- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Enhanced Ribonucleoprotein Immunoprecipitation (RIP) Technique for the Identification of mRNA Species in Ribonucleoprotein Complexes
Published: Vol 12, Iss 19, Oct 5, 2022 DOI: 10.21769/BioProtoc.4526 Views: 2877
Reviewed by: Chiara AmbrogioDavide BottaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2022
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
RNA binding proteins (RBPs) are critical regulators of cellular phenotypes, and dysregulated RBP expression is implicated in various diseases including cancer. A single RBP can bind to and regulate the expression of many RNA molecules via a variety of mechanisms, including translational suppression, prevention of RNA degradation, and alteration in subcellular localization. To elucidate the role of a specific RBP within a given cellular context, it is essential to first identify the group of RNA molecules to which it binds. This has traditionally been achieved using cross-linking-based assays in which cells are first exposed to agents that cross-link RBPs to nucleic acids and then lysed to extract and purify the RBP-nucleic acid complexes. The nucleic acids within the mixture are then released and analyzed via conventional means (e.g., microarray analysis, qRT-PCR, RNA sequencing, or Northern blot). While cross-linking-based ribonucleoprotein immunoprecipitation (RIP) has proven its utility within some contexts, it is technically challenging, inefficient, and suboptimal given the amount of time and resources (e.g., cells and antibodies) required. Additionally, these types of studies often require the use of over-expressed versions of proteins, which can introduce artifacts. Here, we describe a streamlined version of RIP that utilizes exclusion-based purification technologies. This approach requires significantly less starting material and resources compared to traditional RIP approaches, takes less time, which is tantamount given the labile nature of RNA, and can be used with endogenously expressed proteins. The method described here can be used to study RNA-protein interactions in a variety of cellular contexts.
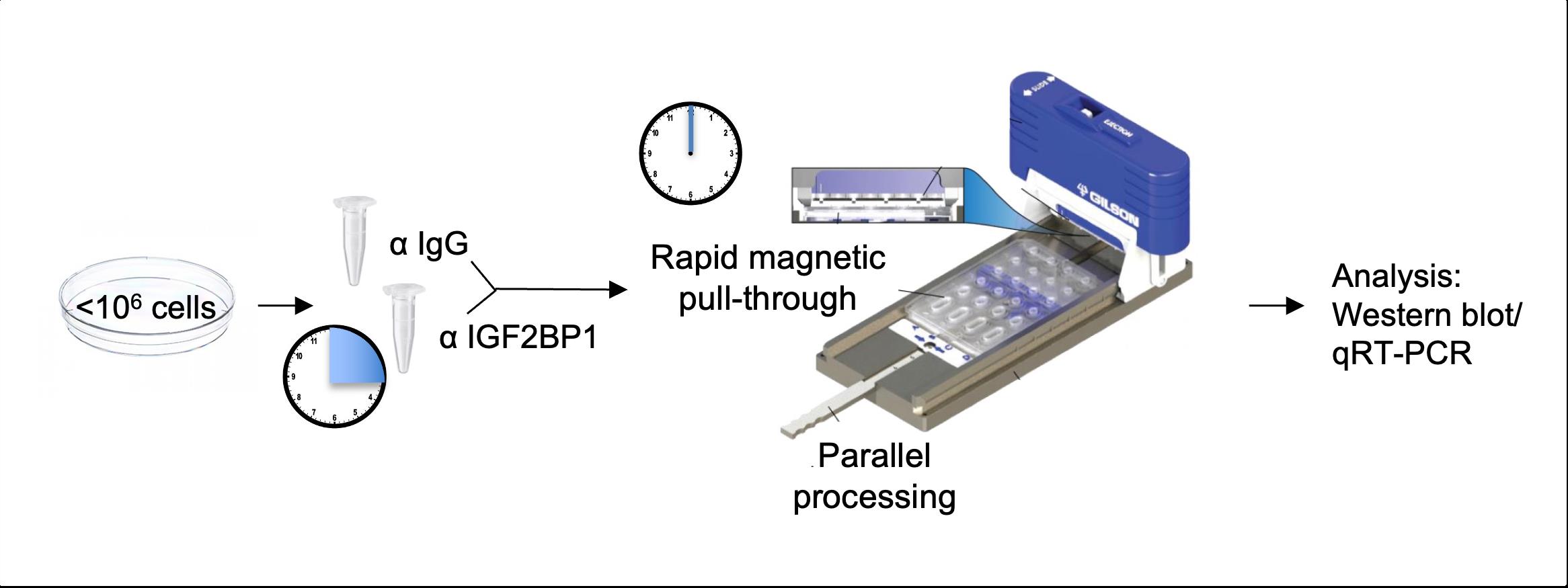
Graphical abstract:
Background
Post-transcriptional regulation of gene expression enables rapid systematic cellular responses to various stimuli and stressors (Chua et al., 2020; Weskamp et al., 2021). Post-transcriptional regulation can be mediated by various cis- and trans-acting elements, including regulatory elements on the mRNA itself as well as microRNAs and RNA-binding proteins (RBPs) (Theil et al., 2018; Furlan et al., 2021; Ma et al., 2022). In cells, RBPs often exist within large complexes, known as ribonucleoprotein (RNP) complexes, that include the RBPs themselves alongside their mRNA binding partner(s) and other proteins/molecules. Through these interactions in RNP complexes, RBPs can regulate the expression, translation, and/or localization of the mRNA species to which they bind (Dvir et al., 2018; Formicola et al., 2019).
Importantly, a single RBP can bind to and regulate the expression of many mRNAs (Hentze et al., 2018). Thus, deregulation of RBP expression has wide-ranging consequences for cellular health and survival. Notably, deregulation of RBP activity has been implicated in numerous human pathologies, including neurological disorders, kidney disease, and various types of cancer (Chatterji and Rustgi, 2018; Wang et al., 2018; Shi et al., 2020; Kinoshita et al., 2021; Weskamp et al., 2021). In order to begin probing the mechanism via which deregulation of RBP expression leads to disease, it is essential to first identify the group of mRNA binding partners to which the RBP binds. By identifying a list of mRNA binding partners for a given RBP, researchers can begin identifying candidate mRNA partner(s) from the list with potential links to the observed phenotype(s) and can thus begin elucidating the mechanism via which deregulated RBP expression leads to a diseased state (Fakhraldeen et al., 2022).
Given the importance of identifying mRNA binding partners of RBPs, research efforts have focused on developing methodologies that can reproducibly purify and identify mRNAs from RNP complexes (Zhao et al., 2022). The most common methodologies developed thus far rely upon cross-linking of often over-expressed RBPs prior to immunoprecipitation (IP). Once the RNP complexes are purified via manual IP, the cross-links are reversed, and the RNA is extracted and then analyzed via the researchers’ preferred method (e.g., qRT-PCR, microarray analysis, Northern blot, or RNA-Seq). Multiple versions of this so-called ribonucleoprotein immunoprecipitation (RIP) procedure have been developed, including RIP-Chip (manual RIP followed by microarray analysis), CLIP (cross-linking and immunoprecipitation), iCLIP (individual nucleotide resolution CLIP), PAR-CLIP (photoactivatable ribonucleoside-enhanced CLIP), HiTS-RAP (high throughput sequencing–RNA affinity profiling), and various adaptations therefrom (Galgano and Gerber, 2011; Jain et al., 2011; Danan et al., 2016, 2022; Haecker and Renne, 2014; Spitzer et al., 2014; Guerreiro et al., 2014; Ozer et al., 2015; Sutandy et al., 2016; Diaz-Munoz et al., 2017; George et al., 2017; Perconti et al., 2019; Lewinski et al., 2020; Benhalevy and Hafner, 2020; Baldini and Labialle, 2021). These approaches to RIP have several drawbacks. First, introduction of exogenously expressed RBPs can affect the mRNA binding profile of the RBP. Second, the processes of cross-linking and reverse cross-linking themselves can introduce artifacts by inducing interactions that were previously absent/not physiologically relevant, or by not capturing all interactions. Third, performing the IP procedure manually is time consuming and may promote irreproducibility based on the handler. Finally, any added benefits of potentially being able to identify specific binding sites of RBPs with their mRNA binding partners using these techniques are overshadowed by the complex computational approaches required in order to perform such analyses (Jens, 2016; Huessler et al., 2019; Busch et al., 2020; Zhao et al., 2022).
The method described here relies on exclusion-based sample preparation (ESP) technologies to perform RIP (Berry et al., 2015; Pezzi et al., 2018; Fakhraldeen et al., 2022). Specifically, we describe a procedure for performing RIP using the EXTRACTMAN device, which is an adaptation of the ESP-based Sliding Lid for Immobilized Droplet Extraction (SLIDE) technology. Not only does this method require less starting material, but it also offers significantly faster processing times, which is of particular relevance to RIP given the labile nature of RNA. Finally, the use of this sensitive method precludes the need to over-express the RBP of interest or to cross-link the lysate and instead allows for purification of the endogenous RBP in its native state. We describe the successful application of this method to identify mRNA binding partners of the RBP IGF2BP1 in the human embryonic kidney cell line HEK293T as well as in the human breast cancer cell lines MCF7 and MDA-MB-231 (Fakhraldeen et al., 2022). The extracted RNA was analyzed by qRT-PCR, but it can be analyzed using microarray analysis, RNA-Seq, or any endpoint of interest for RNA identification/profiling.
Materials and Reagents
293T human embryonic kidney cells (ATCC, catalog number: CRL-3216)
MCF7 cells (ATCC, catalog number: HTB-22)
MDA-MB-231 cells (ATCC, catalog number: CRM-HTB-26)
10 cm dishes (Fisherbrand, catalog number: FB0875712)
Cell scrapers (Fisherbrand, catalog number: 08-100-241)
DMEM, high glucose, HEPES (Thermo Fisher Scientific, Gibco, catalog number: 12430104)
DMEM, low glucose, pyruvate, HEPES (Thermo Fisher Scientific, Gibco, catalog number: 12320032)
Fetal bovine serum (FBS) [various vendors; pre-screened on mammary epithelial cells to identify lots that promoted cell growth without cell death (floating cells) over the course of four passages]
Penicillin/streptomycin (Thermo Fisher Scientific, Gibco, catalog number: 15070063)
1× phosphate buffered saline (PBS), pH 7.4 (Thermo Fisher Scientific, Gibco, catalog number: 10010023)
HEPES (1 M) (Gibco, Thermo Fisher Scientific, catalog number: 15630080)
MgCl2 (Merck/Sigma-Aldrich, catalog number: 208337)
KCl (Merck/Sigma-Aldrich, catalog number: P9541)
NonidetTM P 40 substitute (Fluka Biochemika, catalog number: 74385)
DTT (1,4-dithiothreitol; MilliporeSigma, catalog number: 111474)
RNaseOUTTM recombinant ribonuclease inhibitor (Thermo Fisher Scientific, Invitrogen, catalog number: 10777-019). Store at -20 °C and thaw on ice prior to use
Halt protease and phosphatase inhibitor single-use cocktail (Thermo Fisher Scientific, catalog number: 78442)
Dynabeads® Protein G (Novex by Life Technologies, catalog number: 10003D). Store at 4°C
Tween-20 (Merck/Sigma-Aldrich, catalog number: P9416)
RNeasy® mini kit (Qiagen, catalog number: 74104)
QuantiTect reverse transcription kit (Qiagen, catalog number: 205311)
Bradford reagent (Thermo Fisher Scientific, catalog number: 23238)
Transfer membrane Immobilon-P polyvinylidene difluoride (PVDF) (Merck/Millipore, catalog number: IPVH00010)
PreciseTM 4%–20% Tris-HEPES-SDS protein gels, 4.5 mm (Thermo Fisher Scientific, catalog number: 25244) or any pre-cast gel of choice
Anti-IMP-1 (D33A2) rabbit monoclonal primary antibody (Cell Signaling, catalog number: 8482)
Anti-vinculin mouse primary antibody, clone V284 (MilliporeSigma, catalog number: 05-386)
Anti-GAPDH (14C10) rabbit monoclonal primary antibody (Cell Signaling, catalog number: 2118)
Horseradish peroxidase (HRP) donkey anti-mouse polyclonal secondary antibody (Jackson ImmunoResearch, catalog number: 715-035-151)
HRP goat anti-rabbit polyclonal secondary antibody (Invitrogen, catalog number: G-21234)
HRP mouse anti-rabbit IgG (conformation-specific) (L27A9) secondary antibody (Cell Signaling, catalog number: 5127)
Non-immune rabbit IgG whole molecule control (Jackson ImmunoResearch, catalog number: 011-000-003)
Milk non-fat, dry milk powder (Kroger or equivalent)
Polysome lysis buffer (see Recipes)
RIP wash/elution buffer (see Recipes)
5% milk in TBS-Tween (see Recipes)
Equipment
EXTRACTMAN device (Gilson)
Spectrophotometer (Thermo Fisher Scientific, model: NanoDrop 2000)
Gel documentation system (Bio-Rad, Gel Doc XR+ Gel Documentation System)
Refrigerated centrifuge that can reach speeds of ≥12,000 rpm (must accommodate 50 mL tubes spun at 3,000 × g and 1.5 mL tubes spun at 12,000 rpm)
Plate centrifuge (Beckman, model: Allegra X15R)
Sonicator (Fisher Scientific 100)
Tube rotator that can hold 1.5 mL tubes; a standard rotisserie tube rotator with adaptors for Eppendorf tubes was used
qPCR machine (Applied Biosystems, ABI7900HT Fast Real-Time PCR System)
Procedure
Cell culture
Plate cells in 10 cm dishes at ≥50% confluence in appropriate media. For 293T cells, plating 1.5 × 106 cells in a 10 cm dish and harvesting within 48 h should yield approximately 2 mg of protein, which should suffice for the endpoint(s) of interest.
Note: One 10 cm dish yields 500 µL of lysate, or the equivalent of two samples for RIP (200 µL of lysate are needed per RIP reaction). If cells express high endogenous levels of RBP of interest, one 10 cm dish should provide sufficient material for RIP; if cells express low endogenous levels of RBP of interest, plan on combining two 10 cm dishes into 500 µL of lysate; if endogenous expression levels of RBP are unknown, we recommend starting with the more concentrated lysate (i.e., two 10 cm dishes combined into 500 µL of lysate). Take into account the need for 2× 200 µL of lysates for each RIP: one lysate to be used for purification with an antibody targeting the RBP of interest and one lysate to be used for purification with an anti-IgG control antibody. The purified eluate from each RIP reaction should provide sufficient material for both protein and RNA analyses.
Incubate cells at 37 °C.
Proceed to the next step 24–48 h post plating.
Cell lysis for RNP lysate preparation
Note: Perform all these steps on ice.
Remove media from dish by aspiration.
Rinse cells with 5 mL of ice-cold PBS twice.
Add 5 mL of fresh ice-cold PBS.
Scrape cells from dish using scraper.
Transfer scraped material to 50 mL conical tubes.
Pellet cells by centrifugation at 3,000 × g and 4 °C for 5 min.
Aspirate and discard the supernatant.
Add polysome lysis buffer (PLB) to pellet (500 µL of PLB per 10 cm dish or 500 µL of PLB per two 10 cm dishes for more concentrated lysate) and pipette up and down to resuspend the pellet completely.
Transfer lysate to 1.5 mL Eppendorf tubes.
Store lysate in PLB at -80 °C or proceed directly to step 11.
Sonicate lysates (10 pulses at 4–5 W) in a cold room.
Spin lysates at approximately 12,000 rpm for 30 min at 4 °C.
Transfer supernatant into new Eppendorf tubes and spin two more times using the same conditions (three total centrifugation cycles with identical conditions).
Aliquot approximately 200 µL of lysate per Eppendorf tube and store at -80 °C until ready to perform RIP procedure or proceed immediately to the next step.
RNP immunoprecipitation (RIP)/EXTRACTMAN
Thaw lysates on ice (if frozen).
Add approximately 1.2 µg of primary antibody to 200 µL of RIP lysate (containing approximately 0.8 mg of protein) and add matched IgG control to another 200 µL of RIP lysate.
Note: The quantity of antibody will vary depending on the antibody affinity or endogenous cellular RBP levels and will need to be optimized (1–10 µg of antibody is a typical range).
Add 5 µL of washed protein G–bound paramagnetic Dynabeads to the same tube(s) containing RIP lysate + primary antibody.
Note: Dynabead washing procedure is described in manufacturer’s specification sheet.
Incubate mixture on rotator at low speed (approximately 30 rotations per minute) and 4 °C for preferred amount of time.
Note: Approximately 25% of protein was pulled through with 30 min incubation; however, pull-through efficiency will vary depending on the antibody affinity or RBP levels and will need to be optimized (30 min–16 h should be a reasonable range for incubation times).
Prepare EXTRACTMAN device.
Note: EXTRACTMAN device setup is presented in video form at the following link: https://www.gilson.com/extractman-starter-kit.html.
Set up new disposable microplate and bead collection strip on the device.
Note: Each plate contains four “columns” labeled A–D, with each column accommodating one RIP reaction. Thus, each plate can be used to run up to four RIP reactions simultaneously.
Place the magnet locator at the correct starting position as specified in the EXTRACTMAN manual.
For each RIP reaction that will be run, add 100 µL of wash/elution buffer to each of the five small wells per column in the microplate (this will include the wash wells as well as the bound elution well).
Note: Add less volume to the elution well if a more concentrated lysate is required.
Add 200 µL of the RIP mixture (lysate + primary antibody + paramagnetic Dynabeads) to the large well(s) in the microplate.
Proceed with device operation as described in the EXTRACTMAN manual. Briefly, alternatively proceed with shifting the handle and then the magnet locator to each successive stop on the device sequentially with a brief (< 5 s) pause at each stop.
Collect the remaining volume in each input well following the procedure and put in tube(s) (approximately 200 µL volume; this is what is labeled as the “unbound” fraction).
Collect the volume in the last well per column in the microplate and put in tube(s) (approximately 100 µL volume; this is what is labeled as the “bound” fraction).
Collect the volumes in the middle wash wells and put in tubes (approximately 100 µL per well).
Note: This step is not required, but the collected volume can be used to check for any losses associated with the washing procedure if deemed necessary.
Store the unbound, bound, and wash fractions (if applicable) at -20 °C for downstream analyses or proceed directly to steps D or F for RNA and/or protein analysis, respectively.
RNA isolation
Note: Prior to beginning work, all benchtops, pipettes, handlers’ gloves, etc. should be wiped down carefully with RNase Away or an equivalent solution containing RNase inhibitor(s). Additionally, all tubes, tips, and other consumables should be certified RNase/nuclease-free. Cell culture media, PBS, and other solutions (e.g., lysis buffer components) should be dedicated for RIP and kept separately from the same solutions used for other experimental purposes. This is to avoid compromising the RNA analysis endpoints by preventing degradation of the RNAs within the RNP complexes being purified.
Add 350 µL of lysis buffer provided in the Qiagen RNeasy mini kit directly into the unbound/bound/wash fraction volumes.
Follow manufacturer’s instructions for remainder of processing steps.
Note: The Qiagen RNeasy mini kit was used for the work described here, but other RNA extraction kits/methods will likely yield similar results if procedures for RNA isolation from cells are followed as per manufacturer’s instructions.
qRT-PCR
Determine RNA concentration and quality using approach of choice [e.g., load approximately 1–2 µL of unbound/bound fractions onto NanoDrop device and record RNA concentration and 260/280 (ideal range: 1.7–2.0) and 260/230 ratios (ideal range: 1.8–2.2)].
Set up RT-PCR using kit of choice (e.g., QuantiTect reverse transcription kit) and appropriate thermal cycler settings.
Note: Run each reaction at least in duplicates.
Run qPCR using cDNA generated in previous step and primers of choice with appropriate temperature and time settings for each cycle.
Note: Other endpoints such as Northern blot, microarray, or RNA-Seq analysis can also be applied here.
Western blotting
Determine protein concentration in each of the unbound, bound, and wash fractions (if applicable) using approach of choice (e.g., addition of Bradford reagent followed by colorimetric reading)
Load equal quantities of protein lysate from each of the unbound fractions onto polyacrylamide gel to enable quantitative visualization of the protein of interest. Load appropriate quantities of bound fractions assuming 20%–100% recovery of the RBP of interest (depending on antibody affinity). This loaded fraction should be recorded and then used to calculate the pull-through efficiency.
Perform Western blotting using apparatus, run/transfer settings, and primary and secondary antibodies of choice.
Data analysis
RIP procedures can be used to identify mRNA binding partners of RBPs. Here, we describe the fast and efficient purification of RNPs containing the RBP IGF2BP1 from human cells using the EXTRACTMAN device. RNA can then be extracted from the purified RNPs and subjected to qRT-PCR analysis. Note that qRT-PCR analysis could be used in the context of the work described here because some mRNA binding partners of IGF2BP1 were already identified in previous analyses. If no mRNA binding partners of the RBP of interest are known, then more exploratory RNA identification approaches such as microarray analysis or RNA-Seq should be applied.
Assessment of RBP enrichment efficiency during the purification procedure is performed using Western blot analysis of the purified fraction (bound), and on a portion of the input lysate following the purification procedure (unbound). Analysis of the protein content in the wash fractions might also be useful. The approximate protein yield in lysates from 293T cells was on the order of 4 µg/µL (approximately 2 mg total protein) from one 10 cm dish. The intensity of the bands observed on the Western blot was quantified using the Image Lab software provided with the Bio-Rad gel documentation system. Band intensity values were used for assessment of pull-through efficiency, knowing the linear range of response for the signals. Specifically, dividing the band intensity of the bound fraction by that of the unbound fraction generates a “% pull-through” readout, which can be used as a proxy for purification efficiency (Figure 1).
Importantly, purification efficiency analysis must be performed on blots of both the RBP of interest as well as control proteins (vinculin or GAPDH were used here). While the % pull-through efficiency may vary for the RBP of interest depending on various factors such as antibody affinity, the % pull-through for the control protein should be negligible. Significant % pull-through of the control protein may indicate non-specific immunoprecipitation conditions, which could be modified by increasing the stringency of the buffer binding conditions by altering the salt or detergent concentrations.
The same analysis should also be performed on the unbound and bound fractions of RIP reactions using an IgG control antibody, with the expectation that this will show minimal pull-through of either the RBP or control protein (e.g., GAPDH and vinculin).
In the analysis performed here, the % pull-through efficiency of the RBP CRD-BP following overnight incubation with primary CRD-BP antibody and standard RIP procedures was 37%, compared to a 60% pull-through efficiency when using the EXTRACTMAN-based RIP technique (Figure 1A). Importantly, no measurable amount of either CRD-BP or the control protein vinculin was detected following incubation with IgG control antibody when using either the standard or EXTRACTMAN-based RIP techniques.
The image lab software from Bio-Rad was used to quantify the relative band intensities, which were then used to calculate the % pull-through efficiency. The software instructs the user to select a reference band (in this case, the band in the unbound fraction for each respective RIP reaction). The reference band intensity is then set to 1, and the band intensity of the test/experimental condition (in this case, the band in the bound fraction for each respective RIP reaction) is quantitated relative to the reference band. Thus, the relative band intensity values for the unbound fraction are shown in Table 1 (the band intensity values for the bound fraction are always set to 1). The % pull-through efficiency is then calculated as the band intensity of the unbound fraction divided by the sum of the band intensities for both the bound and unbound fractions multiplied by 100.
Table 1. Relative Western blot band intensity values.
The relative band intensity values for the bands observed on the Western blots shown in Figure 1 were quantified using the Bio-Rad Image Lab software. O/N: overnight.
| Antibody | RIP fraction | O/N | 2 h | 30 min |
| CRD-BP | Unbound | 1 | 1 | 1 |
| Bound | 1.47 | 0.49 | 0.31 | |
| IgG | Unbound | 1 | 1 | 1 |
| Bound | 0.59 | 0.24 | 0.15 |
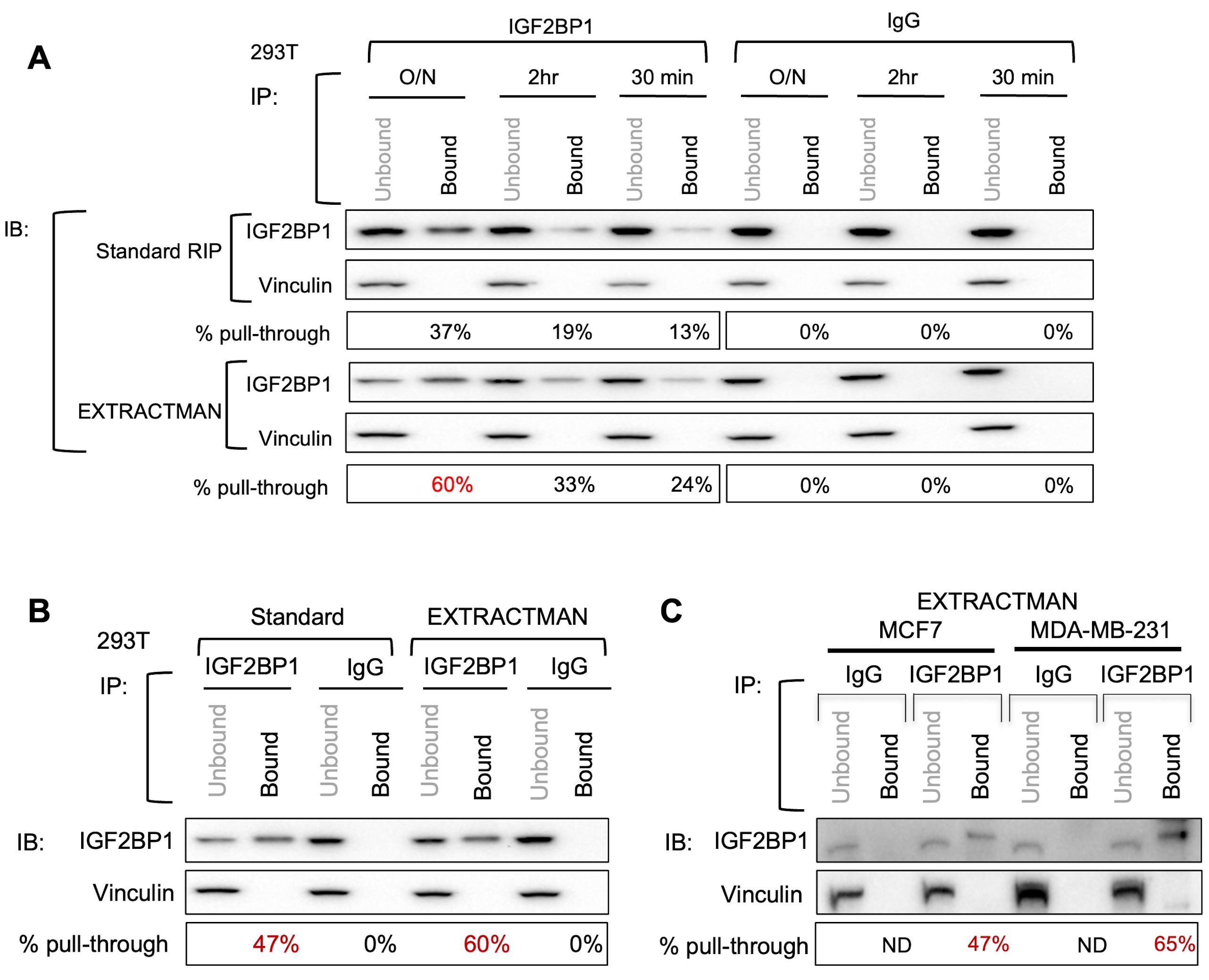
Figure 1. Western blot analysis of pull-through efficiency following RIP of IGF2BP1 in (A–B) 293T cells and (C) MCF7 and MDA-MB-231 cells. (A) Analysis of pull-through efficiency following RIP procedures performed with varying IGF2BP1 primary antibody incubation times [30 min, 2 h, and overnight (O/N)]. (A–B) IGF2BP1 RIP was performed in 293T cells using both standard procedures and EXTRACTMAN, and efficiencies (expressed as percentages) were compared between the two techniques. (C) Analysis of pull-through efficiency following IGF2BP1 RIP procedures performed on human breast cancer cells using EXTRACTMAN.
Once the specificity of the RIP procedure has been confirmed by Western blot of the proteins of interest, analysis of the RNA content within the purified material can be performed. Specifically, similar to protein analysis, RNA is extracted from both the unbound and bound RIP fractions. The extracted RNA is first assessed for quality and quantity, for example, using a NanoDrop device. Then, equal quantities of RNA from each fraction are reverse transcribed for analysis by qPCR. RNA yields from 293T cells were on the order of 400 ng/µL (approximately 12 µg of RNA total) from a single confluent 10 cm dish. Yields varied between cell types but generally fell within the range of 7–15 µg per confluent 10 cm dish. Following the RIP procedure with an antibody targeting the protein of interest using the EXTRACTMAN device, RNA yields in the output fraction ranged approximately from 0.2 to 0.6 µg (or the equivalent of approximately 2%–10% pull-through) (Figure 2).
Reactions are set up using primers for target mRNAs as well as control mRNAs (housekeeping genes and/or genes known to not being associated with your RBP of interest). Reactions are run at least in duplicate. Following the qPCR run, cycle threshold (Ct) values are recorded for each of the unbound and bound fractions from the duplicate set of lysates for each set of primers/gene being investigated. The delta_Ct (∆Ct=Ctbound-Ctunbound) is then calculated for each replicate for each set of primers/gene. The fold change is then calculated as 2-∆Ct. For a given set of primers, if the fold change between the unbound and bound fractions of the RBP pull-through is significantly different from that of the IgG control pull-through complex, this RNA is designated as enriched. To obtain a % mRNA pull-through value, the fold change is divided by a value equivalent to the fold change + 1, i.e.,
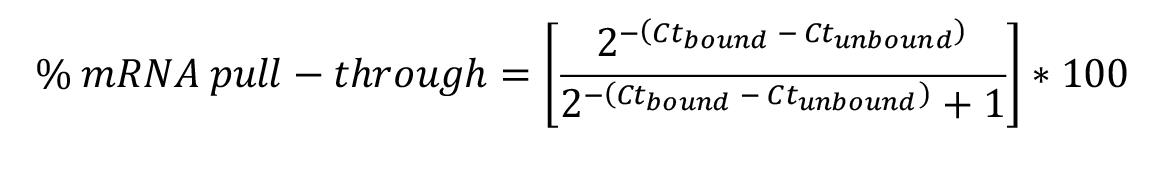

Figure 2. qPCR validation of RNA species pulled through following IGF2BP1 RIP procedures in (A) 293T cells and (B) MCF7 cells. qPCR was performed on RNA isolated from IGF2BP1- and IgG-enriched RIP lysates using primers targeting specific mRNA species that were previously identified in IGF2BP1-containing ribonucleoprotein complexes.
Total % RNA pull-through can be calculated by dividing the total amount of RNA in the bound fraction by the sum of the total amount of RNA in the bound and unbound fractions and multiplying by 100. The total amounts of RNA can be obtained by multiplying the concentration of RNA in each respective fraction by the total volume of that fraction.
Recipes
Polysome lysis buffer
KCl powder (m.w.: 74.5 g/mol) 100 mM, 0.7 g (745 mg)
HEPES solution 10 mM, pH 7.0, 1 mL of 1 M HEPES solution (pH 7.0)
MgCl2 powder (m.w.: 95.2 g/mol) 5 mM, 0.05 g (47.6 mg)
Nonidet P-40 0.5%, 0.5 mL
*Halt protease and phosphatase inhibitor single-use cocktail (100× stock) 1×
*RNaseOUTTM (40 U/µL) 2 U/µL
*DTT 1 mM
Total: 100 mL
* Add fresh prior to each use (only prepare volume required for specific number of lysates needed per experiment; therefore, volume will vary); remaining ingredients can be mixed and prepared as a stock solution in an RNase/nuclease-free tube and stored at 4 °C for up to one month.
5% milk in Tris-buffered saline (TBS)-Tween
Milk 5%, 5 mL
TBS-Tween (0.01%), 95 mL
Total: 100 mL
RIP wash/elution buffer (0.01% Tween)
Tween-20 0.01%, 1 mL
PBS, 99 mL
Total: 100 mL
Acknowledgments
Ildiko Kasza and Joshua Martin offered technical assistance. Gilson, Inc (Madison, WI) kindly shared the EXTRACTMAN device. The work was jointly supported by the Kuwait Foundation for the Advancement of Sciences (project number 2013-6302-03, awarded to SAF), Department of Defense Scholar Award (grant number W81XWH-06-1-0491, awarded to CMA), National Cancer Institute (grant number RO1CA186134, to DJB), and by pilot funding from the National Institutes of Health/National Cancer Institute (grant number P30 CA014520–University of Wisconsin Comprehensive Cancer Center Support). This protocol was originally described in Fakhraldeen et al. (2022).
Competing interests
S. M. B. holds equity in and is employed by Salus Discovery LLC, which has licensed technology described in this paper. D. J. B. holds equity in BellBrook Labs LLC, Tasso Inc, Stacks to the Future LLC, Lynx Biosciences LLC, Onexion Bio-systems LLC, and Salus Discovery LLC. All other authors declare that they have no competing interests with the content of this article.
References
- Baldini, L. and Labialle, S. (2021). Using Native RIP, UV-CLIP or fCLIP to Address Protein-RNA Interactions In Vivo. Methods Mol Biol 2300: 89-98.
- Benhalevy, D. and Hafner, M. (2020). Proximity-CLIP Provides a Snapshot of Protein-Occupied RNA Elements at Subcellular Resolution and Transcriptome-Wide Scale. Methods Mol Biol 2166: 283-305.
- Berry, S. M., Pezzi, H. M., Williams, E. D., Loeb, J. M., Guckenberger, D. J., Lavanway, A. J., Puchalski, A. A., Kityo, C. M., Mugyenyi, P. N., Graziano, F. M., et al. (2015). Using Exclusion-Based Sample Preparation (ESP) to Reduce Viral Load Assay Cost. PLoS One 10(12): e0143631.
- Busch, A., Bruggemann, M., Ebersberger, S. and Zarnack, K. (2020). iCLIP data analysis: A complete pipeline from sequencing reads to RBP binding sites. Methods 178: 49-62.
- Chatterji, P. and Rustgi, A. K. (2018). RNA Binding Proteins in Intestinal Epithelial Biology and Colorectal Cancer. Trends Mol Med 24(5): 490-506.
- Chua, B. A., Van Der Werf, I., Jamieson, C. and Signer, R. A. J. (2020). Post-Transcriptional Regulation of Homeostatic, Stressed, and Malignant Stem Cells. Cell Stem Cell 26(2): 138-159.
- Danan, C., Manickavel, S. and Hafner, M. (2016). PAR-CLIP: A Method for Transcriptome-Wide Identification of RNA Binding Protein Interaction Sites. Methods Mol Biol 1358: 153-173.
- Danan, C., Manickavel, S. and Hafner, M. (2022). PAR-CLIP: A Method for Transcriptome-Wide Identification of RNA Binding Protein Interaction Sites. Methods Mol Biol 2404: 167-188.
- Diaz-Munoz, M. D., Monzon-Casanova, E. and Turner, M. (2017). Characterization of the B Cell Transcriptome Bound by RNA-Binding Proteins with iCLIP. Methods Mol Biol 1623: 159-179.
- Dvir, S., Argoetti, A. and Mandel-Gutfreund, Y. (2018). Ribonucleoprotein particles: advances and challenges in computational methods. Curr Opin Struct Biol 53: 124-130.
- Fakhraldeen, S. A., Berry, S. M., Beebe, D. J., Roopra, A., Bisbach, C. M., Spiegelman, V. S., Niemi, N. M. and Alexander, C. M. (2022). Enhanced immunoprecipitation techniques for the identification of RNA-binding protein partners: IGF2BP1 interactions in mammary epithelial cells. J Biol Chem 298(3): 101649.
- Formicola, N., Vijayakumar, J. and Besse, F. (2019). Neuronal ribonucleoprotein granules: Dynamic sensors of localized signals. Traffic 20(9): 639-649.
- Furlan, M., de Pretis, S. and Pelizzola, M. (2021). Dynamics of transcriptional and post-transcriptional regulation. Brief Bioinform 22(4): bbaa389.
- Galgano, A. and Gerber, A. P. (2011). RNA-binding protein immunopurification-microarray (RIP-Chip) analysis to profile localized RNAs. Methods Mol Biol 714: 369-385.
- George, H., Ule, J. and Hussain, S. (2017). Illustrating the Epitranscriptome at Nucleotide Resolution Using Methylation-iCLIP (miCLIP). Methods Mol Biol 1562: 91-106.
- Guerreiro, A., Deligianni, E., Santos, J. M., Silva, P. A., Louis, C., Pain, A., Janse, C. J., Franke-Fayard, B., Carret, C. K., Siden-Kiamos, I., et al. (2014). Genome-wide RIP-Chip analysis of translational repressor-bound mRNAs in the Plasmodium gametocyte. Genome Biol 15(11): 493.
- Haecker, I. and Renne, R. (2014). HITS-CLIP and PAR-CLIP advance viral miRNA targetome analysis. Crit Rev Eukaryot Gene Expr 24(2): 101-116.
- Hentze, M. W., Castello, A., Schwarzl, T. and Preiss, T. (2018). A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol 19(5): 327-341.
- Huessler, E. M., Schafer, M., Schwender, H. and Landgraf, P. (2019). BayMAP: a Bayesian hierarchical model for the analysis of PAR-CLIP data. Bioinformatics 35(12): 1992-2000.
- Jain, R., Devine, T., George, A. D., Chittur, S. V., Baroni, T. E., Penalva, L. O. and Tenenbaum, S. A. (2011). RIP-Chip analysis: RNA-Binding Protein Immunoprecipitation-Microarray (Chip) Profiling. Methods Mol Biol 703: 247-263.
- Jens, M. (2016). A Pipeline for PAR-CLIP Data Analysis. Methods Mol Biol 1358: 197-207.
- Kinoshita, C., Kubota, N. and Aoyama, K. (2021). Interplay of RNA-Binding Proteins and microRNAs in Neurodegenerative Diseases. Int J Mol Sci 22(10): 5292.
- Lewinski, M., Bramkamp, Y., Koster, T. and Staiger, D. (2020). SEQing: web-based visualization of iCLIP and RNA-seq data in an interactive python framework. BMC Bioinformatics 21(1): 113.
- Ma, Q., Long, S., Gan, Z., Tettamanti, G., Li, K. and Tian, L. (2022). Transcriptional and Post-Transcriptional Regulation of Autophagy. Cells 11(3): 441.
- Ozer, A., Tome, J. M., Friedman, R. C., Gheba, D., Schroth, G. P. and Lis, J. T. (2015). Quantitative assessment of RNA-protein interactions with high-throughput sequencing-RNA affinity profiling. Nat Protoc 10(8): 1212-1233.
- Perconti, G., Rubino, P., Contino, F., Bivona, S., Bertolazzi, G., Tumminello, M., Feo, S., Giallongo, A. and Coronnello, C. (2019). RIP-Chip analysis supports different roles for AGO2 and GW182 proteins in recruiting and processing microRNA targets. BMC Bioinformatics 20(Suppl 4): 120.
- Pezzi, H. M., Guckenberger, D. J., Schehr, J. L., Rothbauer, J., Stahlfeld, C., Singh, A., Horn, S., Schultz, Z. D., Bade, R. M., Sperger, J. M., et al. (2018). Versatile exclusion-based sample preparation platform for integrated rare cell isolation and analyte extraction. Lab Chip 18(22): 3446-3458.
- Shi, Q., Lee, D. Y., Feliers, D., Abboud, H. E., Bhat, M. A. and Gorin, Y. (2020). Interplay between RNA-binding protein HuR and Nox4 as a novel therapeutic target in diabetic kidney disease. Mol Metab 36: 100968.
- Spitzer, J., Hafner, M., Landthaler, M., Ascano, M., Farazi, T., Wardle, G., Nusbaum, J., Khorshid, M., Burger, L., Zavolan, M., et al. (2014). PAR-CLIP (Photoactivatable Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation): a step-by-step protocol to the transcriptome-wide identification of binding sites of RNA-binding proteins. Methods Enzymol 539: 113-161.
- Sutandy, F. X., Hildebrandt, A. and Konig, J. (2016). Profiling the Binding Sites of RNA-Binding Proteins with Nucleotide Resolution Using iCLIP. Methods Mol Biol 1358: 175-195.
- Theil, K., Herzog, M. and Rajewsky, N. (2018). Post-transcriptional Regulation by 3' UTRs Can Be Masked by Regulatory Elements in 5' UTRs. Cell Rep 22(12): 3217-3226.
- Wang, Z. L., Li, B., Luo, Y. X., Lin, Q., Liu, S. R., Zhang, X. Q., Zhou, H., Yang, J. H. and Qu, L. H. (2018). Comprehensive Genomic Characterization of RNA-Binding Proteins across Human Cancers. Cell Rep 22(1): 286-298.
- Weskamp, K., Olwin, B. B. and Parker, R. (2021). Post-Transcriptional Regulation in Skeletal Muscle Development, Repair, and Disease. Trends Mol Med 27(5): 469-481.
- Zhao, W., Zhang, S., Zhu, Y., Xi, X., Bao, P., Ma, Z., Kapral, T. H., Chen, S., Zagrovic, B., Yang, Y. T., et al. (2022). POSTAR3: an updated platform for exploring post-transcriptional regulation coordinated by RNA-binding proteins. Nucleic Acids Res 50(D1): D287-D294.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Fakhraldeen, S. A., Berry, S. M., Beebe, D. J. and Alexander, C. M. (2022). Enhanced Ribonucleoprotein Immunoprecipitation (RIP) Technique for the Identification of mRNA Species in Ribonucleoprotein Complexes. Bio-protocol 12(19): e4526. DOI: 10.21769/BioProtoc.4526.
- Fakhraldeen, S. A., Berry, S. M., Beebe, D. J., Roopra, A., Bisbach, C. M., Spiegelman, V. S., Niemi, N. M. and Alexander, C. M. (2022). Enhanced immunoprecipitation techniques for the identification of RNA-binding protein partners: IGF2BP1 interactions in mammary epithelial cells. J Biol Chem 298(3): 101649.
Category
Cell Biology > Cell-based analysis
Cancer Biology > General technique > Biochemical assays > Other compound
Biological Engineering > Biomedical engineering
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.