- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CRISPR/Cas9-mediated LRP10 Knockout in HuTu-80 and HEK 293T Cell Lines
Published: Vol 12, Iss 19, Oct 5, 2022 DOI: 10.21769/BioProtoc.4521 Views: 3956
Reviewed by: Xi FengAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2021

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Loss-of-function (LoF) variants in the low-density lipoprotein receptor–related protein 10 gene (LRP10) have been recently implicated in the development of neurodegenerative diseases, including Parkinson's disease (PD), PD dementia (PDD), and dementia with Lewy bodies (DLB). However, despite the genetic evidence, little is known about the LRP10 protein function in health and disease. Here, we describe a detailed protocol to efficiently generate a LRP10 LoF model in two independent LRP10-expressing cell lines, HuTu-80 and HEK 293T, using the CRISPR/Cas9 genome-editing tool. Our method efficiently generates bi-allelic LRP10 knockout (KO), which can be further utilized to elucidate the physiological LRP10 protein function and to model some aspects of neurodegenerative disorders.
Graphical abstract:
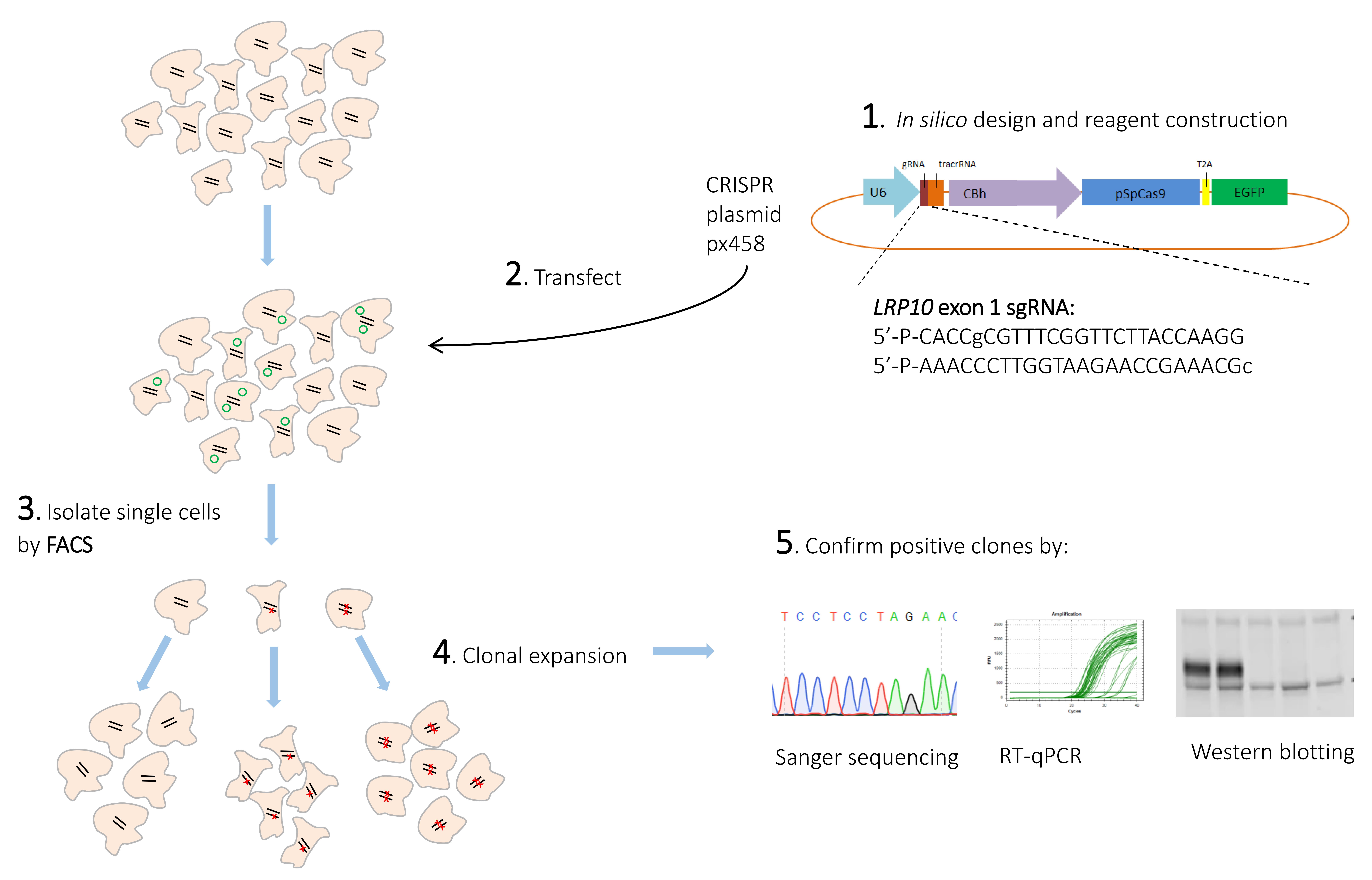
CRISPR/Cas9 workflow for the generation of the LRP10 KO. (1) Designed single guide RNA (sgRNA) is cloned into CRISPR/Cas9 px458 plasmid. (2) Cells are transfected with the CRISPR/Cas9 plasmid containing sgRNA. (3) Two days post transfection, cells are dissociated and sorted as single cells by fluorescence-activated cell sorting (FACS). (4) After several weeks, expanded clonal lines are (5) verified with Sanger sequencing for the presence of INDELs (insertions or deletions), RT-qPCR for the amounts of LRP10 mRNA transcript, and Western blotting for the analysis of the LRP10 protein levels.
Background
Rare, pathogenic variants in the low–density lipoprotein–related protein 10 gene (LRP10) have been identified in patients with familial Parkinson’s disease (PD), PD dementia (PDD), and dementia with Lewy bodies (DLB) (Quadri et al., 2018). Moreover, postmortem analysis of patients carrying distinct LRP10 variants showed a severe burden of alpha-synuclein–associated pathology in the form of Lewy bodies (LB) and Lewy neurites (LN) in the brain stem, limbic, and cortical regions (Quadri et al., 2018). Importantly, functional studies revealed that initially identified LRP10 pathogenic variants affected either LRP10 transcript expression and stability, protein stability, or protein localization, pointing to loss-of-function (LoF) as a shared pathogenic mechanism (Quadri et al., 2018). In addition, earlier studies using LRP10 overexpression models reported that LRP10 participates in intracellular trafficking pathways (Boucher et al., 2008; Brodeur et al., 2009, 2012; Doray et al., 2008). Despite these data, little is known about the endogenous LRP10 function in health and disease, partly due to the lack of in vitro LRP10 knockout (KO) models.
The CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 system is a powerful and precise method for editing the genome in various cell types (Jinek et al., 2012; Cong et al., 2013; Adli, 2018). Gene KO models are often critical for identifying the function of the protein that a particular gene encodes. In the CRISPR/Cas9 system, the endonuclease Cas9 is guided to a specific site in the genome by a single guide RNA (sgRNA) to generate a double-strand break (Adli, 2018). The break is fixed in a process called non-homologous end-joining (NHEJ), which results in INDELs (insertion or deletion) (Sander and Joung, 2014). The introduction of INDELs leads to changes in the reading frame, which disrupts mRNA and protein expression (Sander and Joung, 2014).
Here, we show a step-by-step protocol to efficiently generate a LRP10 KO in the epithelial human cell lines HuTu-80 and HEK 293T using the CRISPR/Cas9 genome-editing tool. Our approach efficiently targeted the first exon of the LRP10 gene using a sgRNA cloned into a plasmid containing Cas9 from Streptococcus pyogenes. We obtained homozygous and compound heterozygous independent LRP10 KO clones carrying INDEL mutations leading to a frameshift and a premature stop codon. These models were used to test the specificity of commercially available and in-house developed antibodies against the endogenously expressed LRP10 protein (Grochowska et al., 2021). Interestingly, HuTu-80 cells highly express both LRP10 and alpha-synuclein, making the LRP10 KO in HuTu-80 cells a suitable model for studying the potential link between LRP10 LoF and alpha-synuclein accumulation in PD, PDD, and DLB. Lastly, given the high targeting efficiency of the LRP10 locus in both cell lines, this protocol holds promise for the generation of LRP10 KO in more relevant in vitro models of neurodegeneration, such as human stem cell–derived neural progenitors that can be differentiated into neurons and glia.
Materials and Reagents
6-well cell culture plates (TC-plate, standard F, Sarstedt, catalog number: 83.3920.005)
48-well cell culture plates (TC-treated, F-bottom, CorningTM CostarTM, catalog number: 3548)
96-well cell culture plates (F-bottom with lid, Greiner Bio-One, catalog number: 655180)
FalconTM round-bottom polystyrene test tubes with cell strainer snap cap, 5 mL (FalconTM, catalog number: 352235)
10 cm Petri dishes (sterile, VWR)
Parafilm® M (Sigma-Aldrich, catalog number: P7793)
DMEM (Sigma-Aldrich, catalog number: D6429), store at 4 °C
DMEM/F-12 (GibcoTM, catalog number: 11320033), store at 4 °C
Fetal bovine serum (FBS, GibcoTM, catalog number: A5256701), aliquot and store at -20 °C
Trypsin–EDTA (0.05%), phenol red (GibcoTM, catalog number: 25300054), aliquot and store at -20 °C
Penicillin–streptomycin (10,000 U/mL, GibcoTM, catalog number: 15140122), aliquot and store at -20°C
DPBS, no calcium and no magnesium (DPBS-/-, GibcoTM, catalog number: 14190144), store at room temperature
pSpCas9-(BB)-2A-GFP (Addgene plasmid #48138; http://n2t.net/addgene:48138; RRID: Addgene_48138)
BbsI (10,000 units/mL; NEB, catalog number: R0539S), aliquot and store at -80 °C
NEBufferTM r2.1 (NEB, catalog number: B7030S), store at 4 °C
T4 DNA ligase (400,000 units/mL, NEB, catalog number: M0202S), store at -20 °C
T4 DNA ligase reaction buffer (NEB, catalog number: B0202S), store at -20 °C
TE buffer solution 1× (TRIS–EDTA buffer) pH 8,0 (VWR, J75793.AE), store at room temperature
Ampicillin (Sigma-Aldrich, 69-52-3)
BD BACTOTM agar (BD, catalog number: 214010)
Tryptone (Millipore, catalog number: T7293)
Yeast extract (Sigma-Aldrich, catalog number: Y1625)
NaCl (Sigma-Aldrich, catalog number: S7653)
Trizma® base (Sigma-Aldrich, catalog number: TRIS-RO)
cOmpleteTM (Roche, catalog number: 11836145001)
Pefabloc® SC (Roche, catalog number: 11585916001)
IGEPAL® CA-630 (Sigma-Aldrich, catalog number: 18896)
SDS (Sigma-Aldrich, catalog number: 71729)
Bromophenol blue (Thermo Fisher Scientific, catalog number: A18469.18)
Glycerol (Sigma-Aldrich, catalog number: G5516)
DTT (Dithiothreitol, Thermo Fisher Scientific, catalog number: R0862)
TWEEN® 20 (Sigma-Aldrich, catalog number: P1379)
Agarose powder (Sigma, catalog number: A9539)
GeneJuice® transfection reagent (Merck Millipore, catalog number: 70967), store at 4 °C
Hoechst 33342, trihydrochloride, trihydrate, 10 mg/mL solution in water (InvitrogenTM, catalog number: H3570)
One ShotTM TOP10 chemically competent E. coli (InvitrogenTM, catalog number: C404010), store at -80 °C
HuTu-80 cells (CLS, catalog number: 330218), expand and cryopreserve cell stocks in liquid nitrogen
HEK 293T (ATCC® CRL-3216TM), expand and cryopreserve cell stocks in liquid nitrogen
DMSO (Sigma-Aldrich, catalog number: W387509), aliquot and store at -20 °C
Sanger sequencing kit (BigDye Terminator v3.1 Cycle Sequencing Kit and ExoSAP-IT, A38073), store the components according to the manufacturer’s specifications
NucleoBond PC, mini kit for transfection-grade plasmid DNA (MACHEREY-NAGEL, REF 740571.100), store the components according to the manufacturer’s specifications
Blood & cell culture DNA mini kit (QIAGEN, catalog number: 13323), store the components according to the manufacturer’s specifications
RNeasy mini kit (QIAGEN, catalog number: 74004), store the components according to the manufacturer’s specifications
SuperScript® III first-strand synthesis system for RT-PCR (Invitrogen, catalog number: 18080-051), store the components according to the manufacturer’s specifications
iTaqTM Universal SYBR® green supermix (Bio-Rad, catalog number: 172-5121), store the components according to the manufacturer’s specifications
4–15% Criterion TGX precast midi protein gel (Bio-Rad, catalog number: 5671085), store at 4 °C
Trans-blot turbo midi 0.2 µm nitrocellulose membranes (Bio-Rad, catalog number: 1704159), store at 4 °C
Blotto, non-fat dry milk (Santa Cruz, catalog number: sc-2325), store at room temperature
Donkey anti-rabbit IgG (H+L), Alexa FluorTM Plus 800 (Thermo Fisher Scientific, catalog number: A32808)
Alexa Fluor® 680 AffiniPure donkey anti-sheep IgG (H+L) (Jackson ImmunoResearch, catalog number: 713-625-147)
1× PBS, store at room temperature
Growth media for HuTu-80 cells (see Recipes), store at 4 °C
Growth media for HEK 293T cells (see Recipes), store at 4 °C
Lysogeny broth (LB; Miller formulation, see Recipes), store at 4 °C
LB agar plates (see Recipes), store at 4 °C
10× Tris-buffered saline (TBS) solution (see Recipes)
Protein lysis buffer, store at 4 °C (see Recipes)
4× sample buffer, store at 4 °C (see Recipes)
Equipment
Thermal cycler (Bio-Rad, model: C1000TouchTM)
Horizontal gel electrophoresis system
Molecular Imager® GelDoc XR System (Bio-Rad)
3790 series genetic analyzer (Thermo Fisher Scientific)
Benchtop orbital incubator shaker (New Brunswick Scientific, model: Innova® 40/40R)
Eppendorf centrifuge 5810 equipped with the A-4-81 rotor (Eppendorf)
Cell culture CO2 incubator (Sanyo, model: MCO-19AIC)
Benchtop EVOS M5000 imaging system (AMF5000) equipped with a GFP light cube (AMEP4951)
Water bath (Precision – CIR 35, Fisher Scientific)
High sensitivity flow cytometer BD FACSAriaTM III (BD Biosciences)
CFX Opus 96 Real-Time PCR (Bio-Rad)
Heraeus FrescoTM 17 microcentrifuge equipped with 24 × 1.5/2.0 mL rotor with ClickSealTM biocontainment lid (Thermo Scientific)
Trans-Blot® TurboTM transfer system (Bio-Rad)
Odyssey CLx imaging system (LI-COR Biosciences)
Software
CHOPCHOP (version 3, https://chopchop.cbu.uib.no/)
TIDE: Tracking of Indels by Decomposition (http://shinyapps.datacurators.nl/tide/)
CFX Maestro Software for CFX Real-Time PCR Instruments (Bio-Rad)
Image StudioTM Lite Ver 5.2 (LI-COR Biosciences)
Procedure
sgRNA in silico design for the LRP10 locus
Determine the optimal CRISPR/Cas9 target sites using the CHOPCHOP web tool (http://chopchop.cbu.uib.no) (Labun et al., 2019) or another preferable CRISPR/Cas9 design tool. Pick sgRNA (20 bp) with the highest predicted efficiency and the lowest predicted off-targets. The sequence of the optimal sgRNA is used to design sense and antisense oligonucleotides to clone the sgRNA into pSpCas9-(BB)-2A-GFP (see step 2). We recommend testing the targeting efficiency of several sgRNA designed against different exons of the gene of interest.
To clone sgRNA into pSpCas9-(BB)-2A-GFP (Ran et al., 2013), order sense and antisense oligonucleotides (Integrated DNA Technologies) with proper BbsI overhangs. These oligonucleotides need to be phosphorylated at the 5’ ends. Importantly, the pSpCas9-(BB)-2A-GFP allows the expression of the sgRNA under the control of the human U6 promoter, which requires a “G” base at the transcription start site. Therefore, add “G” at the start of the sgRNA sequence. For the LRP10 KO, we used sgRNA targeting the first exon of the LRP10 gene (ordered oligonucleotides: 5′-P-CACCGCGTTTCGGTTCTTACCAAGG and 5′-P-AAACCCTTGGTAAGAACCGAAACGC). The target genomic sequence for the generation of the LRP10 KO was chosen based on the score of the online CHOPCHOP tool and the prior testing of multiple sgRNA targeting different exons of the LRP10 gene.
Oligo design template:
sgRNA-oligo-FW: 5’-P-CACC(G)NNNNNNNNNNNNNNNNNNNN-3’
sgRNA-oligo-RV: 5’-P-AAACNNNNNNNNNNNNNNNNNNNN(C)-3’
P, phosphorylated
Cloning of sgRNA into pSpCas9-(BB)-2A-GFP plasmid
Oligonucleotide annealing. Adjust the concentration of the oligonucleotides to 100 µM using sterile demineralized water (dH2O) or TE buffer and combine in the annealing reaction (Table 1). Incubate the reaction in the thermal cycler at 95 °C for 5 min. Next, ramp down to 25 °C at 5 °C/min. The annealed product can be stored at -20 °C for several months.
Table 1. Components of oligonucleotide annealing reaction
Reagent Amount (µL) Final concentration sgRNA-oligo-FW 1 10 µM sgRNA-oligo-RV 1 10 µM 10× T4 ligation buffer 1 1× dH2O 7 - Total volume 10 Set up the restriction enzyme digestion for pSpCas9-(BB)-2A-GFP plasmid with BbsI enzyme (Table 2). Incubate for 90 min at 37 °C. Inactivate the restriction enzyme for 20 min at 65 °C. Load 3 µL of the digested vector on 0.8% agarose gel to check the restriction reaction efficiency. BbsI enzyme cuts the plasmid at two positions, removing 22 base pairs from the plasmid. The uncut plasmid DNA on the agarose gel has a multiple-band pattern (three bands running above 7 kbp) due to several plasmid conformations. The BbsI-digested plasmid DNA shows a single-band pattern running around 9 kbp.
Note: Typically, high cutting efficiency of BbsI is achieved and, therefore, purification of the linearized vector from the gel is not required.
Table 2. pSpCas9-(BB)-2A-GFP digestion setup
Reagent Amount (µL) Final concentration 1 µg pSpCas9-(BB)-2A-GFP 1 50 ng/µL BbsI 1 1,000 units NEBufferTM r2.1 2 1× dH2O 16 - Total volume 20 Set up the ligation reaction (Table 3). Before the ligation, dilute annealed oligonucleotides 1:250 in sterile dH2O to a final concentration of 40 nM. Incubate the ligation reaction at 16 °C overnight or at room temperature for 10 min. Heat inactivate at 65 °C for 10 min. Chill on ice and transform 1–5 μL of the reaction into 50 μL competent cells according to your in-house bacterial transformation protocol. Plate the transformed bacteria on LB agar plates containing 50 µg/mL of ampicillin (see Recipes). Place plates overnight in a 37 °C incubator.
Table 3. Ligation reaction setup
Reagent Amount (µL) Final concentration Linearized pSpCas9-(BB)-2A-GFP X ~50 ng Annealed oligonucleotides (1:250 dilution) 2 8 nM T4 DNA ligase 0.5 20,000 units 10× T4 ligation buffer 1 1× dH2O Fill with dH2O to the total volume of 10 µL - Total volume 10 Next day, pick the colonies (at least six) and inoculate them in a 3 mL bacterial growth medium (LB) containing 50 µg/mL of ampicillin at 37 °C and 200 rpm overnight. Next, perform plasmid mini preparations using a NucleoBond PC mini kit following the manufacturer’s specifications or an in-house protocol. Verify correct clones by Sanger sequencing using a hU6-FW primer (5’- GAGGGCCTATTTCCCATGATT) with BigDye Terminator v3.1 Cycle Sequencing Kit or an in-house protocol. Select one correct clone to proceed to the next step.
Note: Make sure that your plasmid DNA is transfection-grade. DNA preparations obtained from the NucleoBond PC mini kit are suited for molecular-grade experiments and can be directly used for cell transfections.
Cell culture and transfection
Maintain HuTu-80 cells in growth medium (see Recipes) at 37 °C and 5% CO2. Split the cells when 80% confluent. Maintain HEK 293T cells in growth medium (see Recipes) at 37 °C and 5% CO2. Split the cells when 80% confluent.
The day before transfection, seed the following quantities of cells in one well of a 6-well plate: 0.5 million HEK 293T cells and 0.3 million HuTu-80 cells. The next day, cells will reach 30%–40% confluence and are ready for transfection.
Transfect the cells in one well of a 6-well plate. Perform the transfection with pSpCas9-(BB)-2A-GFP containing sgRNA using GeneJuice® transfection reagent according to the manufacturer’s specifications.
Forty-eight hours later, check the transfection efficiency using a benchtop imaging system equipped with the appropriate light cube to visualize GFP-positive cells. Transfected cells express GFP. Transfection efficiency in HuTu-80 cells is low to moderate (10%–30%). Transfection efficiency in HEK 293T cells is high (approximately 80%).
Prepare the transfected cells for fluorescence-activated cell sorting (FACS) 48 h post-transfection.
96-well plate preparation
On the day of FACS, prepare three 96-well plates (flat bottom) by adding 150 µL of HuTu-80 or HEK 293T growth medium to each well.
Incubate the plates before FACS at 37 °C and 5% CO2 for at least 30 min to equilibrate the medium.
FACS
For single-cell sorting, aspirate the medium from the well of the 6-well plate and wash it once with DPBS-/-.
Add 1 mL of trypsin–EDTA to the well and incubate it at 37 °C and 5% CO2 for 5 min.
Add 1 mL of growth medium to the well, gently resuspend to dislodge the cells from the plate, and transfer the cells to a 15 mL Falcon tube.
Spin the cells at 1,000 rpm (198 × g) for 5 min.
Aspirate and discard the supernatant. Next, resuspend the cell pellet in 1 mL of DPBS-/- containing 4% FBS.
Transfer 1 mL of the cell solution to a FalconTM round-bottom polystyrene test tube with a cell strainer snap cap.
Add 1 µL of sterile Hoechst 33342 solution to label dead cells and mix gently by inverting the tube.
Sort GFP-positive cells into three 96-well plates (1 cell per well) by gating out doublets (forward and sideward scatter-based) and dead cells (Hoechst 33342 positive cells). Select the population of cells with high GFP expression. Figure 1 represents the FACS settings on BD FACSAriaTM III for CRISPR/Cas9-mediated LRP10 KO in HuTu-80 cells. Optionally, include a negative control sample (non-transfected and non-Hoechst–stained cells) to optimally determine the gating of the positive cell population.
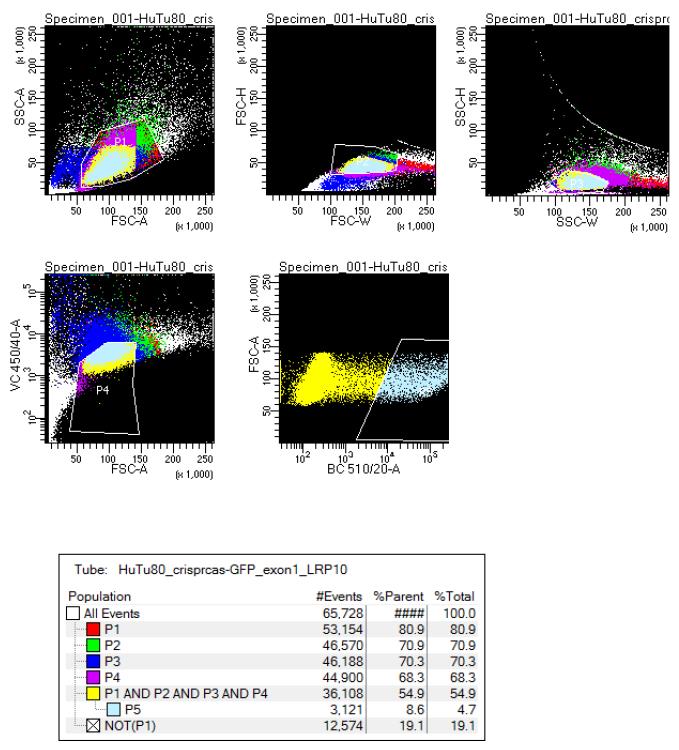
Figure 1. Example of FACS sorting gating settings for CRISPR/Cas9-mediated KO in HuTu-80 cell line.
Expansion of single cells post-sorting
Immediately after sorting, incubate plates at 37 °C and 5% CO2 for 72 h.
Perform the first medium change with 150 µL of growth medium.
Perform subsequent media changes every 72 h until colonies are observed under the light microscope.
The colonies start to emerge 14 days after sorting. You can expect a minimum of 10 clones from one 96-well plate for the HuTu-80 cells. You can expect a minimum of 30 clones from one 96-well plate for the HEK 293T cells.
When colonies reach 80% confluence, aspirate the media from the 96-well plates, wash with DPBS-/-, add 50 µL of trypsin–EDTA per well, and incubate at 37 °C and 5% CO2 for 5 min.
Next, add 150 µL of growth media per well, resuspend to dislodge the cells, and transfer each clone to separate 15 mL Falcon tubes. Spin at 1,000 rpm (198 × g).
Discard the supernatant and resuspend the cell pellet of each clone in fresh 150 µL of growth media. Add one-half (75 µL) of the cell suspension to a new well of a 48-well plate (PCR processing plate) and fill that well with an additional 225 µL of growth media. Add another half (75 µL) of the cell suspension to a new 48-well plate (propagation plate) and fill that well with an additional 225 µL of growth media.
Processing of plates and verification of positive clones
PCR processing plate. Grow clones until they reach 80% confluence. When ready, extract genomic DNA with an in-house protocol or using a blood & cell culture DNA mini kit. Amplify the DNA fragment of interest by PCR using primers flanking LRP10 exon 1 (FW: CAAAGTTTGGCCCGAAGAGG, RV: GGGCAGGCAGGATAGAGTGC), and perform Sanger sequencing following your in-house protocols. Perform INDEL tracking manually or with a freely available web tool (TIDE, http://shinyapps.datacurators.nl/tide/) to identify targeted clones (Brinkman et al., 2014).
Propagation plate. Grow clones until they reach 80% confluence and then cryopreserve as follows: aspirate the media, wash with DPBS-/-, add 50 µL of trypsin–EDTA per well, and incubate at 37 °C and 5% CO2 for 5 min. To each well, add 300 µL of growth media containing 10% DMSO. Wrap the plates with ParafilmTM wrapper and store them in a zippered plastic bag in the freezer at -80 °C. The cells will stay frozen during the screening. As you identify positive clones, thaw and expand the desired clones according to your in-house protocol. Additionally, thaw and expand three unedited clones (LRP10 WT) that underwent the same genome-editing procedure. These clones can be used as a control condition for the downstream experiments. The amount of LRP10 at the transcript and protein level for each clone can be further characterized by RT-qPCR and Western blotting. Below, we briefly describe the recommendations and kits used for the RT-qPCR and Western blotting experiments.
RT-qPCR
Recommendations
For RT-qPCR, we recommend the following primer sets for the LRP10:
Set 1 (LRP10-N-terminal): FW: ACTGCACCTGGCTCATCC and RV: GAGATCAGTGGCTGGAGAGG
Set 2 (LRP10-C-terminal): FW: CCAGGAGTACAGCATCTTTGC and RV: TAGCAGAGAACGCAGGTTGC
Relative LRP10 mRNA levels can be determined after normalization to the geometric mean of the following housekeeping genes:
CLK2 (FW: TCGTTAGCACCTTAGGAGAGG, RV: TGATCTTCAGGGCAACTCG)
COPS5 (FW: CCAGGAACCATTTGTAGCAG, RV: GTAGCCCTTTGGGTATGTCC)
RNF10 (FW:GCATCTGTGAACTGGCTTTG, RV: CTGACGTTTCCTCTTCTCAATG)
Procedure
Extract RNA from cells using RNeasy mini kit following the manufacturer’s specifications.
For each sample (clonal line), perform first-strand cDNA synthesis with random hexamers using SuperScript® III first-strand synthesis system, followed by RNase H digestion. Follow the manufacturer’s specifications. Use 1 µg of RNA per cDNA reaction.
Next, use iTaqTM Universal SYBR® green supermix to prepare RT-qPCR reactions. Follow the manufacturer’s specifications. For each cDNA sample, prepare five separate reactions (five different primer sets). Each reaction contains iTaqTM Universal SYBR® green supermix, a primer set, and 100 ng of cDNA. Each reaction should be performed in triplicate (technical replicates for the RT-qPCR).
Load the PCR tubes or plate into the RT-PCR instrument and start the PCR run. Our thermal cycling protocol is optimized for CFX Opus 96 Real-Time PCR with the following conditions: 3 min at 95 °C (initial denaturation), 40 cycles of 5 s at 95 °C, and 30 s at 60 °C.
The relative normalized LRP10 mRNA expression can be quantified using CFX Maestro Software for CFX Real-Time PCR Instruments.
Western blotting
Recommendations
For Western blotting, we recommend using the LRP10-NT (1-440 aa) (Sino Biological, 13228-T16) and LRP10-CT (463-713 aa) (MRC PPU Reagents, DA058) antibodies at the concentration of 1:500 (Grochowska et al., 2021). We recommend the following fluorescently conjugated secondary antibodies: donkey anti-rabbit IgG (H+L) highly cross-adsorbed secondary antibody, Alexa FluorTM Plus 800 (Thermo Fisher Scientific, A32808), and Alexa Fluor® 680 AffiniPure donkey anti-sheep IgG (H+L) (Jackson ImmunoResearch, 713-625-147) at the concentration of 1:1,000.
Procedure
Before protein lysis, wash the cells once with 1× TBS (see Recipes). Subsequently, add the protein lysis buffer (see Recipes) that contains freshly added protease inhibitors, 1× cOmpleteTM, and 1 mM Pefabloc® SC.
Snap-freeze the lysates. Next, thaw the lysates and clear them by centrifugation at 13,000 rpm (1,628 × g) and 4 °C for 10 min.
Mix the lysates with 4× sample buffer supplemented with 100 mM DTT and incubate for 10 min at 95 °C.
Separate the proteins on 4%–15% Criterion TGX precast gel. Transfer the proteins to the trans-blot turbo midi 0.2 µm nitrocellulose membrane using the Trans-Blot® TurboTM transfer system.
Block the membrane for 1 h at room temperature using the blocking buffer [5% non-fat dry milk (Blotto) and 0.1% v/v TWEEN® 20 in 1× TBS].
Perform primary antibody incubation overnight at 4 °C in blocking buffer.
Wash the membrane three times for 5 min with 1× TBS containing 0.1% v/v TWEEN® 20.
Perform secondary antibody incubation for 1 h at room temperature with fluorescently conjugated secondary antibodies in blocking buffer.
Wash the membrane three times for 5 min with 1× TBS containing 0.1% v/v TWEEN® 20.
Image the blots with Odyssey CLx imaging system. Images can be analyzed with Image StudioTM Lite Ver 5.2.
Notes
After CRISPR/Cas9 genome editing, we extracted DNA from 43 clonally expanded HEK 293T lines, performed PCR using primers flanking LRP10 exon 1, and Sanger-sequenced the obtained PCR products. Using the TIDE web tool (Brinkman et al., 2014), we decomposed electropherograms of individual clones to track INDELs located at the expected double-stranded break site and subsequently genotyped all clonal lines. We demonstrate that our CRISPR/Cas9 approach targeted both alleles in 58.1% of the clonally expanded HEK 293T lines (Figure 2A; homozygous and compound heterozygous clones combined). From all analyzed clones, 4.7% were not targeted, 2.3% were targeted on one of the alleles, and 34.9% were predicted to carry more than two INDELs (Figure 2A). One base pair insertion was the most common mutation introduced after the double-stranded break repair with NHEJ (Figure 2B). Importantly, the insertion of one base pair leads to a frameshift and often creates a premature stop codon. The designed sgRNA also strongly prefers to insert thymidine over other DNA nucleotides (Figure 2C, D, E). The 1 bp homozygous insertion in HEK 293T (KO-1 and KO-2, Figure 2D) and HuTu-80 (KO-1 and KO-2, Figure 2E) was predicted to lead to a frameshift and a premature stop codon at the position p.Gly12Trp fs*18. Similarly, the compound heterozygous mutation with a 1 bp insertion on one DNA strand and 2 bp deletion on the other DNA strand in HEK 293T and HuTu-80 clones was predicted to lead to a frameshift and a premature stop codon at the positions p.Gly12Trp fs*18/p.Gly12Arg fs*17 (KO-3, Figure 2D and E).
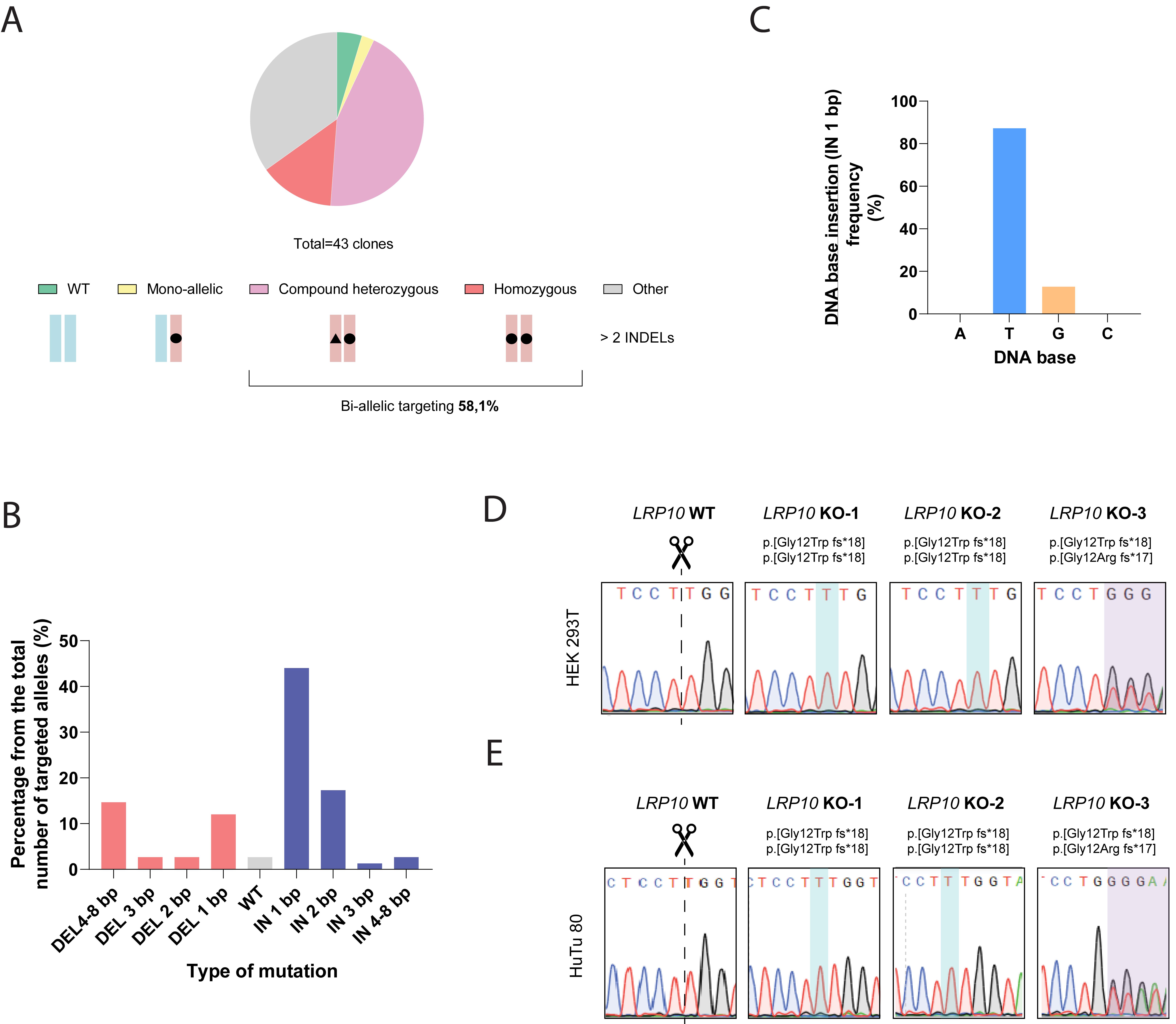
Figure 2. CRISPR/Cas9-mediated LRP10 KO is highly efficient in HEK 293T and HuTu-80 cell lines. (A) LRP10 targeting efficiencies in clonally expanded HEK 293T lines. Genotypes derived from 43 clonal lines were analyzed. WT, unedited LRP10 sequence. (B) Percentage of each mutation type (DEL: deletion; IN: insertion; bp: base pair) from the total number of targeted alleles. (C) Frequency of A, T, G, and C insertions analyzed in all “1 bp insertion” (IN 1 bp) alleles. (D), (E) Electropherograms of LRP10 DNA sequences from three independent clones showing bi-allelic targeting in HEK 293T (D) and HuTu-80 (E) cell lines. Sanger sequencing analysis confirms on-target mutagenesis with alleles containing INDELs. Clone 1 (KO-1) and 2 (KO-2) carry an insertion of a thymidine (c.33dupT) and clone 3 (KO-3) carries an insertion and deletion on two different alleles (c.33dupT and c.32_33insG). All three genotypes are predicted to induce frameshifts leading to a premature stop codon (p.Gly12Trp fs*18 or p.Gly12Arg fs*17).
Recipes
Growth media for HuTu-80 cells
DMEM/F-12, 445 mL
FBS 10%, 50 mL
Penicillin–streptomycin 1%, 5 mL
Total: 500 mL
Growth media for HEK 293T cells
DMEM, 445 mL
FBS 10%, 50 mL
Penicillin-streptomycin 1%, 5 mL
Total: 500 mL
LB (Miller formulation)
Tryptone 1% (w/v), 10 g
Yeast extract 0.5% (w/v), 5 g
NaCl 1% (w/v), 10 g
dH2O, add up to 1 L
Prepare liquid LB medium, followed by autoclaving (120 °C) and cooling down to room temperature.
LB agar plates
BD BACTOTM agar, 7.5 g
LB (Miller formulation), 400 mL
Mix and autoclave at 120 °C. When cooled down to approximately 55 °C, add 50 µg/mL of ampicillin. Prepare Petri dishes on a sterile bench. Carefully pour a thin layer of solution into the Petri dishes (approximately 15 mL per plate). Avoid creating bubbles. Leave the plates to set before storage in the fridge. Plates can be kept in the fridge for a maximum of two weeks.
10× Tris-buffered saline (TBS) solution
Trizma® base, 24 g
NaCl, 88 g
Dissolve in 900 mL dH2O
Adjust pH to 7.6 with HCl
Adjust the final volume to 1 L with dH2O
Protein lysis buffer
1 M Tris-Cl (pH 7.4, stock made from Trizma® base) 50 mM, 25 mL
NaCl 100 mM, 2.92 g
IGEPAL® CA-630 1.0%, 5 mL
Add dH2O up to 500 mL
Store in the fridge. Before use, add 1× cOmplete and 1 mM Pefabloc® SC from the stocks. To prepare a 25× cOmpleteTM stock, dissolve one cOmpleteTM tablet in 2 mL of deionized water or 10 mM phosphate buffer, pH 7.0. The stock solution is stable for at least 12 weeks at -15 to -25 °C. To prepare 100 mM Pefabloc® SC, dissolve 100 mg of Pefabloc® SC in 4.18 mL of deionized water. The concentrated 100 mM stock solution prepared in deionized water is acidic and stable for 1–2 months at -15 to -25 °C if stored in aliquots.
4× sample buffer
1 M Tris-Cl (pH 6.8, stock made from Trizma® base), 3.125 mL
20% SDS solution, 20 mL
Glycerol (v/v), 10 mL
Bromophenol blue, 2 mg
Make 800 µL aliquots and store them in the fridge. Before use, add 200 µL of 1 M DTT to obtain 4× sample buffer. To prepare a 1 M DTT solution, dissolve 1.55 g of DTT powder in 10 mL of deionized water.
Acknowledgments
This work was supported by research grants from the Stichting ParkinsonFonds (Netherlands) and Alzheimer Nederland. pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Addgene plasmid # 48138; http://n2t.net/addgene:48138; RRID:Addgene_48138).
Competing interests
The authors declare no conflicts of interest or competing interests.
References
- Adli, M. (2018). The CRISPR tool kit for genome editing and beyond. Nat Commun 9(1): 1911.
- Boucher, R., Larkin, H., Brodeur, J., Gagnon, H., Thériault, C. and Lavoie, C. (2008). Intracellular trafficking of LRP9 is dependent on two acidic cluster/dileucine motifs.Histochem Cell Biol 130(2): 315-327.
- Brodeur, J., Larkin, H., Boucher, R., Theriault, C., St-Louis, S. C., Gagnon, H. and Lavoie, C. (2009). Calnuc binds to LRP9 and affects its endosomal sorting. Traffic 10(8): 1098-1114.
- Brodeur, J., Theriault, C., Lessard-Beaudoin, M., Marcil, A., Dahan, S. and Lavoie, C. (2012). LDLR-related protein 10 (LRP10) regulates amyloid precursor protein (APP) trafficking and processing: evidence for a role in Alzheimer's disease. Mol Neurodegener 7: 31.
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A. and Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121): 819-823.
- Doray, B., Knisely, J. M., Wartman, L., Bu, G. and Kornfeld, S. (2008). Identification of acidic dileucine signals in LRP9 that interact with both GGAs and AP-1/AP-2.Traffic 9(9): 1551-1562.
- Grochowska, M. M., Carreras Mascaro, A., Boumeester, V., Natale, D., Breedveld, G. J., Geut, H., van Cappellen, W. A., Boon, A. J. W., Kievit, A. J. A., Sammler, E., et al. (2021). LRP10 interacts with SORL1 in the intracellular vesicle trafficking pathway in non-neuronal brain cells and localises to Lewy bodies in Parkinson's disease and dementia with Lewy bodies. Acta Neuropathol 142(1): 117-137.
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096): 816-821.
- Labun, K., Montague, T. G., Krause, M., Torres Cleuren, Y. N., Tjeldnes, H. and Valen, E. (2019). CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res 47(W1): W171-W174.
- Quadri, M., Mandemakers, W., Grochowska, M. M., Masius, R., Geut, H., Fabrizio, E., Breedveld, G. J., Kuipers, D., Minneboo, M., Vergouw, L. J. M., et al. (2018). LRP10 genetic variants in familial Parkinson's disease and dementia with Lewy bodies: a genome-wide linkage and sequencing study. Lancet Neurol 17(7): 597-608.
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A. and Zhang, F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8(11): 2281-2308.
- Sander, J. D. and Joung, J. K. (2014). CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32(4): 347-355.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Grochowska, M. M., Bonifati, V. and Mandemakers, W. (2022). CRISPR/Cas9-mediated LRP10 Knockout in HuTu-80 and HEK 293T Cell Lines. Bio-protocol 12(19): e4521. DOI: 10.21769/BioProtoc.4521.
Category
Cell Biology > Cell engineering > CRISPR-cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.