- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro Fluorescence Imaging–based Actin Bundling Assay
Published: Vol 12, Iss 18, Sep 20, 2022 DOI: 10.21769/BioProtoc.4518 Views: 2777
Reviewed by: David PaulSeham EbrahimMoriah R Beck
Original research article
The authors used this protocol in:

Mar 2022


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Understanding the molecular and structural mechanisms that govern the assembly and organization of higher-order actin architecture requires the use of in vitro actin binding and bundling assays. Crosslinking of actin filaments into bundles can be monitored in vitro via several techniques, including negative staining/electron microscopy, low-speed co-sedimentation assay/SDS-PAGE, and fluorescence staining/confocal microscopy. We and others have previously characterized the N-BAR domain of ASAP1, an ADP-ribosylation factor GTPase-activating protein, as an actin-bundling module; we further identified key lysine residues responsible for actin cross-linking. Here, we use the ASAP1 BAR domain as an example and describe a detailed procedure for observing the actin bundle formation by confocal microscopy. This protocol requires small reaction volumes and takes advantage of bright commercially available fluorescent phalloidins, making it an ideal choice for medium-throughput screening of mutants or domain truncations in their ability to bundle actin.
Graphical abstract:
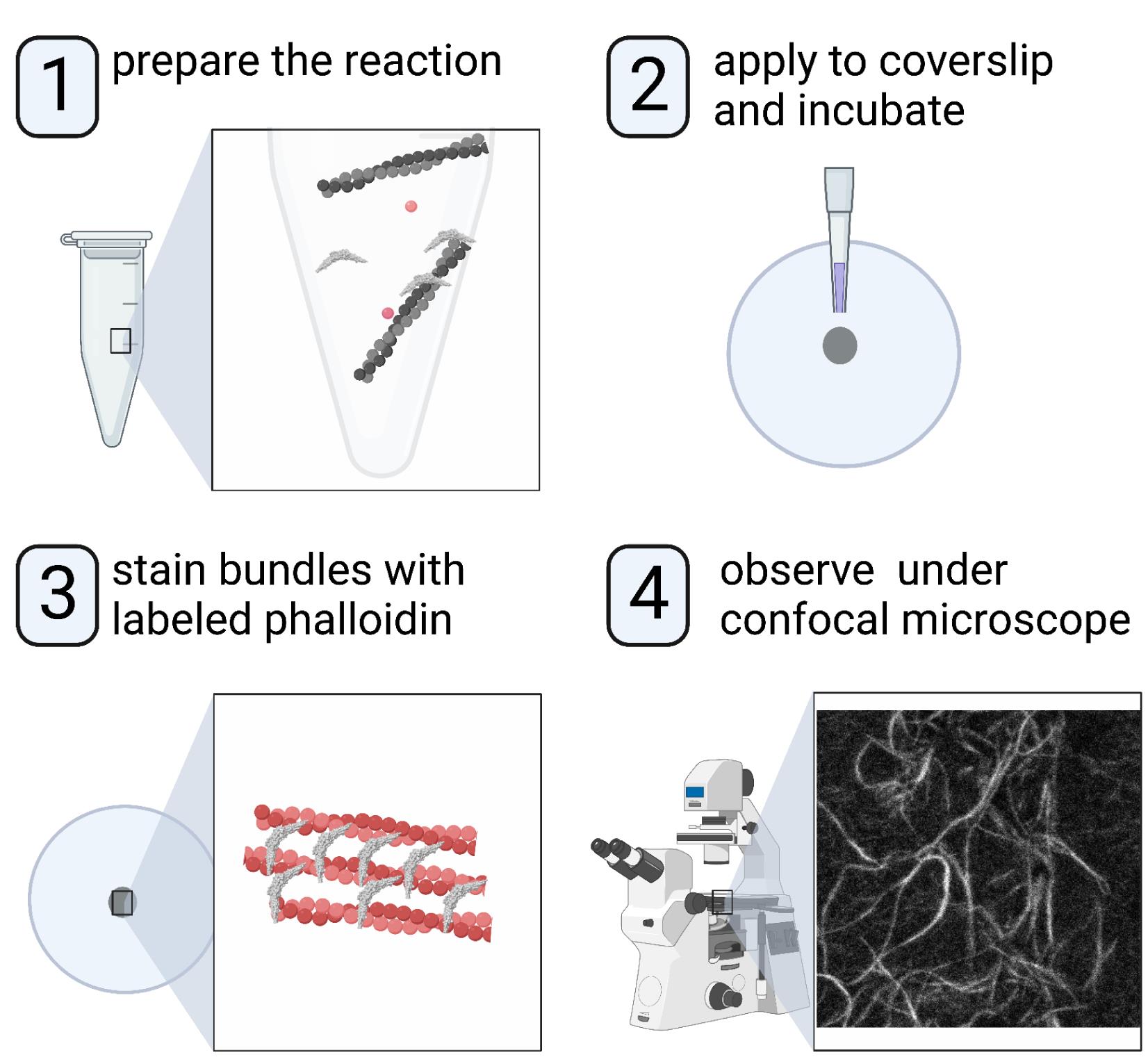
Background
Actin function in normal physiology and pathology depends on the assembly of actin filaments into higher-order structures. The regulation of higher-order actin structures is still being discovered. Advances in our understanding of these dynamics are facilitated by information-dense assays with quantifiable results. ASAP1 is an ADP-ribosylation factor GTPase-activating protein (GAP) that regulates the dynamics of filamentous actin–based structures, including stress fibers, focal adhesions, and circular dorsal ruffles (Randazzo et al., 2000; Oda et al., 2003; Bharti et al., 2007). We and others have previously found that the N-BAR domain of ASAP1 binds and bundles actin filaments and that its actin bundling activity is under auto-inhibitory regulation by its GAP and SH3 domains (Gasilina et al., 2019; Chen et al., 2020). We then set out to identify structural determinants on ASAP1 BAR domain that control actin bundling activity (Gasilina et al., 2022), which required careful quantification. Here, we detail a method to analyze the actin bundling activity of ASAP1 BAR-PH, which is applicable to other proteins with hypothesized actin-bundling activity. Unlike low-speed sedimentation assay, this method requires small quantities of actin filaments and test proteins and allows for simultaneous processing of several samples, which facilitates the evaluation of several concentrations, point mutants, domain truncations, or ionic strengths in a relatively short amount of time. In addition, because the filaments are directly visualized, information such as bundle width and length can be determined, which is not possible with the sedimentation assays; quantification is robust with methods that have been developed for analysis of images.
Materials and Reagents
Circular coverslips, German glass, 12 mm diameter, #1.5 thickness (Electron Microscopy Sciences, catalog number: 72291-02)
24-well flat bottom cell culture plate (Corning Costar, Corning, catalog number: 3526, or similar)
1.5–1.7 mL microcentrifuge tubes (Thomas Scientific, catalog number: 1138W14)
0.5 mL open-top thickwall polycarbonate tube (Beckman Coulter, catalog number: 343776)
50 mL conical tubes (Corning, catalog number: 352070)
0.22 µm syringe filters (Millex, Millipore-Sigma, catalog number: SLGPR33RS)
50 mL Luer tip syringes (BD Luer-Lok Tip, Fisher Scientific, catalog number: 14-820-11)
10 µL pipette tips, ends cut off to make a larger orifice
Frosted microscope slides (Fisherbrand, Fisher Scientific, catalog number: 12-550-343)
Ethanol/dry ice bath or liquid nitrogen for snap-freezing protein
Phosphate buffered saline (PBS) (Gibco, ThermoFisher Scientific, catalog number: 20012027)
Poly-L-lysine, 0.01% solution (Sigma-Aldrich, MilliporeSigma, catalog number: P4707-50ML)
Rabbit muscle G-actin, >99% pure (Cytoskeleton, Inc., catalog number: AKL99)
Note: Using actin prepared in-house from rabbit skeletal acetone powder is also fine, but care needs to be taken to verify that all bundling protein contaminants have been removed.
ASAP1 N-BAR recombinant protein, expressed and purified as described previously (Gasilina et al., 2019, 2022)
α-actinin (Cytoskeleton, Inc., catalog number: AT01)
Note: It is highly recommended to use a bona fide actin bundling protein, such as α-actinin or vinculin tail, as a positive control.
Bovine serum albumin (BSA) (Sigma-Aldrich, Millipore Sigma, catalog number: B8667). Aliquot into 100 µL volumes and snap freeze. Store at -80 °C.
Methanol ≥99.9% (J.T. Baker, VWR International, catalog number: 9093-02)
Fluorescently labeled phalloidin [rhodamine phalloidin (Invitrogen, ThermoFisher Scientific, catalog number: R415), or Alexa Fluor 488 phalloidin (Invitrogen, ThermoFisher Scientific, catalog number: A12379)]. Reconstitute in 1.5 mL of 100% methanol and store at -20 °C.
Note: Although any bright and photostable fluorescent phalloidin conjugate can be used to stain and visualize bundles, the use of fluorophores in the visible spectra will facilitate finding and focusing on the sample during data acquisition.
Adenosine 5’-triphosphate disodium salt (ATP) (Cytoskeleton, Inc., catalog number: BSA04). Reconstitute with 1 mL of cold 100 mM Tris, pH 7.5 (Recipe 7), for a 100 mM stock, aliquot into 5–100 µL portions and 10–50 µL portions, snap freeze, and store at -80 °C. Note that the ATP from cytoskeleton is lyophilized from a solution buffered to pH 7, and, consequently, the solutions do not require additional adjusting of the pH.
Paraformaldehyde, 16%, methanol-free (Electron Microscopy Sciences, catalog number: 15710)
Dako fluorescence mounting medium (Agilent, catalog number: S302380-2)
Calcium chloride (CaCl2) (Sigma-Aldrich, Millipore Sigma, catalog number: 223506)
Potassium chloride (KCl) (Sigma-Aldrich, Millipore Sigma, catalog number: P3911)
Magnesium chloride (MgCl2) (Sigma-Aldrich, Millipore Sigma, catalog number: M9272)
Tris-HCl, 1 M, pH 7.5 (KD Medical, catalog number: RGF-3350)
Dithiothreitol (DTT) Cleland’s reagent (MP Biomedicals, VWR International, catalog number: 0219482101)
1× G-actin buffer (see Recipes)
10× actin polymerization buffer (see Recipes)
1× F-actin buffer (see Recipes)
1 M CaCl2 (see Recipes)
1 M KCl (see Recipes)
1 M MgCl2 (see Recipes)
Tris-HCl, 1 M, pH 7.5 (see Recipes)
1 M DTT (see Recipes)
Equipment
P10, P20, and P200 micropipettes
Benchtop microcentrifuge for 1.5/2 mL tubes
Small volume ultra-centrifuge (e.g., Thermo Scientific Sorvall MTX with S120-AT3 rotor)
Confocal microscope equipped with oil-immersion objective, preferably with a tile scanning option (we use a 63× objective, but given the size of the bundles, any objective between 40× and 100× is suitable.)
Software
Fiji/ImageJ (National Institutes of Health, imagej.nih.gov)
Procedure
Preparation of poly-L-lysine-coated coverslips
Note: This procedure assumes that four technical replicates are made for each bundling assay, and four bundling assays are made in total—F-actin alone, F-actin with ASAP1 BAR-PH, F-actin with BSA as a negative control, and F-actin with α-actinin as a positive control—all at a single concentration. If additional proteins or concentrations are to be tested, scale up the number of coverslips accordingly.
To examine four conditions in quadruplicate, place 16 coverslips into a 24-well plate, one in each well (Figure 1A–B). We use the 24-well plate because the size of the wells is ideal for the 12 mm coverslips.
Add 500 µL of 0.01% poly-L-lysine solution to each well, ensuring the coverslips are fully submerged. If coverslips float up, gently lower them back into solution with a pipette tip (Figure 1C–D).
Replace the lid and incubate at room temperature (RT) for 2 h (Figure 1E).
Aspirate poly-L-lysine solution and wash the coverslips three times with 500 µL of distilled water (Figure 1F).
Place the lid on the plate slightly askew and let coverslips dry overnight (Figure 1G). Alternatively, dry the coverslips in a 65 °C oven for 15 min.
Preparation of globular actin (G-actin) stock
Briefly centrifuge a tube containing 250 µg of lyophilized actin to bring powder to the bottom of the tube.
Add 250 µl of cold G-actin buffer, freshly supplemented with 0.2 mM ATP and 0.5 mM DTT (see Recipes). Pipette gently five to six times to resuspend the powder, taking care to avoid bubbles. Do not vortex.
Incubate on ice for 1 h.
This is a 1 mg/mL (~23 µM) stock of G-actin.
Notes:
Actin is not stable in its globular form. Promptly proceed to Step C or aliquot into smaller quantities and store at -80 °C.
We found that high-purity actin from cytoskeleton has very few aggregates and thus does not require high-speed centrifugation post resuspension. If other sources of actin are used, it may be necessary to centrifuge the G-actin stock at 100,000–150,000 × g to remove aggregates and subsequently calculate the protein concentration of the remaining G-actin fraction.
Preparation of actin filaments (F-actin)
Prepare 10× actin polymerization buffer (see Recipes).
Add 25 µL of the 10× actin polymerization buffer (1/10th of the volume) to 250 µL of 1 mg/mL stock of G-actin. Mix well, taking care to avoid bubbles. Do not vortex.
Incubate at RT for 1 h.
Note: F-actin is stable at 4 °C for up to one month. Do not freeze.
Preparation of potential bundling proteins such as ASAP1 and BAR-PH, the negative control BSA, and the positive control α-actinin for bundling assay (optional but highly recommended)
Pre-cool the tabletop ultracentrifuge and rotor to 4 °C.
Rapidly thaw ASAP1 BAR-PH, BSA, and α-actinin in RT water bath. We typically store the proteins in 100 μL aliquots at protein concentrations of approximately 1 mg/mL. For ASAP1 and recombinant proteins derived from ASAP1, the storage buffer is typically 20 mM HEPES, pH 8.0, and 150 mM NaCl.
Distribute proteins into polycarbonate ultracentrifuge tubes.
Place tubes, appropriately balanced with tubes filled with “blank” solution (e.g., protein buffer or water), into the ultracentrifuge rotor. Secure the lid.
Spin at 150,000 × g and 4 °C for 1 h to sediment particulates and aggregates.
Post-centrifugation, open the rotor lid and mark the sides of the tubes facing outward with a dot to indicate the side with the pellet and debris.
Transfer the spun protein into a new pre-labeled microcentrifuge tube, taking care not to disturb the pellet.
Determine the protein concentration using a protein assay, e.g., BCA or Bradford, or other spectrophotometric method.
Note: If the protein concentration is low, it is advisable to purify the protein into the F-actin buffer or exchange the protein into F-actin buffer using PD-10 (Cytiva) or Zeba columns (ThermoFisher).
Actin bundling assay
Label four microcentrifuge tubes, one for each reaction: F-actin alone, F-actin + BSA, F-actin + α-actinin, and F-actin + ASAP1 BAR-PH (Figure 1H–I).
Set up a 15 µL reaction in each tube using F-actin buffer (see Recipes) to compensate for volume differences (Figure 1J–K). Reactions typically contain 1–3 µM actin and 1–3 µM of the test or control proteins. Use 3 µM actin and 3 µM test proteins and respective positive and negative controls for initial estimation of bundling efficiency.
Note: The reactions should be set up within 10 min of each other and promptly transferred to the coverslips to allow for comparable incubation times for each reaction.
Using a 10 µL pipette held vertically and a tip with the end cut off, pipette 3 µL of the reaction directly into the center of each coverslip (Figure 1L–M). Avoid introducing bubbles—this may require depressing the pipette plunger only to the first stop. Repeat for all replicates and reactions.
Note: Placing the reaction spot directly into the center of the coverslip will greatly facilitate the image acquisition process.
Replace the lid of the 24-well plate and incubate the reaction drops at RT for 1 h.
During this time, prepare 20 mL of 4% paraformaldehyde (PFA) in PBS from the 16% stock and chill on ice. Unused 16% PFA can be stored sealed in parafilm for up to two weeks at 4 °C. Alternatively, dilute the entire 10 mL ampule to 4% in PBS (40 mL total) and store unused portion for up to one week at 4 °C.
Gently place the plate on ice and add 500 µL of ice-cold 4% PFA to each well (pipette on the side of the wells to avoid breaking the reaction drops) (Figure 1N). If any coverslips float up, gently lower them back with a pipette tip. Fix for 30 min (fixing adheres the actin bundles to the coverslips, so they are not lost during the staining process).
Rinse the coverslips gently in cold PBS twice for 5 min each. Gently aspirate.
Stain each coverslip in 500 µL of 1:500 dilution of fluorescent phalloidin in PBS for 30 min on ice. Cover the plate with foil during staining (Figure 1O).
In the meantime, pre-label 12 microscope slides (if placing two coverslips per slide) with identifying information (e.g., date, initials, reaction name, and phalloidin conjugate). Gently wipe the slide with a Kimwipe sprayed with 70% alcohol, if necessary (Figure 1P).
After 30 min, gently aspirate the staining solution and add PBS. Incubate for 5 min. Repeat. After the final PBS wash, aspirate and add 500 µL of ddH2O to each well.
Mount two coverslips on each slide using Dako fluorescent medium. Gently aspirate excess mounting medium from the edges of coverslips (see Figure 1Q–T). Allow to dry for at least 1 h at RT in the dark. Samples can be stored at 4 °C or transferred to -20 °C for long-term storage after imaging.

Figure 1. Pictographic Summary of the Procedure. For details and descriptions, refer to the main text.
Image acquisition and image analysis
Image the coverslips using a confocal microscope. For Alexa 488 phalloidin, use an argon 488 laser; for rhodamine phalloidin, use a krypton 564 laser. Using the tile scanning option (optional) allows for acquisition of a large area of the reaction spot. Typical results are shown in Figure 2. Examples of technically failed assays are shown in Figure 3.
Post-acquisition, images can be further analyzed using ImageJ. Bundle length and number or fluorescence intensity can be measured using freely available plugins; use thereof is described in Gasilina et al. (2019, 2022).
Overview of possible analyses
Examining the efficiency of actin bundling by comparing fluorescent intensities of bundles
Plotting line scan intensities of bundles made by different actin bundling proteins can be a quick way to estimate bundling efficiencies. This method can serve as a straightforward tool for initial characterization of mutants or domain truncations, among others.
Please note that direct comparisons of fluorescence intensities among different microscopy images require acquisition using identical settings (objective, light path, laser power, gain, frame size, zoom, and scan speed).
Download and Install Fiji/ImageJ.
Open the microscopy image of an actin bundling assay.
Select line tool and draw a line across the bundle.
Click “Analyze” → “Plot Profile.”
When the graph of the line scan appears, click “List” and copy the contents of the resulting window into a spreadsheet.
Repeat for other regions of interest and images.
Plot line scans in a single graph to visualize differences in fluorescence intensities of the bundles.
Quantifying the number and length of actin bundles using Ridge Detection Plugin (ImageJ/Fiji)
Download and Install Fiji/ImageJ.
Follow the instructions on https://imagej.net/plugins/ridge-detection to download the Ridge Detection Plugin.
Open an Experiment file in Fiji/ImageJ and convert to 8 bit by clicking Image → Type → 8-bit.
Initialize the Ridge Detection Plugin by clicking Plugins → Ridge Detection.
Set “Mandatory_parameters.” To help with accurate determination of mandatory parameter cut-offs, check “Preview” window and modify high and low contrast in “Optional_parameters” to view bundle detection in real time.
Click “OK” to initiate calculation of bundle length and the number of bundles.
The summary window provides the length and a unique identification number for each identified bundle.
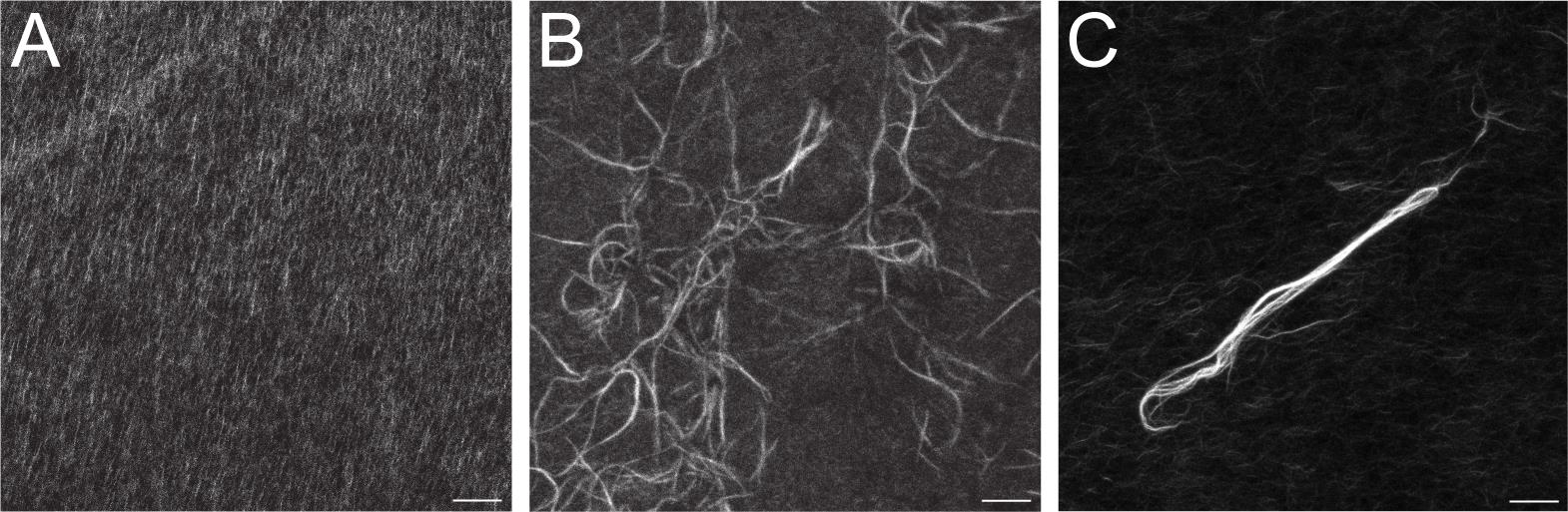
Figure 2. Representative results. (A) “F-actin-only” control sample was stained with fluorescent phalloidin. Note uniform distribution of filaments on the image tile. The negative control protein, e.g., BSA, should appear nearly identical to the F-actin-only control. (B) “F-actin with ASAP1 BAR-PH” sample was stained with fluorescent phalloidin. Note the appearance of thick bundles. (C) “F-actin with α-actinin” positive control sample. α-actinin is a bona fide actin-bundling protein, which robustly cross-links actin filaments into thick bundles. Scale bar = 10 µm.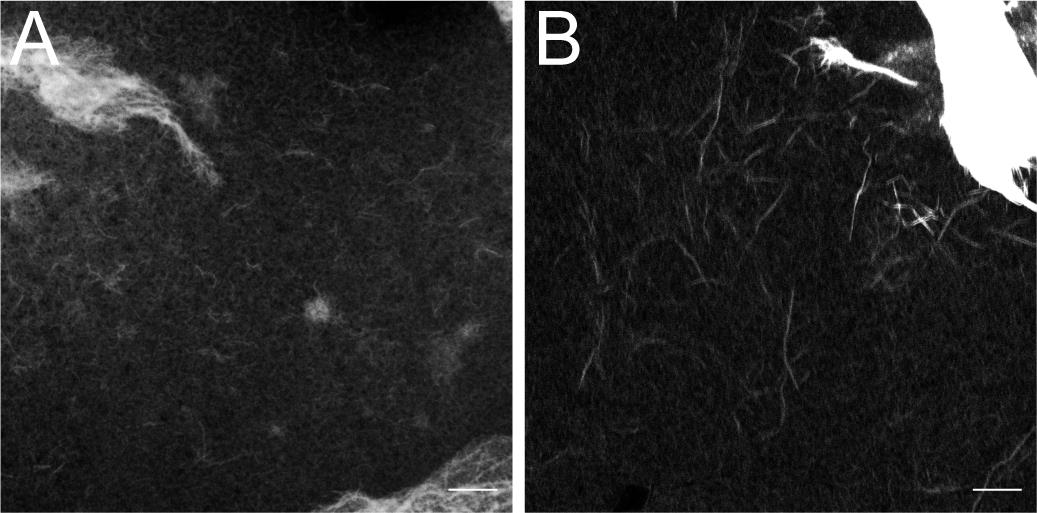
Figure 3. Examples of failed assays. (A) “F-actin-only” control sample was stained with fluorescent phalloidin. Note “clumps” of actin in the upper left and lower right corners. Improper storage of F-actin leads to aggregation of filaments and their subsequent appearance as clumps. Therefore, the rest of the coverslips in this set were discarded. (B) In this particular example, the test protein was not pre-spun before setting up the assay, resulting in debris and particulates being carried through the fixation and staining, which appeared as auto-fluorescent artifacts (upper right). Presence of these strongly fluorescent spots impedes image analysis. Scale bar = 10 µm.
Recipes
1× G-actin buffer (store at 4 °C)
CaCl2 (1 M), 0.2 mM, 10 µL
Tris-HCl (1 M, pH 8.0), 20 mM, 1 mL
ddH2O, 49 mL
Total: 50 mL
Supplement with 0.2 mM ATP and 0.5 mM DTT (final) immediately before use. For each milliliter of G-actin buffer working solution, add 2 µL of the 100 mM ATP stock and 5 µL of the 100 mM DTT stock. Keep on ice. Discard unused stock after 4–5 h.
10× actin polymerization buffer (store at RT)
MgCl2 (1 M), 20 mM, 200 µL
KCl (5 M), 1 M, 2 mL
ddH2O, 7.8 mL
Total: 10 mL
To prepare 100 µL of working solution, mix 10 µL of freshly thawed 100 mM ATP stock with 90 µL of 10× polymerization buffer on ice. Discard unused stock after 4–5 h.
1× F-actin buffer (prepare and use immediately. Keep on ice and discard after 4–5 h)
G-actin buffer + ATP/DTT, 450 µL
10× actin polymerization buffer + ATP, 50 µL
Total: 500 µL
Use the following as a guide (volumes account for pipetting errors): to make G-actin buffer + ATP/DTT, combine 500 µL of G-actin buffer, 1 µL of the 100 mM ATP stock, and 2.5 µL of the 100 mM DTT stock. To make actin polymerization buffer + ATP, combine 10 µL of freshly thawed 100 mM ATP stock and 90 µL of polymerization buffer. To make F-actin buffer, combine 450 µL of G-actin buffer with ATP and DTT and 50 µL of actin polymerization buffer with ATP.
1 M CaCl2
Add 7.35 g CaCl2 to 50 mL of ultra-pure water for a 1 M stock solution. Store in a conical tube at RT.
1 M KCl
Add 18.6 g KCl to 50 mL of ultra-pure water for a 5 M stock solution. Store in a conical tube at RT.
1 M MgCl2
Add 10.2 g MgCl2 to 50 mL of ultra-pure water for a 1 M stock solution. Store in a conical tube at RT.
Tris-HCl, 100 mM, pH 7.5
Add 10 mL of the 1 M Tris-HCl stock to 90 mL of ultra-pure water and filter sterilize, to prepare 100 mL of a 100 mM working solution. Store in two 50 mL sterile conical tubes; keep one tube at RT and store the other at 4 °C.
1 M DTT
Add 1.54 g to 10 mL of ultra-pure water for a 1 M stock. Aliquot 9 mL into nine 1 mL aliquots. Take 1 mL of the 1 M stock and further dilute to 100 mM with 9 mL of ultra-pure water. Aliquot the 100 mM stocks in 500 µL volumes. Store at -20 °C.
Acknowledgments
This work was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health, Department of Health and Human Services (project number BC007365 to P.A.R.). This protocol was adapted from Lin-Jones and Burnside (2007) and our previous work (Gasilina et al., 2019, 2022). The authors thank Natalie M. Badillo for assistance with photography.
Competing interests
The authors declare no competing interests.
References
- Bharti, S., Inoue, H., Bharti, K., Hirsch, D. S., Nie, Z., Yoon, H. Y., Artym, V., Yamada, K. M., Mueller, S. C., Barr, V. A., et al. (2007). Src-dependent phosphorylation of ASAP1 regulates podosomes. Mol Cell Biol 27(23): 8271-8283.
- Chen, P. W., Billington, N., Maron, B. Y., Sload, J. A., Chinthalapudi, K. and Heissler, S. M. (2020). The BAR domain of the Arf GTPase-activating protein ASAP1 directly binds actin filaments. J Biol Chem 295(32): 11303-11315.
- Gasilina, A., Vitali, T., Luo, R., Jian, X. and Randazzo, P. A. (2019). The ArfGAP ASAP1 Controls Actin Stress Fiber Organization via Its N-BAR Domain. iScience 22: 166-180.
- Gasilina, A., Yoon, H. Y., Jian, X., Luo, R. and Randazzo, P. A. (2022). A lysine-rich cluster in the N-BAR domain of ARF GTPase-activating protein ASAP1 is necessary for binding and bundling actin filaments. J Biol Chem 298(3): 101700.
- Lin-Jones, J. and Burnside, B. (2007). Retina-specific protein fascin 2 is an actin cross-linker associated with actin bundles in photoreceptor inner segments and calycal processes. Invest Ophthalmol Vis Sci 48(3): 1380-1388.
- Oda, A., Wada, I., Miura, K., Okawa, K., Kadoya, T., Kato, T., Nishihara, H., Maeda, M., Tanaka, S., Nagashima, K., et al. (2003). CrkL directs ASAP1 to peripheral focal adhesions. J Biol Chem 278(8): 6456-6460.
- Randazzo, P. A., Andrade, J., Miura, K., Brown, M. T., Long, Y. Q., Stauffer, S., Roller, P. and Cooper, J. A. (2000). The Arf GTPase-activating protein ASAP1 regulates the actin cytoskeleton. Proc Natl Acad Sci U S A 97(8): 4011-4016.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Gasilina, A. and Randazzo, P. A. (2022). In vitro Fluorescence Imaging–based Actin Bundling Assay. Bio-protocol 12(18): e4518. DOI: 10.21769/BioProtoc.4518.
- Gasilina, A., Yoon, H. Y., Jian, X., Luo, R. and Randazzo, P. A. (2022). A lysine-rich cluster in the N-BAR domain of ARF GTPase-activating protein ASAP1 is necessary for binding and bundling actin filaments. J Biol Chem 298(3): 101700.
Category
Biochemistry > Protein > Imaging
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.