ayers, G. D., Washington, M. K., Kamath, V., et al. (2014). Excess PLAC8 promotes an unconventional ERK2-dependent EMT in colon cancer. J Clin Invest. 124(5): 2172–2187. https://doi.org/10.1172/JCI71103.
- Muffat, J., Li, Y., Yuan, B., Mitalipova, M., Omer, A., Corcoran, S., Bakiasi, G., Tsai, L. H., Aubourg, P., Ransohoff, R. M., et al. (2016). Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat Med. 22(11): 1358–1367. https://doi.org/10.1038/nm.4189.
- Zwaka, T. P. and Thomson, J. A. (2003). Homologous recombination in human embryonic stem cells. Nat Biotechnol. 21(3): 319–321. https://doi.org/10.1038/nbt788.
- Ma, H., de Zwaan, E., Guo, Y. E., Cejas, P., Thiru, P., van de Bunt, M., Jeppesen, J. F., Syamala, S., Dall'Agnese, A., Abraham, B. J., et al. (2022). The nuclear receptor THRB facilitates differentiation of human PSCs into more mature hepatocytes. Cell Stem Cell. 29(5): 795–809 e711. https://doi.org/10.1016/j.stem.2022.03.015.
Article Information
Publication history
Received: Jun 27, 2024
Accepted: Sep 6, 2024
Available online: Oct 15, 2024
Published: Nov 20, 2024
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Ma, H. (2024). Multiplex Genome Editing of Human Pluripotent Stem Cells Using Cpf1. Bio-protocol 14(22): e5108. DOI: 10.21769/BioProtoc.5108.
Category
Stem Cell > Pluripotent stem cell > Regenerative medicine
Cell Biology > Cell engineering > CRISPR-cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.