- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Fluorescence Screens for Identifying Central Nervous System–Acting Drug–Biosensor Pairs for Subcellular and Supracellular Pharmacokinetics
Published: Vol 12, Iss 22, Nov 20, 2022 DOI: 10.21769/BioProtoc.4551 Views: 2826
Reviewed by: Geoffrey C. Y. LauSrinidhi Rao Sripathy RaoAndrew L. EagleZheng Zachory Wei
Original research article
The authors used this protocol in:

Jan 2022

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
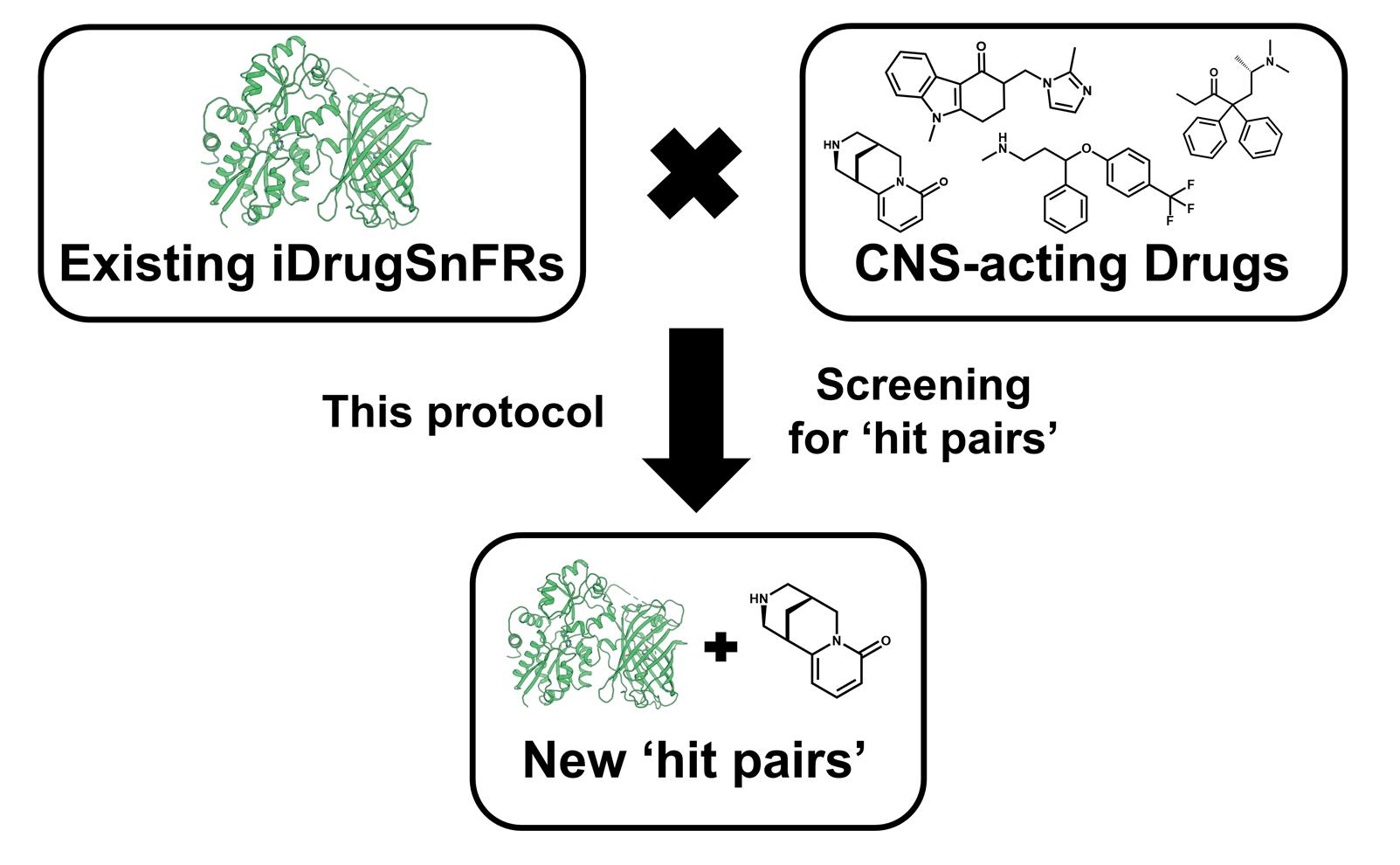
Subcellular pharmacokinetic measurements have informed the study of central nervous system (CNS)–acting drug mechanisms. Recent investigations have been enhanced by the use of genetically encoded fluorescent biosensors for drugs of interest at the plasma membrane and in organelles. We describe screening and validation protocols for identifying hit pairs comprising a drug and biosensor, with each screen including 13–18 candidate biosensors and 44–84 candidate drugs. After a favorable hit pair is identified and validated via these protocols, the biosensor is then optimized, as described in other papers, for sensitivity and selectivity to the drug. We also show sample hit pair data that may lead to future intensity-based drug-sensing fluorescent reporters (iDrugSnFRs). These protocols will assist scientists to use fluorescence responses as criteria in identifying favorable fluorescent biosensor variants for CNS-acting drugs that presently have no corresponding biosensor partner.
Graphical abstract:
Background
Low-molecular-weight central nervous system (CNS)–acting drugs typically bind to receptors, transporters, and ion channels (both ligand- and neurotransmitter-gated). Such CNS-acting drugs have therapeutic uses, but some are also abused (Lester et al., 2012; Henderson and Lester, 2015). Sources for new drug compounds typically include nature (mostly plants) or medicinal chemistry.
The protein targets of CNS-acting drugs are synthesized, assembled, and processed within intracellular exocytotic pathways before eventually reaching the plasma membrane. In some cases, membrane-permeant drugs also interact with their targets in intracellular compartments. For this reason, many papers report on pharmacokinetic characteristics of CNS-acting drugs at the subcellular scale: dynamics and intracellular concentrations. Optical methods provide appropriate time and distance scales, and the genetically encoded fluorescent biosensors we have developed bear strong resemblance to those developed for individual neurotransmitters (Marvin et al., 2018; Unger et al., 2020). The reversibility and linearity of such sensors also render them useful for the more conventional application of monitoring within extracellular biofluids (Muthusamy et al., 2022).
A general term we use to describe our biosensors is iDrugSnFR (intensity-based drug-sensing fluorescent reporter), following the lead of iGluSnFR (an early sensor for the neurotransmitter glutamate) (Marvin et al., 2018). All the iDrugSnFRs described here consist of circularly permuted green fluorescent proteins (cpGFP) inserted into a suitably mutated OpuBC periplasmic choline binding protein (PBP) from Thermoanaerobacter sp513. Upon the development of the first iDrugSnFR (iNicSnFR3a for nicotine), we conducted several screens with the goal of developing iDrugSnFRs for other drugs (Bera et al., 2019; Shivange et al., 2019; Nichols et al., 2022; Muthusamy et al., 2022). The present report shows the protocol for such screens.
In most cases, screening is required to develop a novel biosensor because, at present, we cannot predict atomic-scale details of the interaction between a drug of interest and the PBP site. Some pharmacological structure–activity relations (such as cation-π boxes) that govern the interaction between a CNS-acting drug and its putative binding partner are recapitulated in the drug-iDrugSnFR binding site, while others are not (Bera et al., 2019; Nichols et al., 2022; Muthusamy et al., 2022) (Protein Data Bank files 7S7T, 7S7U, 7S7X, and 7S7Z).
Screens reported here have two classes of molecular input: purified biosensor candidate proteins (13–18 per screen) and drugs of interest (DOIs, 44–84 drugs per screen). The DOIs we have chosen to study typically contain nitrogen (i.e., are alkaloids), have molecular weights (MW) below 500, and are weak bases (6 < pKa < 10). The weakly basic nature of these molecules allows passive diffusion through membranes into cells and organelles, enabling us to study their subcellular pharmacokinetics.
We define a hit pair as a drug–biosensor pair that displays an acceptable fluorescence signal (ΔF/F0 > 1). This definition of a hit arises from our experience that ΔF/F0 > 1 is required for subsequent directed evolution of an optimal biosensor variant. This report describes screens with single DOI concentrations and validations with full dose-response relations (several concentrations of the DOI) (Bera et al., 2019).
In general, the biosensor of the hit pair becomes the starting construct for directed evolution of an optimized variant that senses the DOI. Other papers describe the directed evolution of optimized variants (Bera et al., 2019; Shivange et al., 2019; Nichols et al., 2022; Muthusamy et al., 2022). In all cases, the directed evolution also includes optimizing the selectivity against other drugs. This workflow has resulted in iDrugSnFRs for cholinergic compounds, opioids, and the rapidly acting antidepressant S-ketamine (Shivange et al., 2019; Nichols et al., 2022; Muthusamy et al., 2022). Reports on additional iDrugSnFRs are in preparation.
Materials and Reagents
Disposable borosilicate glass culture tubes (VWR, catalog number: 47729-578)
10 mL syringes (Becton Dickinson, catalog number: 309605)
Sterile syringe filters with 0.2 µm cellulose acetate membrane (VWR, catalog number: 28145-477)
PrecisionGlideTM 18G 11/2 needles (Becton Dickinson, catalog number: 305196)
360 µL 96-well plates (Corning, catalog number: 3915)
2.2 mL 96-square-well storage plates (Thermo Scientific, catalog number: AB0932)
15 mL conical centrifuge tubes (Falcon, catalog number: 14-959-53A)
50 mL Amicon Ultra centrifugal filter tubes (30 kDa cutoff) (Sigma-Aldrich, catalog number: UFC903096)
50 mL conical centrifuge tubes (Falcon, catalog number: 14-432-22)
1.5 mL Premium microcentrifuge tubes (Fisher Scientific, catalog number: 05-408-129)
0.5 mL tubes (Fisher Scientific, catalog number: AB0350)
Sterile film (AeraSeal) (Sigma-Aldrich, catalog number: A9224)
Sterile glass beads
Ice/ice bucket
Dewar flask
Dimethyl sulfoxide (DMSO, C2H6OS) (Fisher Scientific, catalog number: BP231100)
Methanol (CH3OH) (EMD Millipore, catalog number: 179337)
0.5 M sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: 221465)
Ethylenediaminetetraacetic acid (EDTA) (C10H16N2O8) (Sigma-Aldrich, catalog number: E9884)
Sodium dodecyl sulfate (SDS) [CH3(CH2)11SO4)] (Sigma-Aldrich, catalog number: 436143)
Ethanol (EtOH, C2H5OH) (DeConLabs, catalog number: V1016)
Nickel sulfate (NiSO4(H2O)6) (Fisher Scientific, catalog number: AC415611000)
Buffer A (1× PBS, pH 7.4, 10 mM imidazole) [made from 10× PBS, pH 7.4 (Fisher Scientific, catalog number: 10-010-023)]
Buffer B (1× PBS, pH 7.4, 200 mM imidazole) [made from 10× PBS, pH 7.4 (Fisher Scientific, catalog number: 10-010-023)]
1× PBS, pH 7.0 [made from 10× PBS, pH 7.4 (Fisher Scientific, catalog number: 10-010-023)]
3× PBS, pH 7.0 [made from 10× PBS, pH 7.4 (Fisher Scientific, catalog number: 10-010-023)]
1 M MgSO4 (Sigma-Aldrich, catalog number: 230391)
Ampicillin 1,000× stock (Sigma-Aldrich, catalog number: A9518)
SOC media (MP Biomedicals, catalog number: 3031-012)
Difco LB media (Fisher Scientific, catalog number: DF0402)
BL21(DE3) chemically competent cells (Thermo Fisher Scientific, catalog number: C600003)
ddH2O
LB agar plates (ampicillin, 100 mg/mL)
Liquid nitrogen (N2)
2 mM drug stock solutions
Biosensor plasmid DNA
4× Laemmli sample buffer (Bio-Rad, catalog number: 161-0747)
2-Mercaptoethannol (β-ME) (Bio-Rad, catalog number: 161-0710)
HiTrapTM 5ml IMAC FF Nickel NTA column (Cytiva, catalog number: 17-0921-04)
Mini-PROTEAN TGXTM precast protein gels, 10-well, 30 μL (Bio-Rad, catalog number: 456-1093)
Tris-glycine buffer 10× concentrate (Sigma-Aldrich, catalog number: T4904)
Mini-PROTEAN Gel Box (Bio-Rad, catalog number: 1658005)
Precision Plus protein dual color standards ladder (Bio-Rad, catalog number: 161-0374)
QC Colloidal Coomassie stain (Bio-Rad, catalog number: 161-0803)
50× M (see Recipes)
50× 5052 (see Recipes)
Equipment
Heat block (VWR Scientific, catalog number: 13259-005)
Precision GP 05 water bath (Thermo Fisher Scientific, catalog number: TSGP02)
Excella E25 shaking incubator (New Brunswick Scientific, catalog number: M1353-0000)
G-25 incubator shaker (New Brunswick Scientific, catalog number: 76901-1)
Mini centrifuge (Thermo Fisher Scientific, catalog number: 05-090-100)
Fluorospectrometer (Thermo Scientific, catalog number: ND-3300)
ÄKTA Start protein purification system with Frac30 fraction collector (Cytiva, catalog number: 29023051)
XPR204 scale (Mettler Toledo, catalog number: 30355419)
Vortex mixer (Baxter Scientific, catalog number: S8223-1)
epMotion 5075t liquid handling robot and appropriate tips (Eppendorf, catalog number: 5075006022)
Spark 10M multimode plate reader (Tecan, catalog number: 30086376)
Agilent ELx50 microplate strip washer (BioTek)
Sonicator [Sonifier SFX550 control box (Branson, catalog number: 101-063-969), 20 kHz 102-C converter (Branson, catalog number:101-135-066R), tapered 1/8 inch microtip (Branson, catalog number: 101-148-062)]
Allegra 25R centrifuge (Beckman Coulter, catalog number: 369434)
Nanodrop 1000 (Thermo Scientific)
Orion VersaStar Pro pH meter (Thermo Scientific, catalog number: VSTAR90)
Covered container for gel staining/destaining
Rotary shaker (VWR, catalog number: 89032)
Xplorer variable volume 12 channel motorized pipette (50–1,200 µL) (Eppendorf, catalog number: 4861000830)
PowerPacTM HC high-current power supply (Bio-Rad, catalog number: 1645052)
Software
Tecan v3.1 software
Eppendorf EpMotion software version 40.4.0.38
Microsoft Excel version 16.55
Origin Pro version 9.1 64-bit
Procedure
Expression of biosensor proteins
Chemical transformation of plasmid into BL21(DE3) chemically competent cells
Determine biosensor plasmid to express. Coding regions of each candidate biosensor are given in the iSnFRbase database (a repository for tracking biosensor mutagenesis and drug affinities), which will be deposited on GitHub. Figures 4, 5, and 6 give the iSnFRbase identifiers under nicknames.
Add 300 ng of biosensor of chosen plasmid DNA to 100 µL of BL21(DE3) chemically competent cells on ice for 10–30 min.
After incubation, heat-shock the sample on a 42 °C heat block for 30 s and place back on ice for 2 min.
Add 700 µL of SOC media to the sample, resuspend solution with a pipette, and place the sample in a 37 °C shaker for 1 h at 225 rpm.
Spin the sample in a microcentrifuge for 2 min.
Remove 500 µL of supernatant and resuspend the pellet in the remaining supernatant.
Add 60 µL of resuspended sample to an LB agar plate containing 100 mg/mL ampicillin (or appropriate antibiotic) and spread media on plate using 5–10 sterile glass beads.
Keep the plate in a 37 °C incubator for 16–20 h.
Note: The volume of BL21(DE3) chemo-competent cells plated can be reduced if colony density on LB agar plates is too high after 37 °C incubation.
Inoculation
Pick a single colony from the previously grown LB agar plate and add it to a mixture of 192 mL autoclaved LB (made from Difco LB media) and auto-induction additives (4 mL of 50× M, 4 mL of 50× 5052, 200 µL of ampicillin 1,000× stock, and 40 µL of 1 M MgSO4) (Studier, 2005).
Swirl the flask and place in a 30 °C shaking incubator for 24–30 h at 240 rpm.
Transfer the culture into four 50 mL Falcon tubes and centrifuge for 15 min at 5,300 × g and 4 °C.
Decant samples, gently wash each pellet once with 5 mL of 1× PBS, pH 7.0, and store the tubes at -80 °C for later purification (storage at temperatures above -80 °C may result in degraded, non-functional protein).
Note: Initial auto-induction volume can be increased or decreased depending on protein yield as calculated in step A6h.
Preparation for purification
Thaw 50 mL Falcon tubes containing pelleted biosensor culture on ice.
Add 5 mL 1× PBS, pH 7.0, to each tube and completely resuspend pellet while on ice.
Pool resuspensions into a single 50 mL Falcon tube and place it on ice.
Sonicate the resuspended biosensor at 13% amplitude, 0.7 s time-on, and 0.2 s time-off for 30 s. Repeat sonication three to six times, with 3 min between each sonication to allow for cooling.
CRITICAL STEP Effective sonication is required for maximum protein yield. Increase the number of sonication cycles or sonication amplitude if the yield calculated in Step A6h is low.
Centrifuge the sample for 15 min at 5,300 × g and 4 °C.
Collect the supernatant while being careful to avoid collecting any pellet and transfer it to a new 50 mL Falcon tube. CRITICAL STEP If some pellet is gathered with the supernatant, this will markedly lengthen the filtration step in Step A3h.
Note: An additional 15 min centrifugation and transfer to a new 50 mL Falcon tube may facilitate filtration in later steps.
Attach a 0.2 µm filter to a 10 mL syringe and place over a new 50 mL Falcon tube.
Remove syringe plunger and add the previously collected supernatant to the 10 mL syringe, replace syringe plunger, and depress plunger to filter.
Note: Multiple 0.2 µm filters may be required to filter one sample; these may be replaced on the 10 mL syringe. Ensure that filtered and unfiltered supernatant are not mixed during 0.2 µm filter replacement. CRITICAL STEP Unfiltered supernatant can result in bacterial growth in fast protein liquid chromatography (FPLC) systems.
Repeat filtration step with remaining supernatant until the entire sample is filtered.
Note: Multiple 0.2 µm filtrations may be performed in parallel. Pool 0.2 µm filtered supernatant into one 50 mL Falcon tube (as needed).
FPLC purification
Equilibrate Ni-NTA column according to manufacturer’s guidelines.
Note: For general FPLC setup, see Figure 1.
Equilibrate appropriate FPLC lines with Buffer A and Buffer B.
Run an appropriate protein purification method (example program: seven column volume (CV) applications of 100% Buffer A, sample application 10–30 mL, seven CV washouts of unbound protein with 100% Buffer A, 0%–100% Buffer B gradient elution over 10 CV, five CV applications of 100% Buffer B, and collect 4 mL fractions starting at Buffer B gradient elution step).
When prompted to load the sample, load the filtered biosensor solution using either the 1) sample loop or 2) sample valve as appropriate for the supernatant volume and FPLC model.
When purification program is complete, remove fractions from the FPLC carousel and store at 4 °C.
CRITICAL STEP To ensure minimal cross-contamination of biosensors, column stripping and regeneration is recommended when purifying multiple constructs in succession.
Run an appropriate program to strip the column using 50 mM EDTA and 0.05% SDS, followed by 0.5 M NaOH (example program: five CV applications of EDTA 0.05% SDS, two CV applications of H2O, and five CV applications of 0.5 M NaOH).
Note: If changing bottles between programs, wash the lines with water before equilibrating with the new solution. CRITICAL STEP Direct mixing of buffers without a water wash may result in precipitation of nickel in FPLC lines, affecting FPLC function. TROUBLESHOOTING If precipitation is seen, wash lines extensively with water to remove precipitate.
Run an appropriate program to regenerate the column using 0.2 M NiSO4, followed by storage in 20% EtOH (example program: five CV applications of nickel sulfate, three CV applications of H2O, and five CV applications of 20% EtOH).
Ensure that all lines are stored in 20% EtOH to reduce bacterial growth.

Figure 1. Fast protein liquid chromatography (FPLC) setup. The FPLC machine used for protein purification is shown with lines, waste, and fraction collector.Protein gel electrophoresis
Add 20 µL of 2-mercaptoethanol (β-ME) and 180 µL of 4× Laemmli sample buffer to a 1.5 mL microcentrifuge tube and mix by a brief microcentrifuge spin.
Identify protein fractions to sample from the FPLC purification.
Note: A general rule is to sample the range that covers the fractions of detectable protein elution, with a buffer of several fractions on either end of this range. If needed, sample every third or fourth fraction.
Add 20 µL of each chosen protein fraction from purification to 1.5 mL microcentrifuge tubes.
Add 10 µL of the mixture of β-ME and Laemmli sample buffer made in StepA5a a to each protein fraction sample.
Mix tube contents by a brief microcentrifuge spin.
Place samples in a 95 °C heat block for 10 min.
Add 30 µL 10-well Mini-PROTEAN TGXTM precast protein gel to a gel box and fill the box with Tris-glycine buffer (diluted from 10× concentrate).
After 95 °C incubation is completed, briefly spin down 1.5 mL microcentrifuge tubes and fill lanes 2–10 of the gel with 20 μL of the FPLC-purified fraction solution. Add 1.5 µL of Bio-Rad Precision Plus protein dual color standards to lane 1.
Run the gel for 1 h and 10 min at 110 V (or for the time recommended by manufacturer).
Remove the gel from its casting by crimping, place in a coverable container, add Coomassie stain, and place on a rotary shaker on a light setting overnight.
The next morning, decant the Coomassie stain (which can be saved for future gels) and rinse the gel with water.
Incubate the gel with water on the rotary shaker until it becomes clear (the protein bands should be blue).
Note: To hasten the destaining process, replace the water every 10–15 min.
Once the gel is clear, examine the protein bands in lanes 2–10 and compare these to the protein ladder (lane 1) to determine whether the protein bands match the molecular weight of the desired protein.
Note: Bands of lower or higher molecular weight may be visible along with the desired protein and indicate unwanted proteins. CRITICAL STEP Care should be taken to concentrate biosensor protein fractions with minimal contamination from these other proteins. If desired, size-exclusion chromatography can be performed to further increase protein purity.
Concentration of purified protein
Transfer 8 mL of 1× PBS, pH 7.0, to a 50 mL centrifugal filter (30 kDa cutoff).
Centrifuge the tube for 9 min at 3,400 × g and 4 °C to wash the filter.
Add 11 mL of the chosen FPLC purified protein fractions to the 50 mL centrifugal filter (30 kDa cutoff) and centrifuge using the settings in Step A6b.
Note: Single-chain biosensors that merge OpuBC, cpGFP, and two 4-residue linkers have a molecular weight of approximately 62 kDa. The 30 kDa cutoff is probably appropriate for concentrating all known biosensors that merge a PBP moiety with a fluorescent protein moiety.
Repeat Step A6c until all desired fractions are added to the filter and centrifuged.
Add 8 mL of 1× PBS, pH 7.0, to the centrifugal filter and centrifuge using the settings in Step A6b.
Repeat Step A6e six more times to buffer exchange the biosensor solution and remove all residual imidazole.
When the final centrifugation is reached, determine the volume of the biosensor in the centrifugal filter. If it is 500–1,000 µL, continue to Step A6h. If it is >1,000 μL, centrifuge the biosensor solution at 3,400 × g in 3 min increments until the volume is 500–1,000 µL.
Note: A volume of 500–1,000 µL generally provides a protein concentration of 50–200 μM. 250 μL of biosensor protein at 50 μM is generally sufficient to run several screens and perform the multiple concentration drug–biosensor fluorescence validations.
Measure the A280 of the protein on Nanodrop 1000 (or equivalent UV-Vis spectrophotometer) using the manufacturer’s instructions. After obtaining the A280, determine the concentration using Beer’s Law. The appropriate extinction coefficient for each biosensor being purified is calculated from the amino acid composition.
Note: Purification of protein from 200 mL of autoinduction media generally provides a final yield of 0.2–2.0 mg.
Aliquot the biosensor solution into 0.5 mL tubes and flash freeze in liquid nitrogen.
CRITICAL STEP Multiple freeze-thaw cycles are not recommended, so storage in 20–50 µL aliquots is recommended.
Store the biosensor solutions at -80 °C.
TROUBLESHOOTING If biosensor yield is low (less than 250 μL at 50 μM), the following suggestions are recommended: increase autoinduction time to the 30 h maximum, remake auto-induction solutions and repeat autoinduction, increase auto-induction volume, increase number of sonication rounds, and increase sonication amplitude. Although GFP-based biosensor protein production in E. coli is generally robust, some variability remains.
Creation of drug plate
Assembly of drug library
Choose drugs based on criteria relevant to the project of interest (i.e., controlled substance scheduling, absence of existing biosensors, and clinical importance).
Collect information on each drug and compile in Microsoft Excel for reference and comparison; this also allows tracking of delivery status and storage.
Note: In our case, information was collected on each drug’s commercial availability, pharmacological or therapeutic class, trade name, rationale for study, IUPAC name, stereochemistry, chemical formula, formula weight, CAS number, formulation, structure, additional pharmacological properties, EC50, activity (agonist/antagonist), solubility, pKa, logP (octanol-water partition coefficient) or logDpH7.4 (logP corrected for fraction of uncharged (deprotonated) molecules at pH 7.4), controlled substance scheduling, handling instructions, vendor, price, and purity.
Drug solvation
Choose a solvation method for each drug based on solubility in water and organic solvents.
CRITICAL STEP If using PBP-based biosensors, avoid DMSO when possible (interactions between DMSO and PBP-based biosensors can result in high baseline fluorescence). Use 1× PBS (pH 7.0) as the solvent for water-soluble drugs. TROUBLESHOOTING It is suggested to perform small-scale tests of drug solubility in various solvent systems prior to drug plate formation to ensure complete dissolution. Water, 3× PBS (pH 7.0), ethanol, or methanol are recommended solvation systems.
Calculate desired amount of drug to result in a drug solution at 2 mM, 2–8 mL.
For drugs in powder form, use the following steps:
Measure on a Mettler Toledo XPR204 scale and add the powder to a 15 mL Falcon tube.
Add approximately 80% of the calculated appropriate solvent required by serological pipette and mix by vortexing.
Measure the pH of the resulting solution and adjust to pH 7.0. CRITICAL STEP The pH of a solution affects the baseline fluorescence of iDrugSnFRs (Shivange et al., 2019). This variation amounts to 10-fold per pH unit, or 26% per 0.1 pH unit. Consistency of pH across drug solutions will reduce variability in screen results.
Add the remaining volume required to obtain a 2 mM concentration.
Note: For solutions that do not dissolve fully by vortexing, place in a 42 °C heat bath until solvation is complete. CRITICAL STEP Ensure that the drug is completely dissolved before continuing to drug plate formation.
For drugs in liquid form, use the following steps:
Dilute drug directly into 80% of the calculated appropriate solvent.
Mix drug by vortexing.
Measure the pH of the resulting solution and adjust to pH 7.0. CRITICAL STEP The pH of a solution affects the baseline fluorescence of iDrugSnFRs (Shivange et al., 2019). This variation amounts to 10-fold per pH unit, or 26% per 0.1 pH unit. Consistency of pH across drug solutions will reduce variability.
Add the remaining volume required to obtain a 2 mM concentration.
CRITICAL STEP Ensure that the drug is completely dissolved before continuing to drug plate formation.
Drug plate formatting and compilation
Design a 2.2 mL (deep-well) 96-well drug plate with drugs of interest (Figure 2).
Add three control wells for each solvation condition and intersperse these across the plate to monitor potential cross contamination due to pipetting.
Add 1 mL of the appropriate drug solution or solvent to each well.
Store the remaining drug solution as described in MSDS for future screening plates.

Figure 2. Drug plate design example of a drug–biosensor fluorescence screen. The drug plate design used for a single concentration drug–biosensor fluorescence screen is shown, with drugs organized by class and control wells mixed throughout the plate.
Single concentration drug–biosensor fluorescence screen
Biosensor dilution
Thaw a biosensor protein aliquot on ice.
Dilute the biosensor in 3× PBS, pH 7.0, so that the diluted solution has a concentration of 111 nM and volume of 30–50 mL.
Transfer the solution to a 50 mL Falcon tube and mix by inversion.
Mixing of drug–biosensor solution using epMotion liquid handling robot
Create a program in the Eppendorf ePBlue software for biosensor screening.
Note: This program needs a location for a 50 µL Eppendorf pipette tip box (for distributing drug solution), 300 µL Eppendorf pipette tip box (for distributing biosensor solution), the 2.2 mL 96-well drug plate containing 1 mL drug solutions created in Step B3, three 360 µL 96-well plates (for mixed drug–biosensor solution), and a 50 mL tube containing biosensor solution (Figure 3). The program must allow for 100 µL biosensor solution to be added from the 50 mL tube to each well of the three 360 µL 96-well plates (using 300 µL tips), 11 µL of drug solution to be added from the 2.2 mL 96-well drug plate containing 1 mL drug solution to corresponding wells of the three 360 µL 96-well plates (using 50 µL tips), and a step for the mixing of drug and biosensor solutions. The data from drug-containing wells on these plates will serve as the Drug–Biosensor Fluorescence values for ΔF/F0 calculations in Equation 1 (Data analysis). The data from the solvent control wells from this plate (i.e., 25% DMSO, 3× PBS, pH 7.0) will serve as Baseline Biosensor Fluorescence for ΔF/F0 calculations in Equation 1 (Data analysis).
Run the program outlined in Step C2a.
Note: This step can also be performed manually if a liquid handling robot is not available.
Run the program outlined in Steps C2a-b with no biosensor present (i.e., 3× PBS, pH 7.0 only).
Note: The data from these plates will serve as Baseline Drug Fluorescence for ΔF/F0 calculations in Equation 1 (Data analysis).
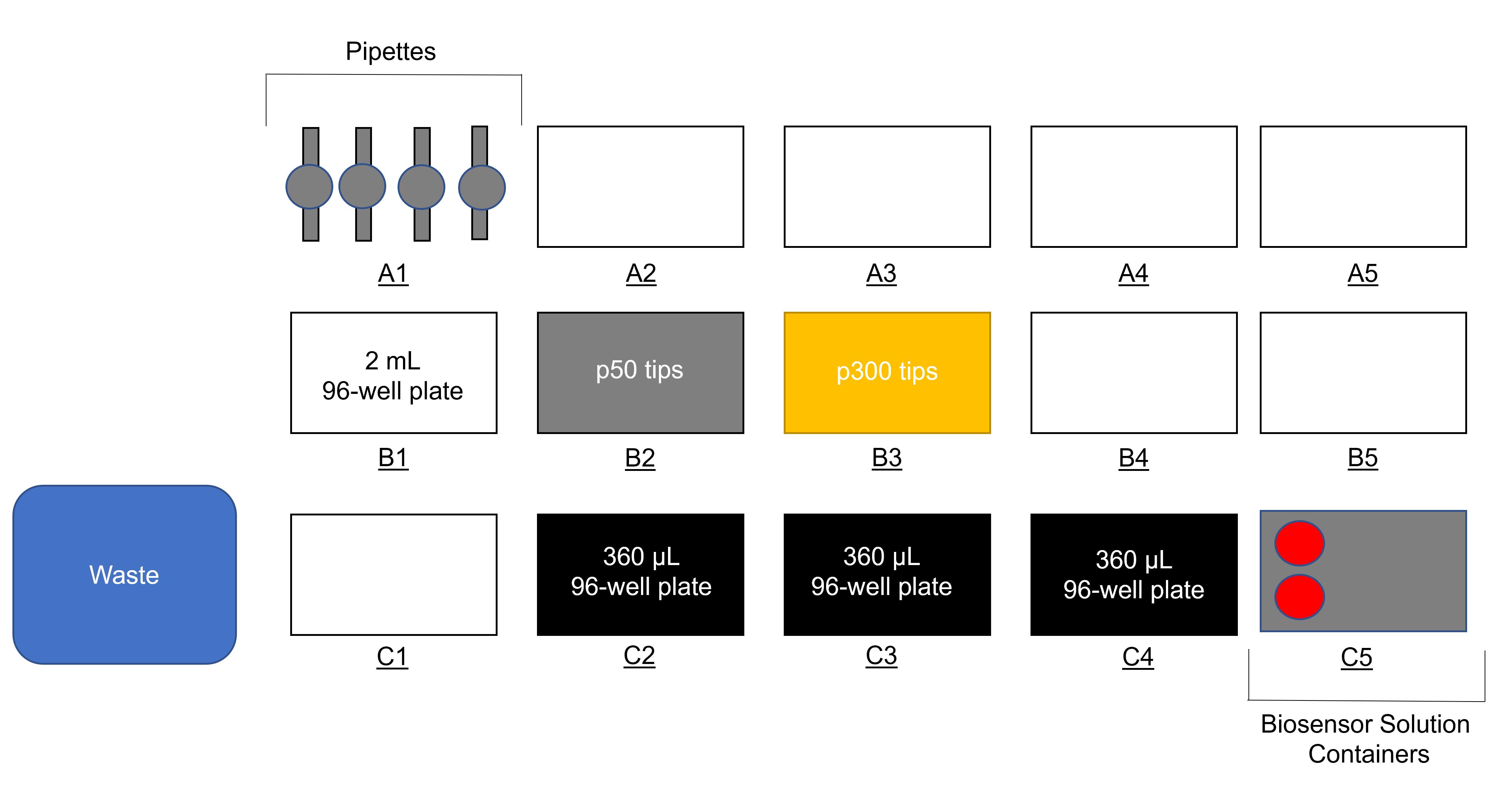
Figure 3. Eppendorf epMotion setup for single concentration drug–biosensor fluorescence screen. The setup to mix drug and biosensor solutions is shown on the ePMotion 5075 liquid handling robot. This shows a 2.2 mL 96-well plate with 1 mL of drug/solvent solutions in position B1, p50 tips in B2, p300 tips in B3, 360 µL 96-well plates in C2–C4, and containers of biosensor solution (2× 50 mL tubes) in C5, as desired.Fluorescence measurements using Spark 10M
Remove 360 µL 96-well plates one at a time from the Eppendorf machine and place into the plate holder of the Spark 10M Fluorescence Reader.
Design a program to measure fluorescence of the drug–biosensor solution. Our program contains the following elements: it shakes the plate for 10 s at an amplitude of 1.5 mm to allow for additional mixing and then measures fluorescence with an excitation wavelength of 480 nm and emission wavelength of 535 nm.
Note: An excitation wavelength of 480 nm and emission wavelength of 535 nm is appropriate for all GFP-based biosensors. If a different fluorescent protein is being used, the excitation and emission wavelengths should be adjusted to match its fluorophore.
Example results:
2017 single concentration drug–biosensor fluorescence screen
In 2017, a single concentration drug–biosensor fluorescence screen was conducted with a primary focus on drugs used to treat psychiatric disorders (Figure 4). Drugs were chosen both based on clinical importance and to encompass a range of pharmacological or therapeutic classes; classes present included opioids, anticholinergics, antipsychotics, antidepressants, benzodiazepines, anti-seizure medications, and sedatives. In total, 84 drugs and 13 biosensors were screened. We also grouped results into general categories based on the most common treatment use: schizophrenia, major depressive disorder, anxiety disorders, epilepsy, and other.
Drug–biosensor fluorescence validation of hit pairs through multiple concentration response experiments
Creation of drug dilution plate
Choose a drug-biosensor hit pair for a multiple concentration response experiment.
Create a drug dilution plate that allows for a triplicate dose-response relation measurement of the desired drug. CRITICAL STEP Accurate drug concentrations in the drug dilution plate are necessary for accurate calculations of the drug–biosensor interaction.
Pipette 1 mL of 2 mM drug solution into wells A1–C1 of a 2.2 mL 96-well plate.
Perform a √1 0
dilution eight times. Transfer 316 µL of solution from A1–C1 wells to A2–C2.
Add 684 µL of 3× PBS, pH 7.0, to wells A2–C2 and mix by pipette.
Transfer 316 µL of solution from wells A2–C2 to wells A3–C3, add 684 µL of 3× PBS, pH 7.0, and mix.
Repeat above steps until wells A9–C9 are filled.
Add 684 µL of 3× PBS, pH 7.0, to wells A10–C10 to serve as a blank.
Biosensor dilution
Thaw a biosensor protein aliquot from Step A6j on ice.
Dilute the biosensor in 3× PBS, pH 7.0, so that the diluted solution has a concentration of 111 nM and volume of 30–50 mL.
Transfer the solution to a 50 mL Falcon tube and mix by inversion.
Mixing of drug–biosensor solution using epMotion liquid handling robot
Create a program in the Eppendorf ePBlue software for validating biosensor hit pairs.
Note: This program needs a location for a 50 µL Eppendorf pipette tip box (for distributing drug solution), 300 µL Eppendorf pipette tip box (for distributing biosensor solution), the 2.2 mL 96-well drug plate containing 1 mL drug solutions created in Step D1, one 360 µL 96-well plate (for mixed drug–biosensor solution), and a 50 mL Falcon tube containing biosensor solution. The program must allow for 11 µL of drug solution to be added from the 2.2 mL 96-well drug plate containing 1 mL drug solutions to corresponding wells of the 360 µL 96-well plate, 100 µL of biosensor solution to be added from the 50 mL Falcon tube to the same wells of the 360 µL 96-well plate, and for these solutions to be mixed (there will only be drug and biosensor solution in wells A–C 1–9 of the plate if the 2.2 mL 96-well drug plate containing 1 mL drug solutions is set up as instructed in step D1). The data from columns 1–9 will serve as the Drug–Biosensor Fluorescence values for ΔF/F0 calculations in Equation 1 (Data analysis). The data from column 10 will serve as Baseline Biosensor Fluorescence for ΔF/F0 calculations in Equation 1 (Data analysis).
Run the program outlined in Step C2a.
Notes:
This step can also be performed manually if a liquid handling robot is not available.
The range of drug concentration sampled can be increased or decreased as needed to sample a full dose-response relation.
Fluorescence measurements using Spark 10M
Once the program on the epMotion liquid handling robot is complete, remove the 360 µL 96-well plate from the Eppendorf machine and place it into the plate holder of the Tecan Spark 10M Fluorescence Reader.
Utilize the same program as in step C3b.
Repeat steps D3–4 with no biosensor present (i.e., 3× PBS, pH 7.0 only)
Note: The data from these plates will serve as Baseline Drug Fluorescence for ΔF/F0 calculations in Equation 1 (Data analysis).
Example results for future studies:
5-HT3 receptor Antagonists
Four 5-HT3 antagonists were included in the 2019 screen: ondansetron, tropisetron, palonosetron, and alosetron. In this class, 23 hit pairs involved drugs, including several pairs found for ondansetron, tropisetron, and palonosetron. No hit pairs were found for alosetron. Top hit pairs for each drug were validated through multiple concentration drug–biosensor fluorescence dose-response studies as described in step D above (Figure 7).
Additionally, these validations allowed for the calculation of EC50, Hill coefficient (nH, a measure of apparent cooperativity among binding sites), and S-slope (as defined in Data analysis) of the drug–biosensor hit pairs. Validation screens were conducted for the four strongest hit pairs for each DOI. Thus, ondansetron was studied against the biosensors AK1, cc70, v7.1.2, and Scop4; tropisetron was studied against the biosensors AK1, v7.1, v7.1.2, and v8; and palonosetron was studied against the biosensors AK1, v7.1, v7.1.2, and Scop4. From these validation screens, the drug–biosensor pairs with the most favorable profile were chosen (Figure 7). This was determined based on the S-slope, EC50, and Fmax of the drug–biosensor pairs, with a higher Fmax, higher S-slope, and lower EC50 being most desirable.
Several 5-HT3 antagonist × biosensor hit pairs were validated through these dose-response relations. Two strong hit pairs were validated for palonosetron. Palonosetron × v7.1.2 displayed an Fmax of 5.4, and palonosetron × v7.1 displayed an Fmax of 3.2. We expect that both hit pairs have Fmax values large enough (i.e., > 3) to be used directly for cellular imaging. For ondansetron and tropisetron, a single hit pair was identified, with an Fmax of 2 for ondansetron × cc70 and an Fmax of 2.2 for tropisetron × v7.1.2. Because the Fmax values for these pairs are < 3, further directed evolution will be required to optimize a variant for successful use for imaging. For all hit pairs shown, nH is near 1, suggesting that the biosensors would have desired binding properties with these drugs.
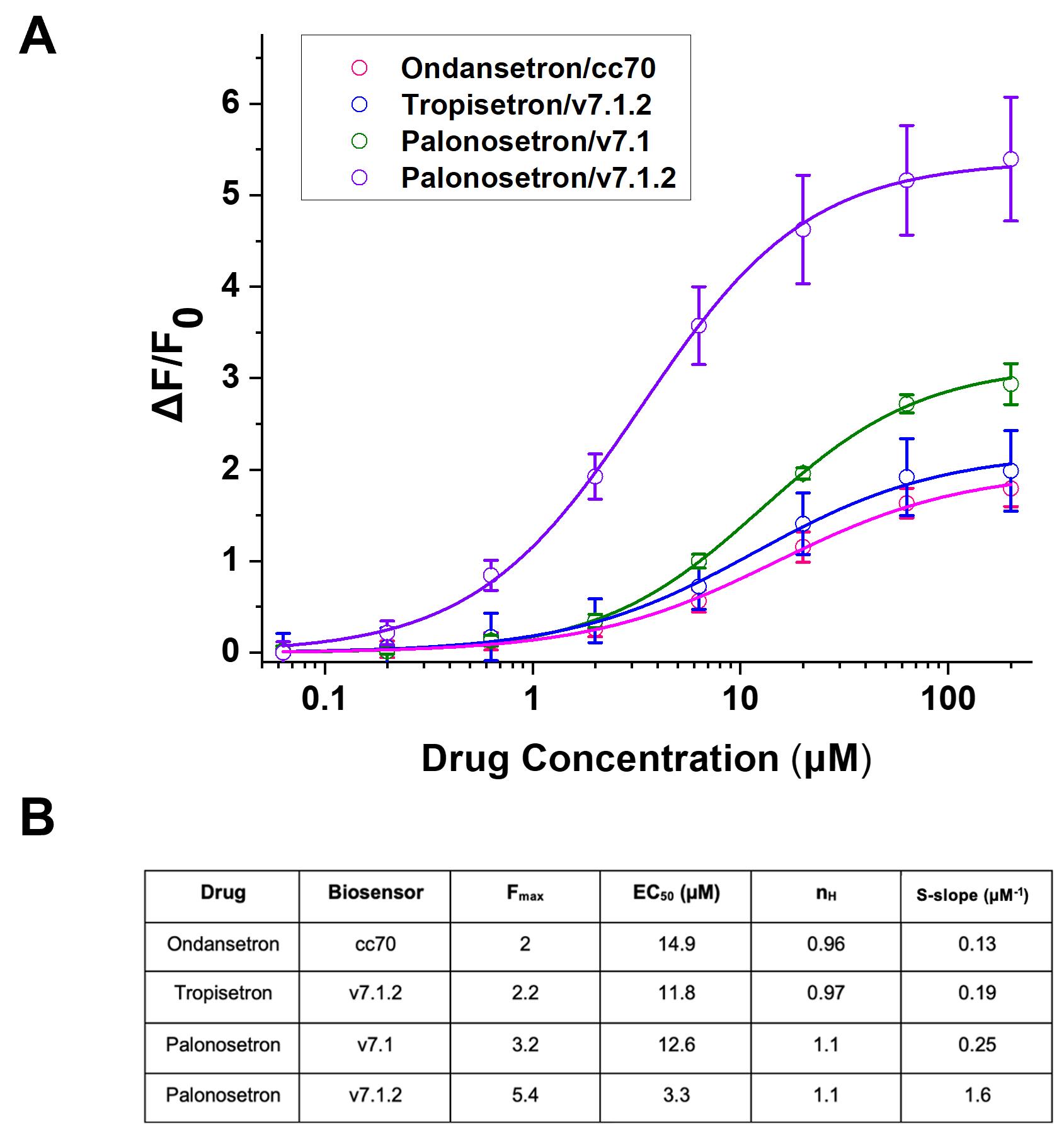
Figure 7. Multiple concentration drug–biosensor fluorescence validations for 5HT3 antagonists. Dose-response relations (A) and analysis [Fmax, EC50, Hill coefficient (nH), and S-slope] (B) for 5-HT3 antagonists. Only the most promising dose-response relation(s) for each drug of interest are shown. Biosensors included are cc70, v7.1.2, and v7.1.CB1/CB2 Ligands
The 2019 screen included nine CB1/CB2 ligands: SLV-319 (+/-), AM 6545, PSNCBAM-1, O-2050, WIN-55 212,22, CP 94545, NIDA 41020, rimonabant, and leelamine. In total, 34 hit pairs were found for drugs in this class, with several hit pairs being found for each of six drugs [SLV-319 (+/-), AM 6545, PSNCBAM-1, WIN-55 212,22, NIDA 41020, and rimonabant]. We found no hit pairs including the remaining three drugs (O-2050, CP 94545, and leelamine).
The top four hit pairs for each drug were validated through multiple concentration drug–biosensor fluorescence dose-response measurements. Thus, SLV-319 (+/-) was measured against the biosensors v4.6, v7A, and L194D1; AM 6545, rimonabant, and PSNCBAM-1 were measured against the biosensors AK1, v4.6, v7A, and L194D1; WIN-55 212,22 was measured against the biosensors v4.6, v4.8.1.2, v7A, and L194D1; and NIDA 41020 was measured against the biosensors v4.6, v4.8.1.2, Scop4, and L194D1.
From these screens, the drug–biosensor pairs with the most favorable profile were chosen (Figure 8). This was determined based on the S-slope, EC50, and Fmax of the drug–biosensor pairs, with a higher Fmax, higher S-slope, and lower EC50 being most desirable. The multiple concentration drug–biosensor validations showed promising data for CB1/CB2 ligands: when calculated as S-slopes ((ΔF/F0)/[Drug]) at 0.6 µM, the sensitivities plotted in Figure 8 range from 1.6 to 6 µM-1. Our limited supplies of the test drugs vitiated full dose-relations at saturated concentrations and Hill equation fits; but these data already suggest that it will be possible to conduct cellular experiments with these ligands at 1 < µM. Further optimization may present the challenge of directed evolution in non-aqueous solvents such as DMSO or methanol.
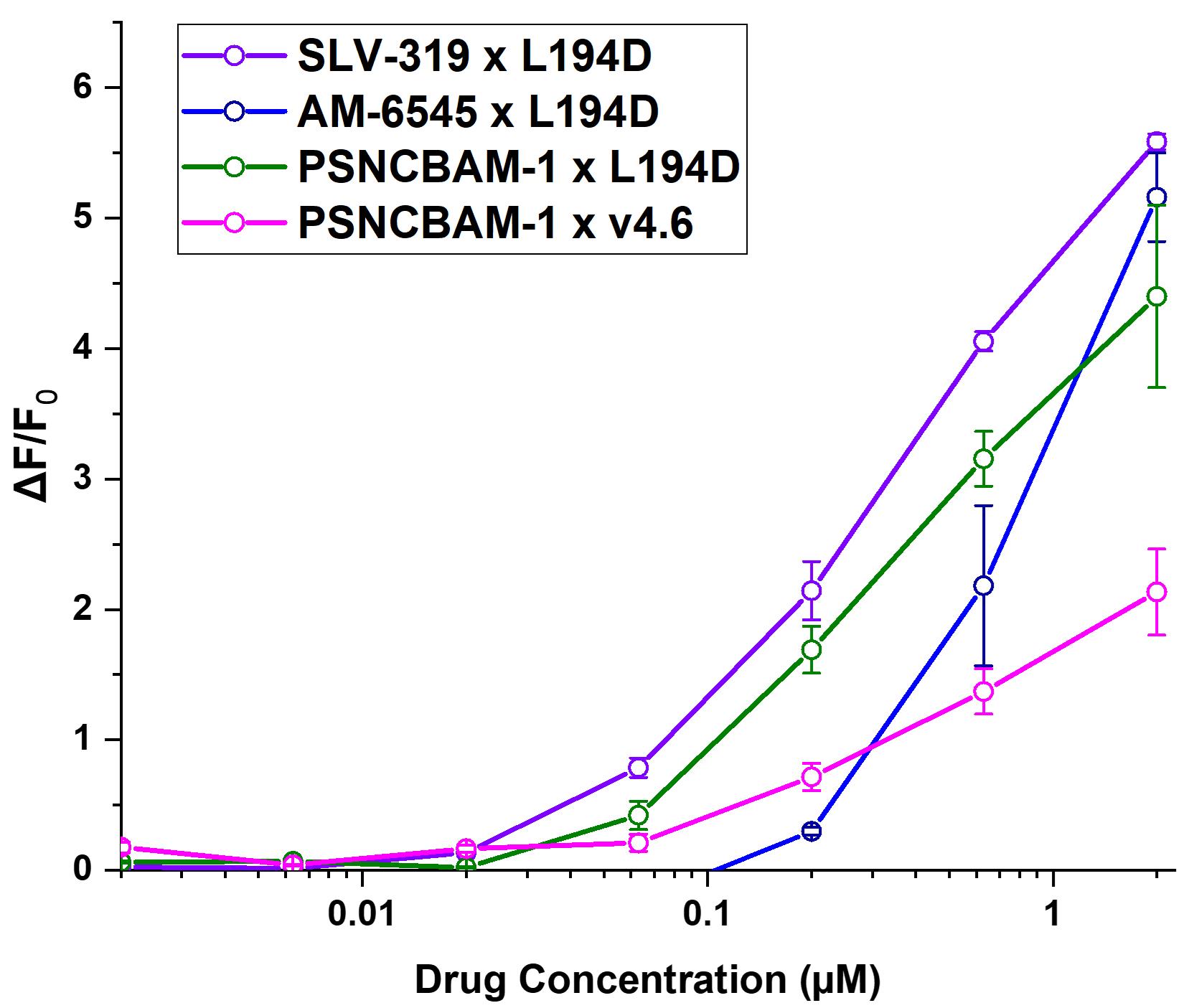
Figure 8. Multiple concentration drug–biosensor fluorescence validations for CB1/CB2 ligands. Dose-response relations for CB1/CB2 ligands. Only the most promising dose-response relation(s) for each drug of interest are shown.
As noted, a hit pair comprises a pair of molecules: drug and biosensor. In total, 190 drug–biosensor hit pairs were identified, with ΔF/F0 ranging from 1.0 to 18.5. From these, 108 hit pairs had 1 < ΔF/F0 < 3 and thus could likely be used to generate optimized biosensor variants through directed evolution. The remaining 82 hits had ΔF/F0 > 3 and thus could likely be used for cellular imaging without further mutations. Hit pairs were concentrated in three clinical use categories (schizophrenia, major depressive disorder, and other), with no hit pairs identified for anxiety and epilepsy drugs. The v7.1.2 biosensor participated in the largest number of hit pairs; these also had the highest ΔF/F0 values (Figure 4).
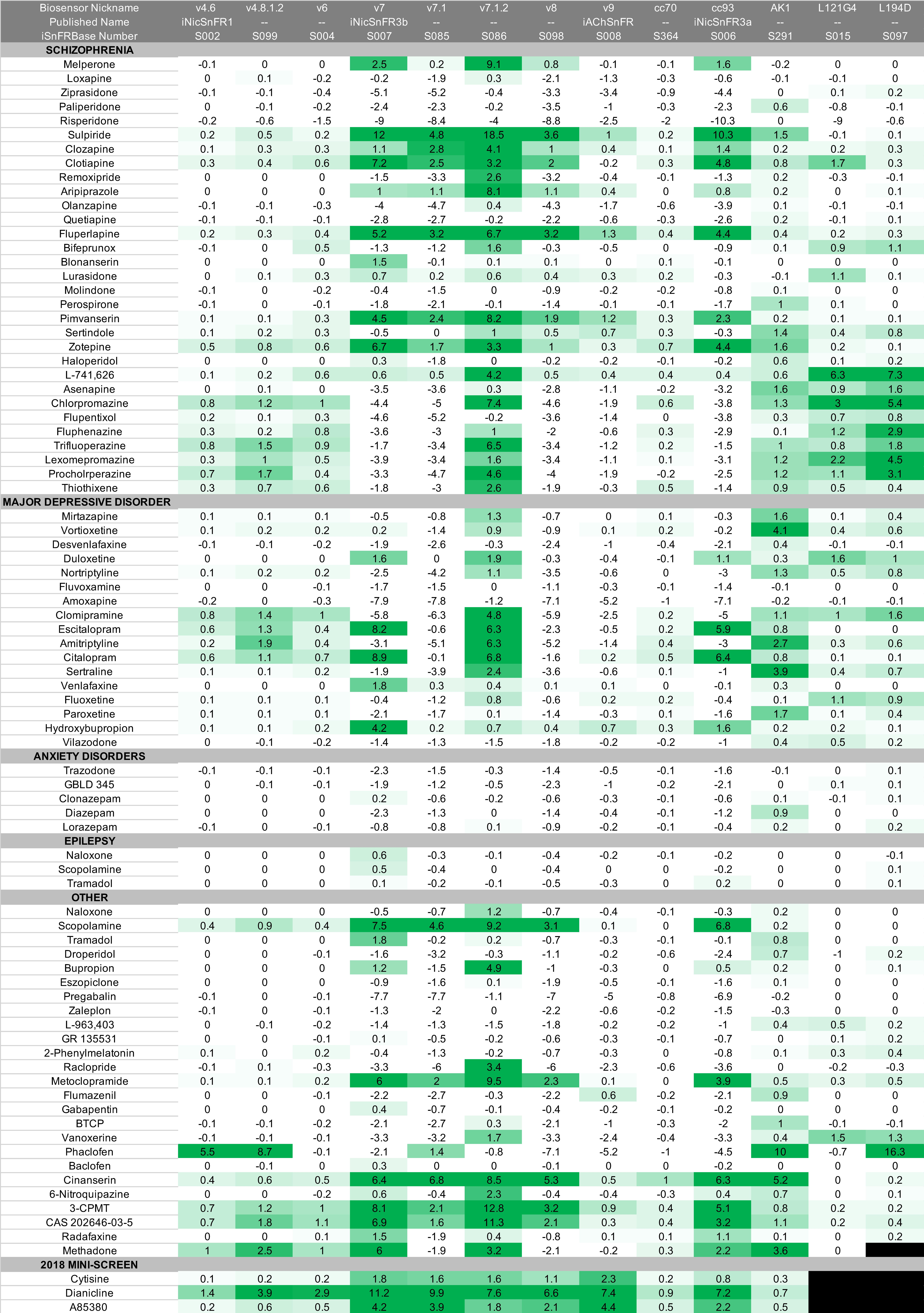
Figure 4. Results map from the 2017 single concentration drug–biosensor fluorescence screen. Fluorescence response (as represented by ΔF/F0) for drug–biosensor pairs included in the 2017 screen. Fluorescent response shading from green (ΔF/F0 ≥ 3) to white (ΔF/F0 ≤ 0), in which stronger hits are represented by darker green, weaker hits by lighter green, and non-hits by white. Drug–biosensor pairs that were not screened are in black.
This screen allowed for the identification of drug–biosensor pairs, from which the biosensor could potentially be evolved for engineering of novel variant biosensors for various neuropsychiatric medications. Biosensors for two nicotinic agonists, cytisine and dianicline, were engineered based on hit pairs obtained from this screen. The directed evolution of these sensors, as well as associated experiments on the subcellular pharmacokinetics of these drugs, are detailed in Nichols et al. (2022). The hit pair involving methadone became the basis for a selective variant, iS-methadoneSnFR (Muthusamy et al., 2022).
Because many of the drugs included in this screen are sparingly water-soluble, DMSO was often used for dissolution. However, this screen showed that DMSO interacts with several biosensor candidates, even in the absence of a DOI. This phenomenon resulted in the presence of many negative ΔF/F0s, as the baseline biosensor fluorescence was higher than would be reasonable if the biosensor and solvent were not interacting. This showed that additional normalizations and corrections would be required when DMSO was used as a solvent with iDrugSnFRs (see Notes). We have not systematically studied the residues responsible for DMSO sensitivity.
The 2017 screen was also informative in showing a failure: we found no hit pairs involving S-ketamine, its enantiomers, or its metabolites. Faute de mieux, we performed site-saturated mutagenesis on a residue, Tyr357, that makes a key cation-π interaction with nicotinic drugs (Shivange et al., 2019; Unger et al., 2020). This resulted in the S-ketamine responsive Tyr357Gly construct termed AK1, and we evolved AK1 into variants that satisfactorily sense S-ketamine. We included these variants in subsequent screens described below.
2018 single concentration drug–biosensor fluorescence screen
The 2018 screen was conducted to identify biosensor–drug hit pairs for opioids of several categories (Figure 5). Drugs included in the screen were chosen based on clinical importance, as well as presence in the existing literature. Most opioids included activate the μ opioid receptor, though several drugs target κ and 𝛿 opioid receptors. An Other category was composed of opioid inhibitors. In total, 48 drugs and 16 biosensors were included. From these pairs, 205 total drug–biosensor hits were identified. Of these, 128 hits had 1 < ΔF/F0 < 3, while 76 had ΔF/F0 > 3. Thirty of the 48 drugs tested participated in at least one hit pair, with most having hit pairs with several biosensors (one drug had a single hit pair). Additionally, each biosensor participated in a hit pair with at least one drug. The proportion of hit pairs varied by drug category. No hit pairs were found for µ opioids ligands, while the largest proportion of hit pairs were found for µ opioid ligands. Hit pairs also varied in fluorescence response, with the weakest pair having a ΔF/F0 of 1.0 (our at least definition for a hit pair) and the strongest pair having a ΔF/F0 of 19.0 (tapentadol × biosensor v7). Generally, the v7 biosensor series (including v7, v7.1, v7.1.2, v8, and v9) had strong responses with μ opioids (Figure 5).
This screen allowed for the identification of drug–biosensor pairs that could lead to the engineering of novel opioid biosensor variants. Furthermore, this screen showed many strong hit pairs involving clinically relevant μ opioid ligands, which could allow for direct cellular imaging or efficient generation of novel biosensors for this drug class.
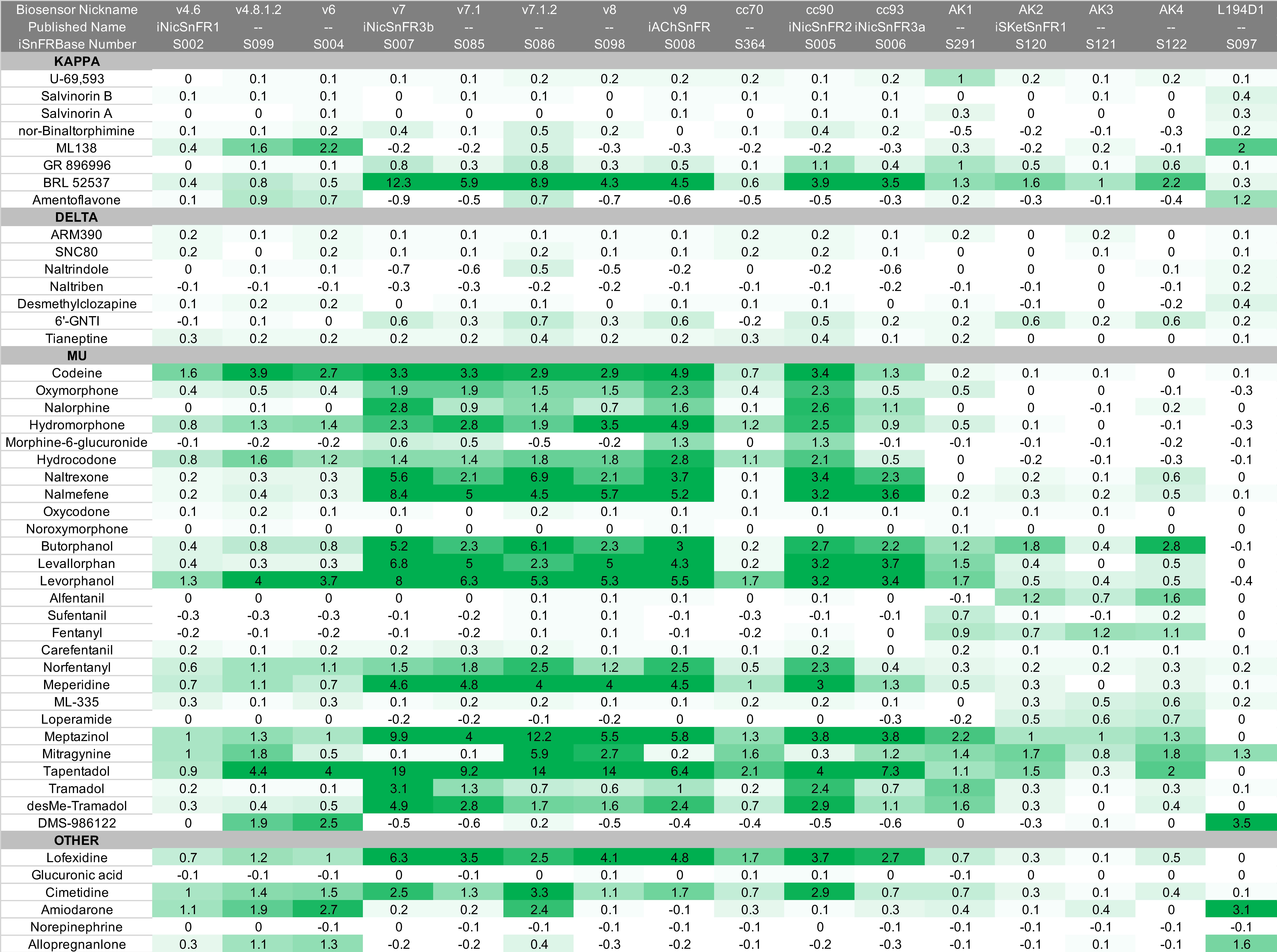
Figure 5. Results map from the 2018 single concentration drug–biosensor fluorescence screen. Fluorescence response (as represented by ΔF/F0) for drug–biosensor pairs included in the 2018 single concentration drug–biosensor fluorescence screen. Drugs are grouped by opioid category. Fluorescent response shading from green (ΔF/F0 ≥ 3) to white (ΔF/F0 = 0), in which stronger hits are represented by darker green, weaker hits by lighter green, and non-hits by white.
2019 single concentration drug–biosensor fluorescence screen
The 2019 screen was completed to identify hit pairs involving previously unscreened classes of CNS-acting drugs (Figure 6). Several drugs were included from each of five classes: 5-HT3 antagonists, anticholinergics, CB1/CB2 ligands, opioids, and neonicotinoids. Individual drugs of interest from a range of classes were also included and designated by an Other category. In total, 44 drugs were included and tested against 18 biosensor proteins. A total of 106 drug–biosensor hit pairs were identified, with 81 having 1 < ΔF/F0 < 3 and 24 having ΔF/F0 > 3. Twenty-one of the 44 drugs chosen participated in a hit pair with at least one biosensor. Additionally, each biosensor had a hit pair with at least one drug. Certain biosensors showed strong responses with several drugs in the same class. For example, L194D1 [a precursor to the biosensor iSeroSnFR, which detects serotonin (Unger et al., 2020)] displayed hit pairs for six CB1/CB2 ligands, with responses ranging from ΔF/F0 of 3.5–5.0. Several other biosensors (v4.6, v.4.8.1.2, v7 436A, and Scop4) also participated in hit pairs with several CB1/CB2 ligands, albeit less strongly (Figure 6).
This screen expanded the number of drug–biosensor hit pairs identified that could be used for engineering of novel variants, as well as the range of drug classes. 5-HT3 antagonists participated in many hit pairs and also showed promising results in validation through dose-response measurements (below).
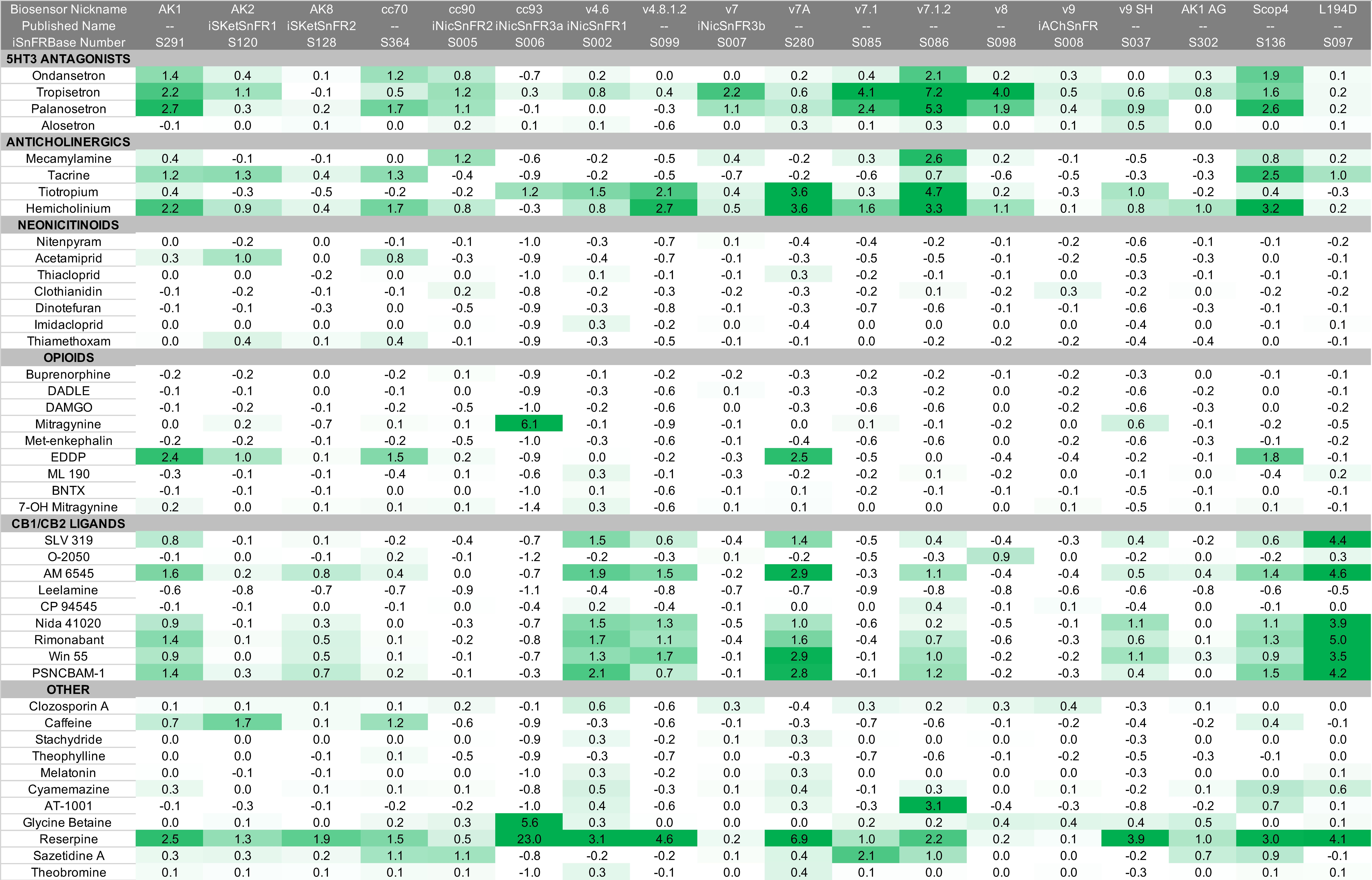
Figure 6. Results map from the 2019 single concentration drug–biosensor fluorescence screen results map. Fluorescence response (as represented by ΔF/F0) for drug–biosensor pairs included in the 2019 single concentration drug–biosensor fluorescence screen. Drugs are grouped by class. Fluorescent response shading from green (ΔF/F0 ≥ 3) to white (ΔF/F0 = 0), in which stronger hits are represented by darker green, weaker hits by lighter green, and non-hits by white.
Data analysis
Single concentration drug–biosensor fluorescence screen
Data analysis for our single concentration drug–biosensor fluorescence screens is relatively straightforward. When fluorescence measurements are obtained using the Tecan fluorescence reader, measurements are obtained for the biosensor alone, the drug of interest alone, and the biosensor when the drug of interest is added. These measurements are automatically output into an Excel file through the Tecan program and can be used to calculate ΔF/F0 for each drug–biosensor pair. This calculation allows for a direct comparison of fluorescence between drug–biosensor pairs with varying drug and biosensor baseline fluorescence. Additionally, it allows for determination of the strength of a hit regardless of whether the involved biosensor has high or low baseline fluorescence. The calculation used for ΔF/F0 is given in Equation 1.
ΔF/F0=[(Drug-Biosensor Fluorescence)-(Baseline Biosensor Fluorescence)-(Baseline Drug Fluorescence)]/(Baseline Biosensor Fluorescence)
EQUATION 1. Relative fluorescence values (ΔF/F0)
Equation used to calculate relative fluorescence values (ΔF/F0) for each drug–biosensor combination in single concentration screens and dose-response relations.
Multiple concentration drug–biosensor fluorescence validation
For our multiple concentration drug–biosensor fluorescence validations, Tecan measurements include biosensor baseline fluorescence, fluorescence of biosensor when the DOI is added at eight concentrations (0.02–2 µM final drug concentration), and drug baseline fluorescence. These measurements yield ΔF/F0 for each drug concentration using Equation 1. A well-behaved dose-response relation suggests that more complex interactions do not occur. Therefore, we wish to obtain values for ΔFmax/F0, EC50, and nH. ΔFmax/F0 represents the estimated maximum fluorescence value as drug concentration approaches infinity. EC50 represents the drug concentration at which ΔF/F0 increases to ΔFmax/2F0. The Hill coefficient is nH. These classical values can be obtained via curve-fitting through Lineweaver-Burke plots or Hill plots; we use the Origin Pro 9.1 program.
One goal of many drug development efforts is to find drugs with high potency, i.e., the lowest possible EC50 and IC50. Because we aim for biosensor variants that respond at pharmacologically relevant drug concentrations, we often work near the linear start of the drug–biosensor dose-response relation. To evaluate sensitivity, we define the S-slope metric as a linear fit to (∆F/F0 )/[ligand] at the beginning of the dose-response relation, with units of (μM)-1. Such measurements do not require a measurement of the maximal response, ΔFmax/F0, and are therefore appropriate for dose-response relations that do not extend to saturating levels (i.e., Figure 8). When a fully characterized dose-response is available (nH is always near 1.0), we also characterize S-slope by combining the values for ΔFmax/F0 and EC50 (Equation 2). Note that S-slope can be increased either by increasing ΔFmax/F0 or by decreasing EC50.
S-slope=(ΔFmax/F0)/EC50
EQUATION 2. S-slopeEquation used to calculate S-slope dose-response relations from maximum fluorescence increase (ΔFmax/F0) and half-maximal concentration (EC50). These values are determined via curve fitting to complete dose-response relations, for instance during multiple concentration drug–biosensor fluorescence validation.
Database Availability
The iSnFRbase numbers in Figures 4, 5, and 6 refer to the posted file, https://github.com/lesterha/lesterlab_caltech/blob/main/iSnFRBase0711%20as%20of%207-15-22_for_%E2%80%8BBeatty_et_al_2022b.xlsx
Notes
Through our single concentration screens, over 500 drug–biosensor hit pairs were identified that show promise for creation of novel biosensors for CNS-acting drugs. While most hit pairs had 1 < ΔF/F0 < 3 (approximately 65%) and thus will likely require further directed evolution before they can be used for imaging with their paired drug, a substantial portion (approximately 35%) had ΔF/F0 > 3 and thus could likely be directly used for cellular imaging. However, one must note that the screened biosensors were engineered to bind another drug, so directed evolution will still need to be conducted to establish selectivity for the new drug of interest. Once hit pairs have been successfully identified, they can be further assessed by multiple concentration validations. This is particularly useful in cases where a single drug participates in multiple hit pairs, and a single sensor needs to be chosen to begin directed evolution.
Several idiosyncrasies underlie the described protocols. First, many iDrugSnFRs of the OpuBC family bind and respond to amine-containing buffers at the tens of mM concentrations used in typical biological and molecular experiments. This is likely due to the amine-binding OpuBC parentage with the cation-π box at the binding site (Bera et al., 2019; Nichols et al., 2022; Muthusamy et al., 2022) (pdb files 7S7T, 7S7U, 7S7X, 7S7Z). Thus, we use only phosphate- and/or bicarbonate-based buffers. Second, most cpGFP-based biosensors are sensitive to pH, which is likely due to the proton candle snuffer mechanism (Barnett et al., 2017; Muthusamy et al., 2022; Nichols et al., 2022). Thus, one is limited to imaging in nearly pH-neutral organelles such as the cytoplasm, endoplasmic reticulum, and cis-Golgi apparatus. Third, many iDrugSnFRs are activated by DMSO alone, which implies that one cannot use commercially available screening libraries formatted in DMSO and must be cautious when using DMSO for water-insoluble drugs. Lastly, one must consider the fact that some drugs may have detectable fluorescence independent of biosensor interaction and require correction for this fluorescence in ΔF/F0 calculations (as outlined in Equation 1 above). Additionally, the protocol has been revised with each screen to increase efficiency.
Given the wide range of drugs for which hit pairs were identified in the screens shown, our fluorescence screening protocol is probably generalizable for drug classes other than those we have studied. There are several rubrics for describing drug classes.
One definition of drug class invokes chemistry. We do not know the upper limit for MW of ligands that could participate in hit pairs; we have not screened drugs with MW > 500. Wild-type OpuBC and its orthologs bind permanently charged quaternary amines such as choline, betaine, and proline betaine. Only a few clinically useful drugs (for instance tiotropium) are quaternary amines. Our screens have concentrated on primary, secondary, and tertiary amines that are partially protonated at neutral pH, i.e., they are weakly basic. We assume that only the protonated form of the drug makes a cation-π interaction with the iDrugSnFR (Bera et al., 2019; Nichols et al., 2022; Muthusamy et al., 2022) (Protein Data Bank files 7S7T, 7S7U, 7S7X, and 7S7Z). In many alkaloids, the nitrogen remains unprotonated at neutral pH. For instance, the iDrugSnFRs that bind nicotinic drugs do not sense cotinine, an amide, and we doubt that OpuBC-based iDrugSnFRs would detect amides. In unpublished work related to the 2018 screen (Figure 5), we found no hit pairs involving opioid peptides (the peptide bond is an amide bind).
Another rubric for drug class invokes target pharmacology (receptor, channel, transporter, or enzyme). As noted in the Introduction, the screens we describe are necessary because little correlation exists between the structure–activity relations that govern, on one hand, target binding and, on the other hand, binding to OpuBC variants, other than the presence of a weakly basic amine.
Other rubrics for drug class invoke the disorder being treated or the type of drug abuse. Within the former rubric, the figures show hit pairs for classical antidepressants, rapidly acting antidepressants, smoking cessation therapeutics, analgesics, antiemetics, antipsychotics, and appetite suppressants. Our unpublished data show hit pairs including the antidementia drug tacrine. Within the latter rubric, we found hit pairs including nicotine dependence, opioid dependence, and dissociative effects. We have not screened US Drug Enforcement Administration Schedule 1 drugs; among these, it is likely that hit pairs could be identified for some psychedelics (especially psilocin and analogs), cocaine, and MDMA.
Hit pairs for other drug classes (however one defines the classes) should probably be approached by merging circularly permuted fluorescent protein GFP with variants of a different PBP (Scheepers et al., 2016) Hit pairs for amino acid drugs might be generated by mutating the binding site in the glutamate-sensing iGluSnFR variants (Marvin et al., 2018). Many drugs that inhibit intracellular enzymes are amides; we have not considered the likeliest PBPs for binding amides. Finally, several scientists have proposed the intriguing challenge of generating PBP-based sensors for peptides.
Recipes
50× M
Reagent Final concentration Amount Na2HPO4 1.25 M 17.75 g KH2PO4 1.25 M 17.0 g NH4Cl 2.5 M 13.4 g Na2SO4
H2O
Total
0.25 M
n/a
n/a
3.55 g
remainder
100 mL
50× 5052
Reagent Final % Amount Glycerol 25% 25 mL Glucose 2.5% 2.5 g Alpha-lactose 10% 10 g H2O
Total
n/a
n/a
remainder
~100 mL
Acknowledgments
We have been supported by The Brain and Behavior Research Foundation (NARSAD Award 2014-23069), California Tobacco-Related Disease Research Program (Aaron L. Nichols, 27FT-0022) (Dennis A. Dougherty, 27IP-0057), The Della Martin Foundation (Kallol Bera), Howard Hughes Medical Institute (Jonathan S Marvin and Loren L Looger), National Institute on Drug Abuse (Henry A Lester, DA043829) (Henry A Lester and Anand K Muthusamy, DA049140), National Institute of General Medical Sciences (Henry A Lester, GM-12358) (Anand K Muthusamy T32-GM7616), National Institute of Mental Health (Henry A Lester, 213MH120823), National Institute of Neurological Disorders and Stroke fellowship (Anand K Muthusamy T32NS105595), the Caltech CI2 program, and Caltech SURF donors Samuel P. and Frances Krown (Zoe G Beatty). This protocol was derived from the original research paper “Fluorescence activation mechanism and imaging of drug permeation with new sensors for smoking-cessation ligands” (Nichols et al., 2022).
Competing interests
Anand Muthusamy, Henry Lester, Loren Looger, and Jonathan Marvin have filed a patent application that includes opioid biosensors. Lin Tian is a co-founder of Seven Biosciences.
Ethics
There are no ethical concerns relevant to this paper.
References
- Barnett, L. M., Hughes, T. E. and Drobizhev, M. (2017). Deciphering the molecular mechanism responsible for GCaMP6m's Ca2+-dependent change in fluorescence. PLoS One 12(2): e0170934.
- Bera, K., Kamajaya, A., Shivange, A. V., Muthusamy, A. K., Nichols, A. L., Borden, P. M., Grant, S., Jeon, J., Lin, E., Bishara, I., et al. (2019). Biosensors Show the Pharmacokinetics of S-Ketamine in the Endoplasmic Reticulum. Front Cell Neurosci 13: 499.
- Henderson, B. J. and Lester, H. A. (2015). Inside-out neuropharmacology of nicotinic drugs. Neuropharmacology 96(Pt B): 178-193.
- Lester, H. A., Miwa, J. M. and Srinivasan, R. (2012). Psychiatric drugs bind to classical targets within early exocytotic pathways: therapeutic effects. Biol Psychiatry 72(11): 907-915.
- Marvin, J. S., Scholl, B., Wilson, D. E., Podgorski, K., Kazemipour, A., Muller, J. A., Schoch, S., Quiroz, F. J. U., Rebola, N., Bao, H., et al. (2018). Stability, affinity, and chromatic variants of the glutamate sensor iGluSnFR. Nat Methods 15(11): 936-939.
- Muthusamy, A. K., Kim, C. H., Virgil, S. C., Knox, H. J., Marvin, J. S., Nichols, A. L., Cohen, B. N., Dougherty, D. A., Looger, L. L. and Lester, H. A. (2022). Three Mutations Convert the Selectivity of a Protein Sensor from Nicotinic Agonists to S-Methadone for Use in Cells, Organelles, and Biofluids. J Am Chem Soc 144(19): 8480-8486.
- Nichols, A. L., Blumenfeld, Z., Fan, C., Luebbert, L., Blom, A. E. M., Cohen, B. N., Marvin, J. S., Borden, P. M., Kim, C. H., Muthusamy, A. K., et al. (2022). Fluorescence activation mechanism and imaging of drug permeation with new sensors for smoking-cessation ligands. Elife 11: e74648.
- Scheepers, G. H., Lycklama A. N. J. A. and Poolman, B. (2016). An updated structural classification of substrate-binding proteins. FEBS Lett 590(23): 4393-4401.
- Shivange, A. V., Borden, P. M., Muthusamy, A. K., Nichols, A. L., Bera, K., Bao, H., Bishara, I., Jeon, J., Mulcahy, M. J., Cohen, B., et al. (2019). Determining the pharmacokinetics of nicotinic drugs in the endoplasmic reticulum using biosensors. J Gen Physiol 151(6): 738-757.
- Studier, F. W. (2005). Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41(1): 207-234.
- Unger, E. K., Keller, J. P., Altermatt, M., Liang, R., Matsui, A., Dong, C., Hon, O. J., Yao, Z., Sun, J., Banala, S., et al. (2020). Directed Evolution of a Selective and Sensitive Serotonin Sensor via Machine Learning. Cell 183(7): 1986-2002 e1926.
Article Information
Copyright
![]() Beatty et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Beatty et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Beatty, Z. G., Muthusamy, A. K., Unger, E. K., Dougherty, D. A., Tian, L., Looger, L. L., Shivange, A. V., Bera, K., Lester, H. A. and Nichols, A. L. (2022). Fluorescence Screens for Identifying Central Nervous System–Acting Drug–Biosensor Pairs for Subcellular and Supracellular Pharmacokinetics. Bio-protocol 12(22): e4551. DOI: 10.21769/BioProtoc.4551.
- Nichols, A. L., Blumenfeld, Z., Fan, C., Luebbert, L., Blom, A. E. M., Cohen, B. N., Marvin, J. S., Borden, P. M., Kim, C. H., Muthusamy, A. K., et al. (2022). Fluorescence activation mechanism and imaging of drug permeation with new sensors for smoking-cessation ligands. Elife 11: e74648.
Category
Neuroscience > Development
Molecular Biology > Protein
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.