- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Optimized Expression and Isolation of Recombinant Active Secreted Proteases Using Pichia pastoris
(*contributed equally to this work) Published: Vol 13, Iss 5, Mar 5, 2023 DOI: 10.21769/BioProtoc.4628 Views: 4256
Reviewed by: Neha NandwaniRitu GuptaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Recombinant proteins of high quality are crucial starting materials for all downstream applications, but the inherent complexities of proteins and their expression and purification create significant challenges. The Pichia pastoris yeast is a highly useful eukaryotic protein expression system. Pichia’s low cost, genetic tractability, rapid gene expression, and scalability make it an ideal expression system for foreign proteins. Here, we developed a protocol that has optimized the expression and isolation of a non-mammalian secreted metalloprotease, where we can routinely generate recombinant proteins that are pure and proteolytically active. We maximized growth and protein production by altering the feeding regime, through implementation of a non-fermentable and non-repressing carbon source during the methanol-induction phase. This approach increased biomass production and yielded milligrams of recombinant protein. Downstream applications involving active, recombinant fungal proteases, such as conjugation to nanoparticles and structure-related studies, are greatly facilitated with this improved, standardized approach.
Graphical abstract

Background
The production of active recombinant proteases at concentrations that allow for their use in downstream applications can be particularly challenging (Colige, 2020). The yeast Pichia pastoris has been successfully used as a eukaryotic expression system for foreign proteins (Macauley-Patrick et al., 2005). Unlike mammalian cells, Pichia does not require complex media or growth conditions, which makes its use cost-effective and scalable since high cell densities can be achieved in minimal media (Macauley-Patrick et al., 2005). Pichia strains are also genetically tractable, and the variety of available promoters and selectable markers facilitate gene expression (Daly and Hearn, 2005). Moreover, the presence of a eukaryotic protein synthesis pathway in Pichia allows high levels of posttranslational modification including glycosylation, proteolytic processing, and disulfide bond formation—processes that are limited in prokaryotic expression systems (Macauley-Patrick et al., 2005; Ahmad et al., 2014; Macek et al., 2019).
Generating large quantities of recombinant proteins is achievable with bioreactors; however, this is not feasible for most and, in general, recombinant proteins are produced in shaking flasks, similar to our approach (Schwettmann and Tschesche, 2001; Fernandez et al., 2013). The relatively small volumes of culture and limited cell densities that are ultimately achieved with this approach have made the isolation of recombinant proteases in large quantities difficult. Our goal was to develop a standardized protocol that would allow for the routine expression and isolation of a proteolytically active secreted metalloprotease, with high purity and in large enough quantities to facilitate downstream applications. We made use of the AOX1 methanol-induced promoter in Pichia that ensures that transcription is tightly regulated by a repression/derepression mechanism (Ahmad et al., 2014). Carbon sources such as glucose or glycerol, which are nutritional requirements for Pichia’s cell growth, cannot be used during the induction phase since both would repress transcription. The challenge arises during induction with methanol, since growth is severely inhibited preventing high levels of biomass production, which can ultimately negatively impact protein production. We found that the addition of sorbitol, a non-repressible and non-fermentable carbon source in yeast, during the methanol-induction phase, significantly improved cell viability and biomass production. The inclusion of sorbitol boosted protein production to milligram quantities. Although implementing non-repressing carbon sources such as alanine, mannitol, trehalose, and sorbitol had previously been explored, it has not been readily adopted (Inan and Meagher, 2001; Celik et al., 2009).
Here, we provide a detailed protocol for the expression and isolation of a proteolytically active metalloprotease from a neurotropic fungal pathogen, which has been applied to our work involving protein-nanoparticle conjugates as drug-delivery platforms for the central nervous system (Aaron and Gelli, 2020).
Materials and Reagents
96-well assay plate with black wall and clear, flat bottom (Corning, catalog number: 3904)
PYREX® Delong shaker Erlenmeyer flask with extra-deep baffles, 250 and 1,000 mL (Corning, catalog numbers: 4450-250, 4446-1L)
1.5 mL microcentrifuge tubes (USA Scientific, catalog number: 1615-5500)
15 mL centrifuge tubes, conical, sterile, polypropylene (VWR, catalog number 89039-664)
Serological pipettes, sterile wrapped (Corning, catalog numbers: 4487, 4488, 4489, 4490)
250 mL centrifuge tube (Corning, catalog number: 430776)
10 kDa NMWL Ultra-4 centrifugal filter unit (Amicon, catalog number: UFC803008)
Peptone (Research Products International, catalog number: P20250); store at room temperature
Yeast extract (Thermo Scientific, catalog number: J23547-A1); store at room temperature
Dextrose (VWR, BDH Chemicals, catalog number: BDH9230); store at room temperature
Sorbitol (Sigma-Aldrich, CAS 50-70-4); store at room temperature
Methanol (Sigma-Aldrich, CAS 67-56-1); store at room temperature
Bacto agar (VWR, Life Sciences, catalog number: J637); store at room temperature
G418 sulfate (Fisher Bioreagents, CAS 108321-42-2); store at 4 °C
PureCube INDIGO Ni-MagBeads (magnetic beads) (Cube Biotech, catalog number: 75225)
Magnet, >835 KA/m (Magneto Inc., Amazon)
Pierce fluorescent protease activity kit (Thermo Fisher Scientific, catalog number: 23266)
FTC-Casein (5 mg/mL in ultrapure water)
TPCK-trypsin (50 mg/mL in tris-buffered saline)
Tris-buffered saline (25 mM Tris, 150 mM NaCl, pH 7.2)
Yeast media (see Recipes)
BMGY
BMMY
YPD
YPD + G418 (4 mg/mL) agar plate
Buffers (see Recipes)
Binding buffer pH 8.0
Wash buffer pH 8.0
Elution buffer pH 8.0
Phosphate-buffered saline, pH 7.4
Solutions (see Recipes)
10% glycerol
5% methanol
1 M potassium phosphate buffer, pH 6.0
10× YNB
500× biotin
Strains
Pichia pastoris GS115 (Invitrogen, catalog number: C18100)
Pichia pastoris GS116 <Cn MPR1> (Aaron and Gelli, 2020)
Equipment
-80 °C freezer
-20 °C freezer
4 °C refrigerator
pH meter
Vortex (Scientific Industries, SKU: SI-0236 or equivalent)
Swing-bucket centrifuge (Beckman Coulter, model: Allegra X-15R; 208 V, 60 Hz)
Innova 4200 incubator shaker (New Brunswick) or equivalent temperature-controlled shaker
Pipetman (Eppendorf, P20, P200, P1000)
Power supply (Bio-Rad, model: PowerPac Universal Power Supply)
Protein gel electrophoresis equipment (Bio-Rad, model: Mini-PROTEAN)
Microplate reader (Molecular Devices, model: SpectraMax M5e)
Procedure
The steps in the following segments were done using a methylotrophic yeast, Pichia pastoris, for the expression of recombinant proteins. The recombinant Mpr16XHIS protein of focus was isolated from a strain of P. pastoris transformed with a construct composed of MPR1 cDNA isolated from Cryptococcus neoformans KN99 with a 6XHIS tag at the C-terminus in a pPIC9K backbone, as described in the main article (Aaron and Gelli, 2020). This plasmid confers resistance to G418, which was added to agar plates (4 mg/mL) in both the original selection of transformed P. pastoris and while expanding frozen (-80 °C) stocks for the current protocol. We handled open cultures in a sterile field under an open flame throughout this protocol, checking for bacterial contamination at least once every 24 h by brightfield microscopy. The volumes and concentrations described below were optimal for our purposes but may need to be scaled for other experiments. The protocol below can be divided into three stages: (1) growth, induction, and expression; (2) isolation and purification; and (3) verification and storage.
Expression of recombinant Mpr1 protein
Streak the Pichia pastoris GS116 <Cn MPR1> strain (Aaron and Gelli, 2020) on a YPD + G418 (4 mg/mL) agar plate. Incubate the plate at 30 °C for 72 h.
Note: Any gene of interest can be subcloned in expression vectors for P. pastoris.
Select a single colony from the G418 plate and transfer directly to a 250 mL baffled Erlenmeyer flask containing 50 mL of BMGY media (see Recipes) using a flame-sterilized inoculating loop.
Note: Using a baffled Erlenmeyer flask greatly increases biomass production compared to a non-baffled Erlenmeyer flask.
Incubate the P. pastoris culture at 30 °C for 24 h, shaking at 300 rpm. Cover the flasks loosely with aluminum foil allowing for gas exchange.
After 24 h, decant cultures into 50 mL centrifuge tubes and spin down at 3,724 × g for 5 min.
Note: The culture is easily contaminated due to the nutrient-rich BMGY medium; thus, examine the culture for any possible contamination after every 24 h period. In our case, the cultures were examined by brightfield microscopy to rule out contamination.
Discard the supernatant and wash the pellet 1–3 times using 25 mL of BMMY (see Recipes) and vortex the pellet until it is fully resuspended. Spin down the pellet at 3,724 × g between washes. Resuspend cells in BMMY and measure the optical density at 600 nm (OD600).
Note: Following steps 1–5, we typically see an OD600 of approximately 25 in 20 mL of BMMY.
Add the cell mixture to the desired volume of BMMY, in order to dilute the culture to a final OD600 = 1. Cover the flask with sterile cheesecloth, allowing for appropriate gas exchange.
Incubate the induced culture at 30 °C, shaking, for 24 h.
Add sorbitol and methanol to the culture to a final concentration of 50 g/L and 0.5%, respectively. Incubate at 30 °C, shaking, for 24 h.
Note: Sorbitol is a non-repressing carbon source and we found that a co-feeding regime of methanol with sorbitol increases biomass production (Figures 1 and 2).

Figure 1. Visualization of Pichia biomass production. A. Initiating induction in BMMY with OD600 = 1.0. B. Following 24 h induction in BMMY and just prior to co-feeding with 50 g/L sorbitol and 0.5% methanol. C. Following 24 h of co-feeding with sorbitol and methanol. At this stage, measurements of OD600 are typically well over 18.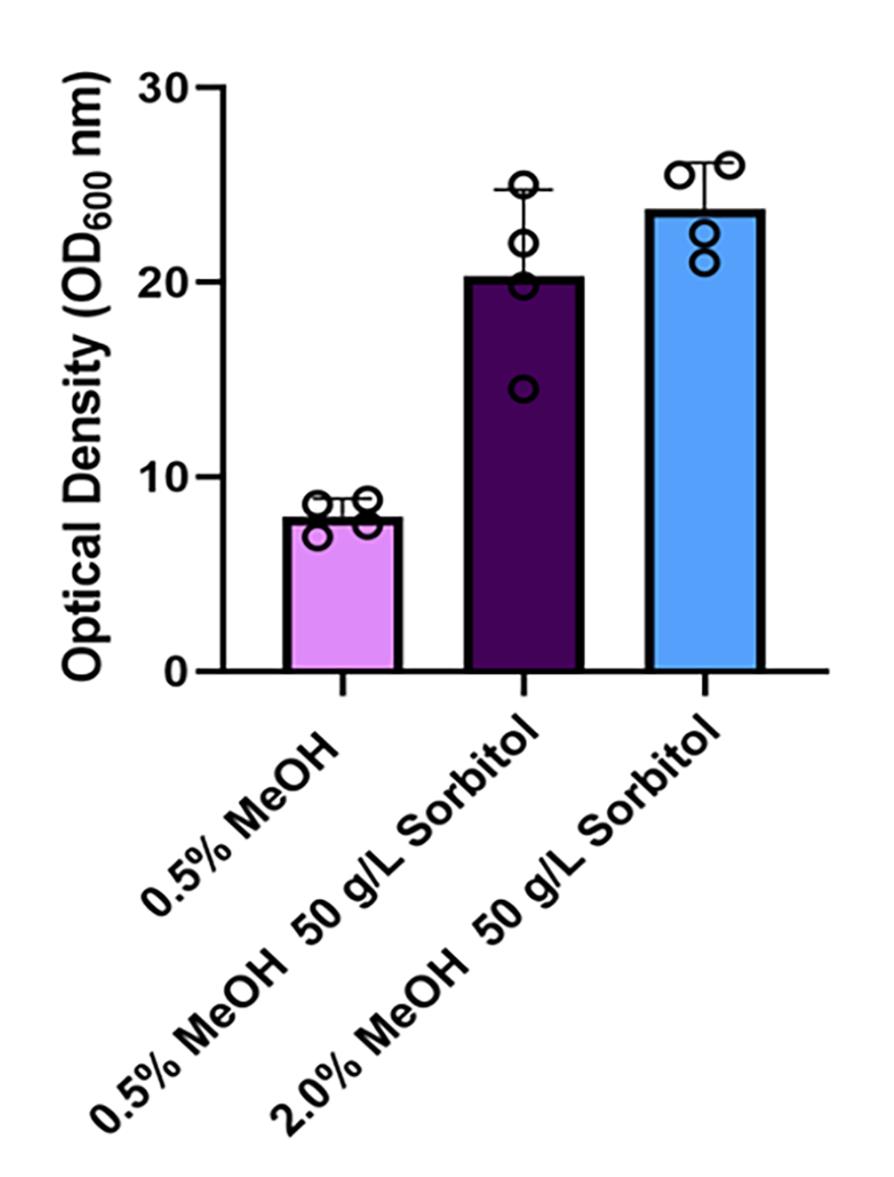
Figure 2. Optical densities of Pichia cultures supplemented with sorbitol. A comparison of the optical densities (OD600) of cultures with 0.5% methanol (MeOH) or co-fed with 0.5% MeOH and 50 g/L of sorbitol or 2% MeOH and 50 g/L of sorbitol. The cultures were co-fed following 24 h inoculation of the Pichia strain in BMMY media at a starting OD600 of 1. The OD600 measurements of the cultures were recorded 24 h after the co-feeding was initiated.
Isolation and purification of recombinant Mpr1 protein
For PureCube INDIGO Ni-MagBeads, mix and aliquot beads in a 15 mL conical centrifuge tube and briefly centrifuge the beads; then, place the aliquot near a strong magnet in order to remove and replace the storage buffer supernatant with phosphate-buffered saline, pH 7.4. Keep the beads on ice and see protocols provided by the manufacturer (PureCube) for further information.
Note: If inducing a 250 mL culture, use 750 µL of beads from storage. This equals approximately 188 µL of beads (25% of storage solution contains beads with binding capacity of 70 mg/mL).
Autoclave the beads for 15 min on liquid cycle. Allow beads to cool before proceeding.
Measure the final OD600 of the P. pastoris culture. Decant the culture into centrifuge bottles.
Note: The addition of sorbitol with cultures induced with 0.5% methanol (MeOH) significantly increased the optical density (OD600) of the Pichia cultures from ~8 to >20 (Figure 2).
Centrifuge at 3,724 × g for 5 min at 30 °C. Decant and keep the supernatant.
Add 1.5 mL of 25% (v/v) Ni-MagBeads per 500 mL of collected culture supernatant. Incubate at 30 °C, shaking at 250 rpm, for 30 min.
Collect the Ni-MagBeads from the supernatant using a strong magnet. Decant the supernatant into a fresh vessel, then remove the magnet and resuspend the beads in 6 mL of binding buffer. Transfer resuspended beads in a 15 mL conical tube. Repeat at least twice. Wash with 10 mL of binding buffer until all beads have been collected.
Place the collected beads in a centrifuge tube and centrifuge at 3,724 × g for 5 min at 4 °C.
Magnetically pellet the MagBeads and remove binding buffer. Add 10 times the bead volume of wash buffer to the beads. Remove the magnet and vortex for at least 2 min.
Note: Each 250 mL culture volume, receiving 188 µL of beads initially, is washed with 1.88 mL wash buffer at this step. We found that washing once was sufficient to remove any foreign matter, but still washed thrice without appreciable loss of MagBeads.
Briefly centrifuge and collect beads at the bottom of the tube with a magnet to facilitate the exchange of wash buffer for elution buffer. Use twice the initial bead volume of elution buffer. Vortex beads for 5 min and centrifuge at 3,724 × g for 5 min at 4 °C.
Note: Multiple elution steps can be performed at this stage to enrich for Mpr1; however, we found that one elution step was sufficient.
Transfer the protein-containing elution buffer supernatant to a 10 kDa NMWL Ultra-4 centrifugal filter unit.
Centrifuge and concentrate the sample at 4,000 × g for 15 min at 4 °C. Measure protein concentration by the method of Bradford (Bradford, 1976).
Note: Timing for this step may vary depending on desired final concentration and quantity of elution buffer added. In our case, the above conditions yielded approximately 300 µL of protein suspended in elution buffer. PBS was exchanged for elution buffer following step 11, centrifuging for 8 min instead of 15. On average, 2.5 mg of Mpr1 was extracted from a 500 mL BMMY culture (Figure 3).
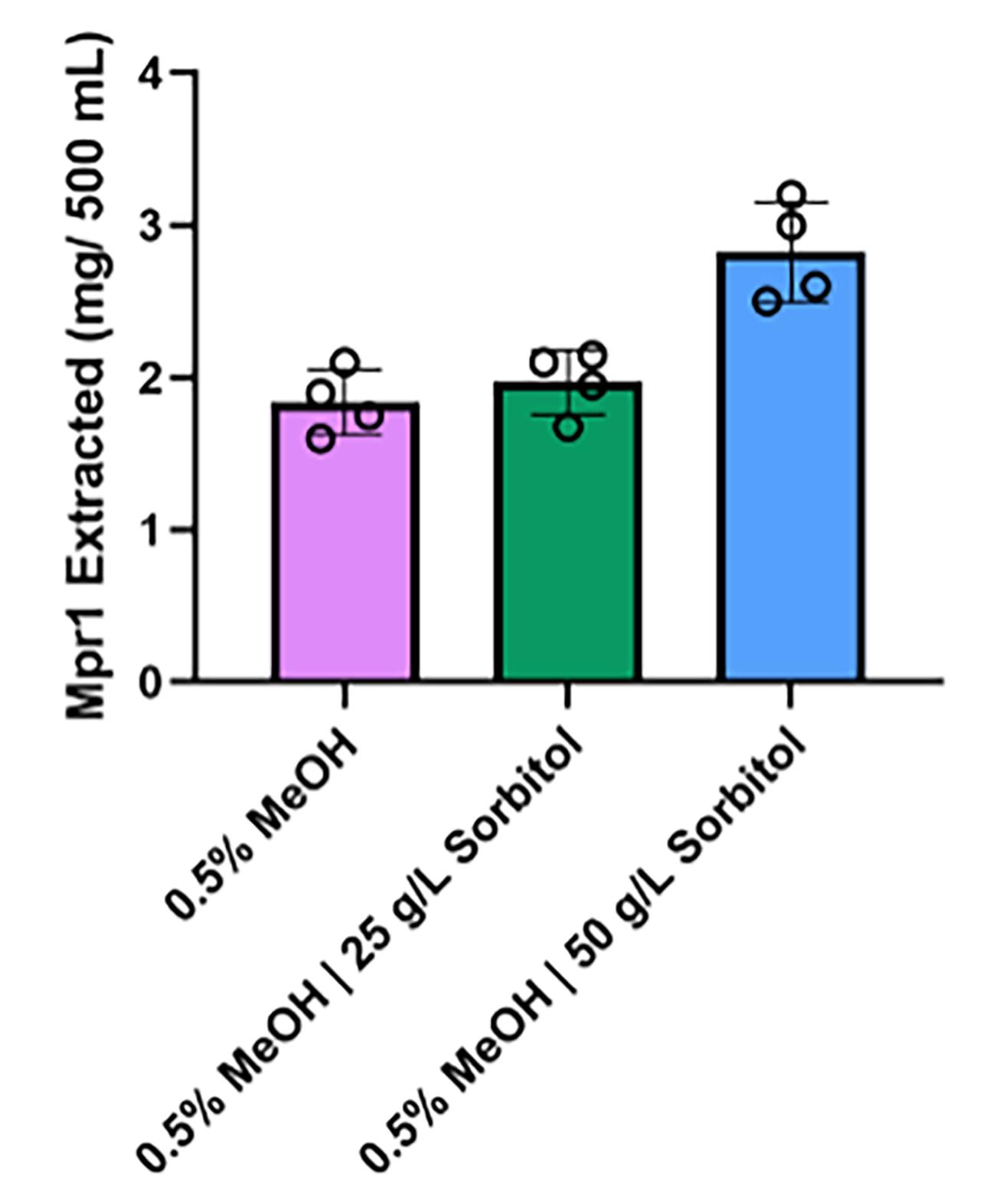
Figure 3. Mpr1 production in media supplemented with sorbitol. A comparison of the quantity of Mpr1 recombinant protein extracted from cultures grown with 0.5% methanol (MeOH), 0.5% MeOH supplemented with 25 g/L of sorbitol, or 0.5% MeOH with 50 g/L of sorbitol. Data represent Mpr1 extractions from four different cultures. The addition of sorbitol boosted Mpr1 protein production when MeOH concentrations were maintained at 0.5%.Assess the purity of the recombinant protein by SDS-PAGE electrophoresis.
Note: Coomassie or silver-stained SDS-PAGE polyacrylamide gels will visualize isolated recombinant proteins. Silver protein stains are an ultra-sensitive method of protein detection, which could be used to examine whether contaminating proteins were present in the isolation. Following step 11, examination of Mpr1 recombinant protein by silver-stained SDS-PAGE electrophoresis yielded a single polypeptide band (Figure 4).
Activity, purity, and storage of Mpr1
After saving a portion of the Mpr1 recombinant protein for proteolytic activity and other assays, flash freeze the remainder in liquid nitrogen and store at -80 °C.
Note: Proteolytic activity determination requires 50 µL or approximately 10% of the final eluted volume. We conducted proteolytic activity assays under various storage conditions (data not shown) and found that cryopreservation in liquid nitrogen produced only a small loss of proteolytic activity compared to protein that had not been subjected to freeze-thaw.
Program the microplate reader to measure fluorescence (485/538 nm excitation/emission) at 5 min intervals over at least one hour, shaking between reads. Sensitivity should be set to medium or low.
Following the manufacturer’s instructions, thaw aliquots of FTC-casein (5 mg/mL) and TPCK-trypsin (50 mg/mL) gradually at 4 °C. Dilute FTC-casein in tris-buffered saline to 10 µg/mL and store temporarily at 4 °C.
Note: 10 mL of 10 µg/mL FTC-Casein is sufficient for a 96-well plate.
Dilute Mpr1 recombinant protein in TBS to 200 µg/mL and 20 µg/mL, such that there is at least 350 µL of each dilution when finished. Set aside 350 µL of the TBS used in these dilutions to serve as a blank.
Dilute a thawed TPCK-trypsin aliquot to 1 µg/mL in TBS.
Add 100 µL of 200 µg/mL Mpr1, 20 µg/mL Mpr1, 1 µg/mL TPCK-trypsin, and TBS blank to triplicate wells in an opaque-walled (black or white), clear-bottom 96-well plate.
Add 100 µL of 10 µg/mL FTC-casein to each well.
Note: This is best done with a multichannel pipette due to the time-sensitivity of this step, although a serological pipette may suffice.
Remove the lid on the plate and place it in the plate reader. Begin the program at 5 min post FTC-casein addition. Proteolytic activity is measured by the decrease in fluorescence resonance energy transfer signal (Figure 5).
Data analysis
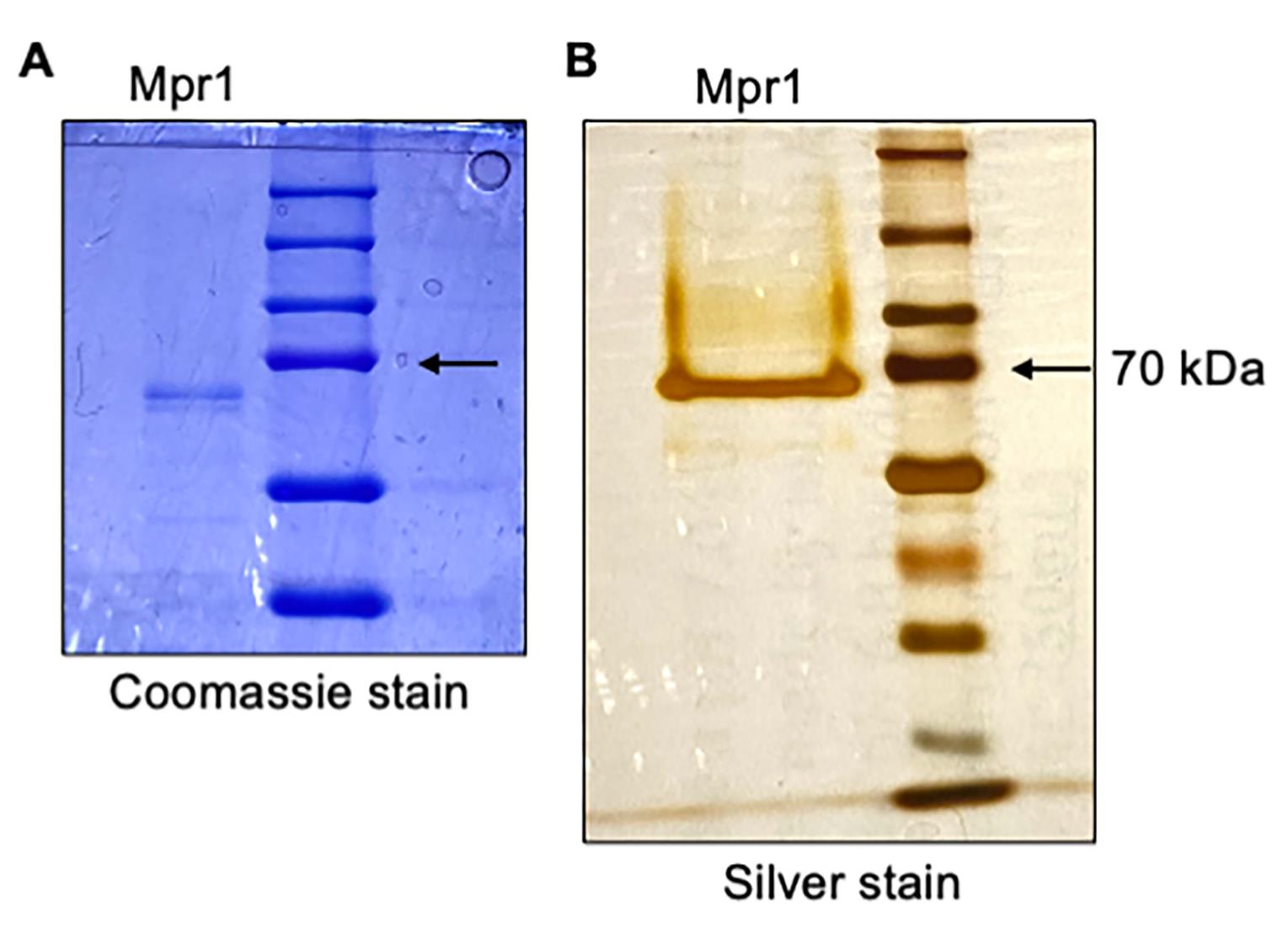
Figure 4. Purification of recombinant Mpr1 metalloprotease. SDS-PAGE analysis of isolated Mpr1 recombinant protein with (A) Coomassie-stained and (B) silver-stained 10% SDS-page gel run under reducing conditions with protein molecular marker (right lane, kDa). Bold arrow indicates recombinant Mpr1 protein as a single band (left lane). 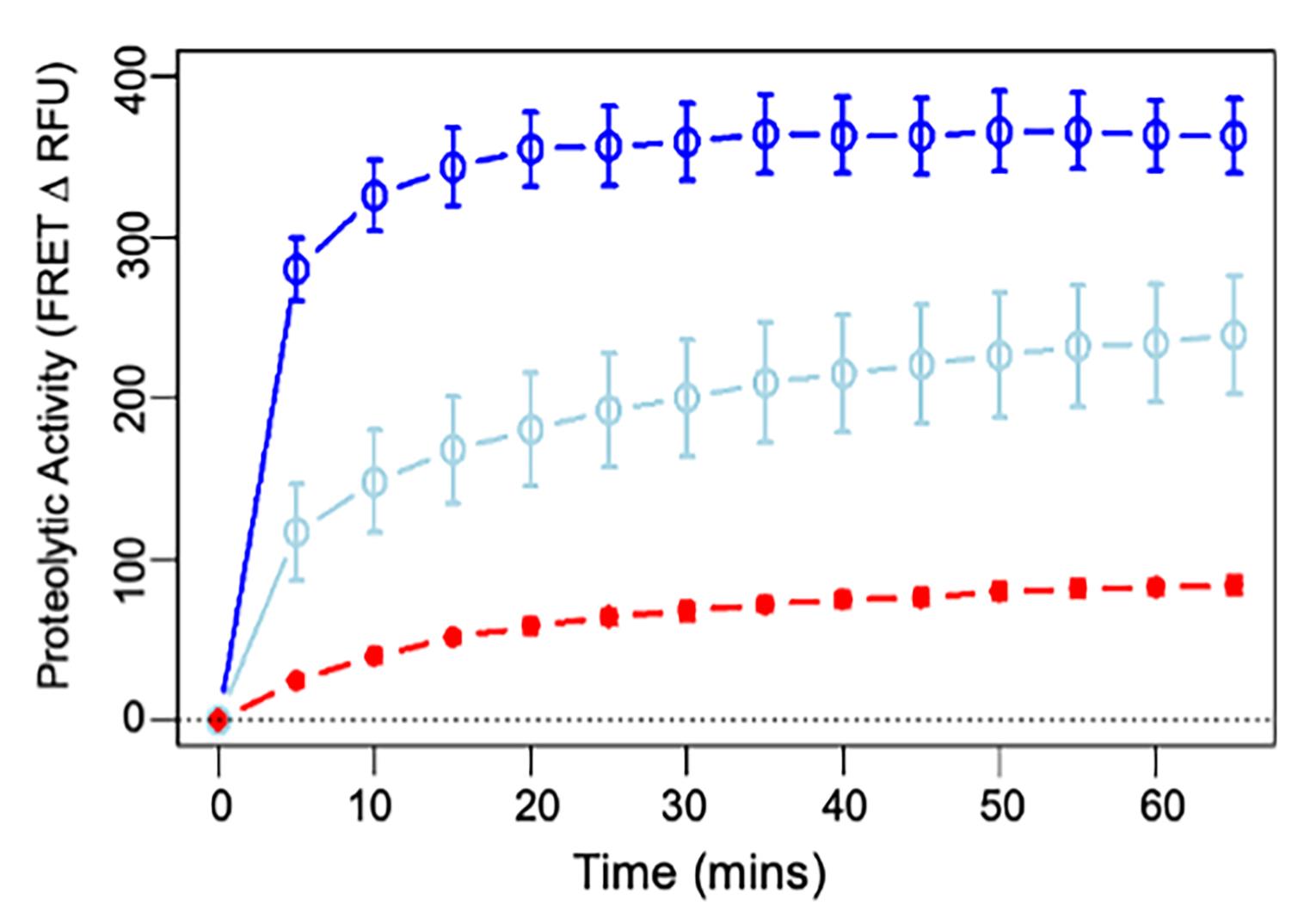
Figure 5. Proteolytic activity of Mpr1 at 100 (dark blue) and 10 µg/mL (light blue) compared with 0.5 µg/mL trypsin (red). Proteolytic activity was measured by the decrease in fluorescence resonance energy transfer (FRET) signal using a Pierce fluorescent proteases assay kit. The average fluorescence intensity in control wells containing only the proteolytic substrate (FITC-conjugated casein) was subtracted at each time point. Each data point represents the average of three readings (bars: SD). This assay was repeated with Mpr1 protein from five different extractions with similar results. Mpr1 retained proteolytic activity after flash-freezing and several weeks in storage (not shown).
Notes
Co-feeding optimization: We optimized sorbitol and methanol concentrations for production of our protein of interest, a metalloprotease with potentially unusual properties. The optimal concentration of sorbitol and methanol for other proteins may well differ from what we report here.
Sterility considerations: The media used during the initial growth of P. pastoris can easily become contaminated; hence, good sterile technique is vital for the success of your protein extraction.
Variability: This protocol is robust to minor deviations. For instance, the duration of incubation periods was not always exactly 24 h.
Environmental impact: Significant plastic waste was generated in the maintenance of sterile conditions throughout this protocol. Alternatives may be available, although such alternatives may have their own environmental drawbacks (e.g., the use of caustic chemicals in cleaning procedures).
Recipes
Yeast media
BMMY media
(1% yeast extract, 2% peptone, 100 mM potassium phosphate pH 6.0, 1.34% YNB, 5% biotin, 0.5% methanol)
Mix 20 g of peptone and 10 g of yeast extract and add deionized H2O to 700 mL.
Sterilize media by autoclave (liquid cycle 121 °C, 20 min).
Under sterile conditions, add 100 mL potassium phosphate, pH 6.0, 100 mL of 10× YNB, 100 mL of 5% methanol, and finally 2 mL of 500× biotin.
BMGY media
(1% yeast extract, 2% peptone, 100 mM potassium phosphate pH 6.0, 1.34% YNB, 5% biotin, 1% glycerol)
Mix 20 g of peptone and 10 g of yeast extract and add deionized H2O to 700 mL.
Sterilize media by autoclave (liquid cycle 121 °C, 20 min).
Under sterile conditions, add 100 mL 1 M potassium phosphate, pH 6.0, 100 mL of 10× YNB, 100 mL of 10% glycerol, and finally 2 mL of 500× biotin.
YPD
(1% yeast extract, 2% peptone, 2% dextrose, 2% agar)
Mix 20 g of peptone and 10 g of yeast extract and add deionized H2O to 950 mL.
Sterilize media by autoclave (liquid cycle 121 °C, 20 min).
For liquid media: add 50 mL of sterile 40% (w/v) glucose and mix.
For agar plates: While stirring the autoclaved mixture on a magnetic stir plate, add 50 mL of sterile 40% glucose per liter of YPD media.
While still warm, pour media into plastic Petri dishes (9 cm diameter)
Allow agar mixture to cool and solidify.
Buffers
Binding buffer pH 8.0
50 mM NaH2PO4, 300 mM NaCl, and 10 mM imidazole
Wash buffer pH 8.0
50 mM NaH2PO4, 300 mM NaCl, and 20 mM imidazole
Elution buffer pH 8.0
(50 mM NaH2PO4, 300 mM NaCl, and 500 mM imidazole)
Solutions
10% glycerol
Mix 100 mL of glycerol with 900 mL of deionized H2O.
Sterilize by autoclaving, store at room temperature.
5% methanol
Mix 50 mL methanol with 950 mL of deionized H2O.
Filter-sterilize and store at 4 °C.
1 M potassium phosphate buffer, pH 6.0
Mix 132 mL of 1 M K2HPO4 with 868 mL of 1 M KH2PO4. Adjust to pH 6.0 with KOH.
Sterilize by autoclaving, store at room temperature.
10× YNB
Weigh 34 g of YNB without ammonium sulfate and amino acids. Then, mix with 100 g of ammonium sulfate and fill up to 1 L of deionized H2O.
Filter-sterilize and store at 4 °C.
500× biotin
Dissolve 20 mg of biotin in 100 mL of deionized H2O.
Filter-sterilize and store at 4 °C.
Acknowledgments
We are acknowledging the original research paper (Aaron and Gelli, 2020) published by our lab from which this protocol was derived. We are grateful to The Hartwell Foundation for their support and to members of the Gelli lab for useful suggestions.
Competing interests
The authors declare that there are no financial or other competing interests.
References
- Aaron, P. A. and Gelli, A. (2020). Harnessing the Activity of the Fungal Metalloprotease, Mpr1, To Promote Crossing of Nanocarriers through the Blood-Brain Barrier. ACS Infect Dis 6(1): 138-149.
- Ahmad, M., Hirz, M., Pichler, H. and Schwab, H. (2014). Protein expression in Pichia pastoris: recent achievements and perspectives for heterologous protein production. Appl Microbiol Biotechnol 98(12): 5301-5317.
- Bradford, M.M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72(1-2):248-254.
- Celik, E., Calik, P. and Oliver, S. G. (2009). Fed-batch methanol feeding strategy for recombinant protein production by Pichia pastoris in the presence of co-substrate sorbitol. Yeast 26(9): 473-484.
- Colige, A. C. (2020). Challenges and Solutions for Purification of ADAMTS Proteases: An Overview. Methods Mol Biol 2043: 45-53.
- Daly, R. and Hearn, M. T. (2005). Expression of heterologous proteins in Pichia pastoris: a useful experimental tool in protein engineering and production. J Mol Recognit 18(2): 119-138.
- Fernandez, D., Russi, S., Vendrell, J., Monod, M. and Pallares, I. (2013). A functional and structural study of the major metalloprotease secreted by the pathogenic fungus Aspergillus fumigatus. Acta Crystallogr D Biol Crystallogr 69(Pt 10): 1946-1957.
- Inan, M. and Meagher, M. M. (2001). Non-repressing carbon sources for alcohol oxidase (AOX1) promoter of Pichia pastoris. J Biosci Bioeng 92(6): 585-589.
- Macauley-Patrick, S., Fazenda, M. L., McNeil, B. and Harvey, L. M. (2005). Heterologous protein production using the Pichia pastoris expression system. Yeast 22(4): 249-270.
- Macek, B., Forchhammer, K., Hardouin, J., Weber-Ban, E., Grangeasse, C. and Mijakovic, I. (2019). Protein post-translational modifications in bacteria. Nat Rev Microbiol 17(11): 651-664.
- Schwettmann, L. and Tschesche, H. (2001). Cloning and expression in Pichia pastoris of metalloprotease domain of ADAM 9 catalytically active against fibronectin. Protein Expr Purif 21(1): 65-70.
Article Information
Copyright
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Turner, A., Lanser, D. and Gelli, A. (2023). Optimized Expression and Isolation of Recombinant Active Secreted Proteases Using Pichia pastoris. Bio-protocol 13(5): e4628. DOI: 10.21769/BioProtoc.4628.
Category
Microbiology > Heterologous expression system > Non-model species
Biochemistry > Protein > Isolation and purification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.