- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Protein Tyrosine Phosphatase Biochemical Inhibition Assays
Published: Vol 12, Iss 18, Sep 20, 2022 DOI: 10.21769/BioProtoc.4510 Views: 3060
Reviewed by: Chiara AmbrogioZheng Zachory WeiKarem A CourtAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2022
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
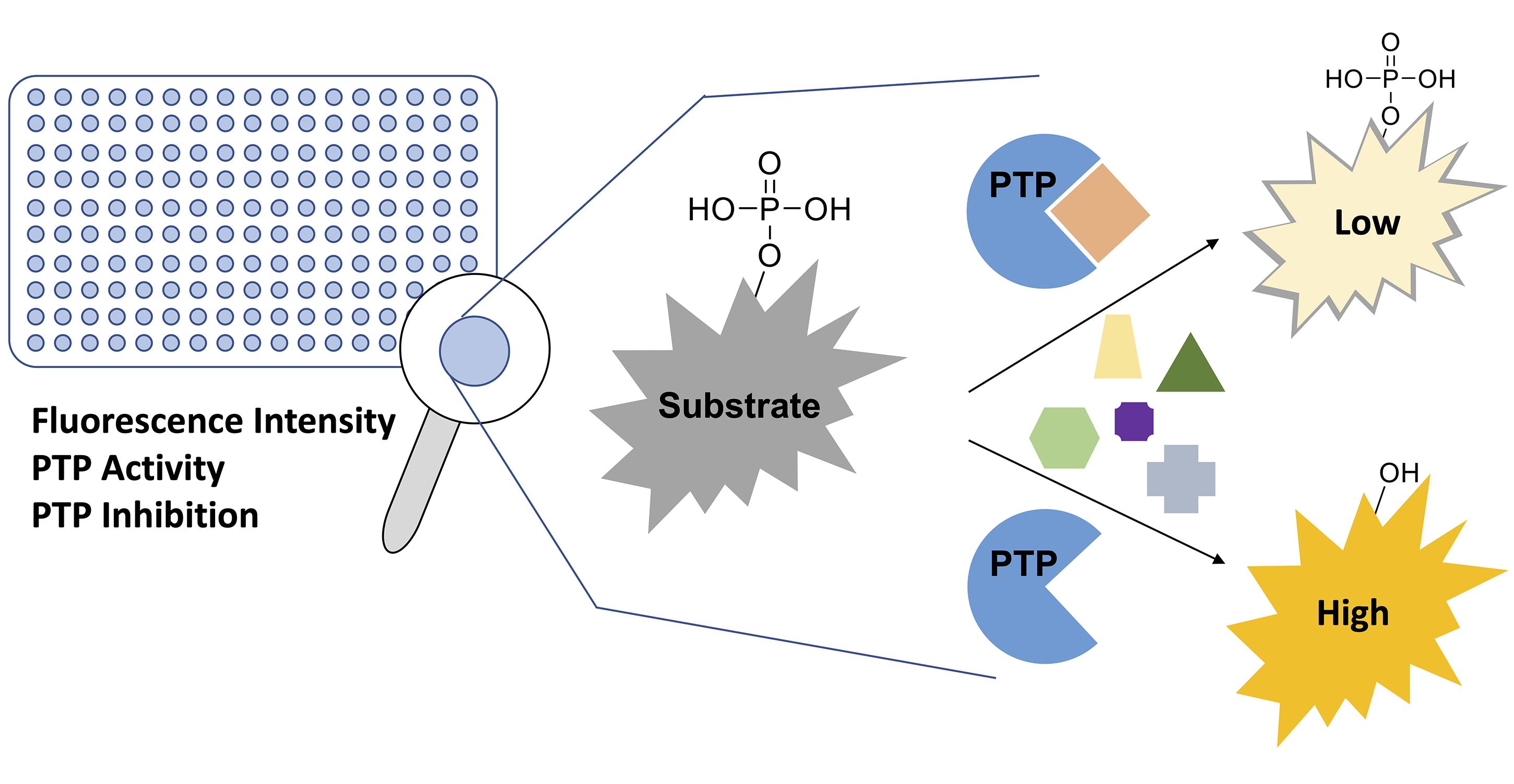
Disturbance of the dynamic balance between protein tyrosine phosphorylation and dephosphorylation, modulated by protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs), is known to be crucial for the development of many human diseases. The discovery of agents that restore this balance has been the subject of many drug research efforts, most of which have focused on tyrosine kinase inhibitors (TKIs), resulting in the development of more than 50 FDA-approved TKIs during the past two decades. More recently, accumulating evidence has suggested that members of the PTP superfamily are also promising drug targets, and efforts to discover tyrosine phosphatase inhibitors (TPIs) have increased dramatically. Here, we provide protocols for determining the potency of TPIs in vitro. We focus on the use of fluorescence-based substrates, which exhibit a dramatic increase in fluorescence emission when dephosphorylated by the PTP, and thus allow setting up highly sensitive and miniaturized phosphatase activity assays using 384-well or 1536-well microplates and a continuous (kinetic) assay format. The protocols cover PTP specific activity assays, Michaelis–Menten kinetics, dose-response inhibition assays, and dose-response data analysis for determining IC50 values. Potential pitfalls are also discussed. While advanced instrumentation is utilized for compound spotting and liquid dispensing, all the assays can be adapted to existing equipment in most laboratories. Assays are described for selected PTP drug targets, including SHP2 (PTPN11), PTP1B (PTPN1), STEP (PTPN5), and VHR (DUSP3). However, all protocols are applicable to members of the PTP enzyme family in general.
Graphical abstract:
Background
Protein tyrosine phosphorylation is a reversible posttranslational modification (PTM) and a fundamentally important mechanism in eukaryotic cell signal transduction and regulation (Hunter, 2009). Perturbations in tyrosine phosphorylation can lead to the development of many human diseases, including cancer, neurological disorders, autoimmunity, and immunodeficiency, as well as cardiovascular, metabolic, and infectious diseases (Tautz et al., 2006; Vang et al., 2008; Labbe et al., 2012; Goebel-Goody et al., 2012; Zhang, Z. Y. et al., 2015; Menegatti, 2022). Targeting protein tyrosine kinases (PTKs) has been a major focus of drug discovery efforts in the last two decades, resulting in more than 50 FDA-approved tyrosine kinase inhibitors (TKIs) (Cohen et al., 2021). On the other hand, the discovery of clinical candidates that target protein tyrosine phosphatases (PTPs) has significantly lagged behind the kinases for multiple reasons (reviewed in Tautz et al., 2013; Tonks, 2013; Stanford and Bottini, 2017). Unquestionably, an inflection point in tyrosine phosphatase inhibitor (TPI) research was the discovery of SHP099, the first truly selective and drug-like inhibitor of the SHP2 (PTPN11) phosphatase (Chen et al., 2016). The compound has since served as a blueprint for several investigational drugs that are currently being tested in phase 1/2 clinical trials for the treatment of solid tumors (Song et al., 2022).
The success in bringing SHP2 inhibitors into the clinic has garnered a new wave of interest in targeting PTPs. Here, we provide protocols for determining the potency of TPIs in enzymatic phosphatase assays. The experiments described cover: 1) PTP activity assays, to determine a suitable enzyme concentration; 2) Michaelis–Menten kinetics, to determine the Michaelis–Menten constant (Km) of the substrate for a specific PTP; and 3) dose-response inhibition assays and dose-response data analysis, to determine IC50 values of potential inhibitors. We utilized advanced instrumentation for automated compound spotting and liquid dispensing. However, the assays described herein can be adapted to existing equipment in most laboratories. While our protocols are applicable to PTPs in general, we show examples that utilize four specific phosphatases with promising therapeutic potential in various diseases, including cancer [SHP2, PTP1B (Vainonen et al., 2021)], type II diabetes [PTP1B (Zhang, Z. Y. et al., 2015)], Alzheimer’s disease [STEP (Lambert et al., 2021)], as well as arterial thrombosis, sepsis, and septic shock [VHR (Tautz et al., 2015; Singh et al., 2015)].
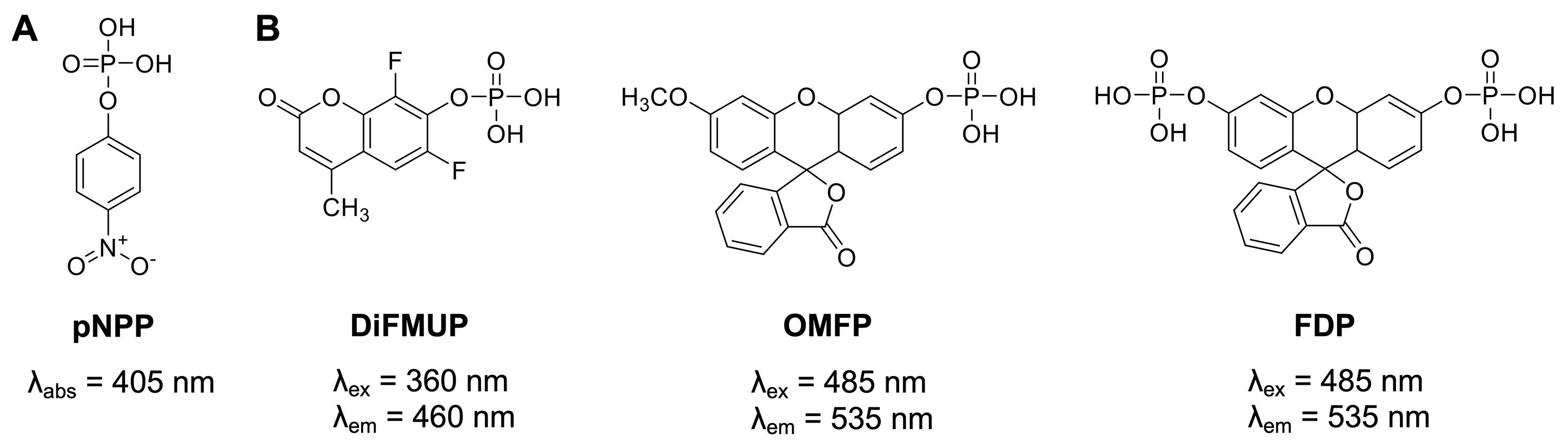
Figure 1. Generic protein phosphatase substrates used for PTP enzymatic assays. (A) The colorimetric substrate p-nitrophenyl phosphate (pNPP). (B) The fluorogenic substrates 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP), 3-O-methylfluorescein phosphate (OMFP), and fluorescein diphosphate (FDP).
Historically, a widely used generic protein phosphatase substrate is p-nitrophenyl phosphate (pNPP) (Bessey et al., 1946) (Figure 1A). Conversion of pNPP generates p-nitrophenol, which can be directly monitored via its absorbance at 405 nm. While the absorbance of p-nitrophenol is linear over a relatively wide range of concentrations (approximately 5–500 μM), colored small molecules of interest can absorb light at similar frequencies, resulting in potential false negative results. Moreover, relatively high concentrations of recombinant PTPs (in the mid nanomolar range) are typically required to produce sufficient p-nitrophenol signal to background (S/B) and signal to noise (S/N) ratios. More recently, fluorogenic protein phosphatase substrates such as 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP), 3-O-methylfluorescein phosphate (OMFP), or fluorescein diphosphate (FDP) (Figure 1B) have been utilized for PTP fluorescence intensity assays (Huang et al., 1999; Welte et al., 2005; Tierno et al., 2007; Tautz and Sergienko, 2013). These substrates have a low fluorescence in the phosphorylated state but become strong fluorophores when dephosphorylated. Typically, PTP assays using fluorogenic substrates are several orders of magnitude more sensitive than comparable pNPP assays, thus requiring significantly less recombinant PTP enzyme (picomolar to low nanomolar concentrations). Additionally, the fluorescence emission of the dephosphorylated products can be measured over a wide range of concentrations (approximately 10 nM to >100 μM) with excellent S/B and S/N ratios for highly reproducible PTP assays in continuous (kinetic) mode, which allows for the most accurate determination of the initial velocity rates (V). PTP assays using fluorogenic substrates can be easily miniaturized and performed in 384-well or 1536-well formats, allowing for efficient dose-response testing of candidate inhibitors. For the protocols provided here, we employ DiFMUP and/or OMFP with standard volume 384-well microplates. These plates do not require automated liquid handling and are amenable to manual liquid transfers using multichannel pipettes. However, laboratories with advanced equipment will experience no difficulty in adapting the protocols to a 1536-well format, as we have successfully performed similar assays using 1536-well microplates in a total assay volume as small as 5 μL.
Notes and Considerations
PTPs typically are most active at a pH between 5.5 and 6 (Groen et al., 2005). Our standard assay buffer that works well for most PTPs is Bis-Tris used at pH 6 (see Recipe 1). Buffer systems closer to physiological pH (e.g., Tris buffer at pH 7.4) may be used as well. For inhibition assays, we recommend not using buffers containing sulfonic acids such as HEPES, which can compete with inhibitor binding at the active site. A reducing agent such as dithiothreitol (DTT) ensures that the PTP catalytic cysteine is in the active, reduced state. It also prevents potentially oxidizing compounds from nonspecifically inhibiting the PTP through oxidation of the catalytic Cys. The addition of a detergent such as 0.01% Tween 20 is highly recommended, as it stabilizes the protein over the course of the assay and reduces the likelihood of promiscuous, aggregate-based inhibition (Feng and Shoichet, 2006). Bovine serum albumin (BSA) or globulin proteins may be used as detergent substitutes. For a detailed description of buffer optimization experiments, we refer to our previous publication (Tautz and Sergienko, 2013).
Fluorogenic substrates such as DiFMUP or OMFP may encounter compound spectral interference, either via compound autofluorescence or compound-induced fluorescence quenching. In our experience, the DiFMUP assay, which relies on the near-UV/blue spectral range (λex = 360 nm, λem = 460 nm), is more prone to such interference than the red-shifted OMFP assay (λex = 485 nm, λem = 535 nm). A pre-read of the assay plate (containing enzyme solution and compound) before addition of the substrate will typically show any potential compound autofluorescence. Likewise, IC50 curves going beyond 100% inhibition may indicate fluorescence quenching. When compound fluorescence interference is suspected, an increase in fluorescent product by using either greater enzyme concentrations or longer reaction times, while still staying within the linear range of substrate conversion, could lessen such interference effects. However, the best approach to mitigate such issues is to retest the suspected compounds using orthogonal substrates (e.g., using OMFP and/or pNPP instead of DiFMUP).
In our protocols, we use acoustic droplet dispensing of compound DMSO stock solutions, allowing for the transfer of nanoliter quantities. Specifically, when we use 384-well standard volume plates and a total assay volume of 25 μL, we transfer 250 nL of compound DMSO stock solution, which results in a final DMSO concentration of 1% that is typically well tolerated by recombinant PTPs. For manual transfer of compound stock solutions using a pipette, we recommend transferring no less than 1 μL to ensure accuracy. In our experience, when using a 1 μL transfer, the corresponding final DMSO concentration of 4% has no considerable effect on PTP stability or activity. In any case, the DMSO content should be kept at an equal amount in all wells, including wells for positive and negative controls.
Whenever feasible, we prefer using OMFP over DiFMUP because of the OMFP red-shifted excitation and emission wavelengths compared to DiFMUP, which lower the chances of compound fluorescence interference. However, one potential issue is the limited aqueous solubility of OMFP that requires initial dissolution in DMSO, resulting in extra DMSO added to the reaction mixtures. Using a 10 mM OMFP stock solution in DMSO (which reaches the limit of OMFP solubility in DMSO), the final DMSO concentration is usually manageable for most PTPs, for which OMFP Km values are in the mid- to low-micromolar range. However, for some PTPs (e.g., SHP2), the OMFP Km is in the high micromolar range, which makes it difficult (or impossible) to keep the final DMSO concentration at the recommended ≤5%, which ensures negligible impact on the stability and activity of the recombinant PTP.
Materials and Reagents
384-well black, flat bottom, standard volume microplates (Greiner Bio-One FLUOTRAC 200, catalog number: 781076)
Aluminum adhesive plate seals (Sigma-Aldrich, catalog number: Z721557)
1.5 mL Eppendorf tubes
15 mL conical tubes
50 mL conical tubes
Recombinant human PTPs with a purity of at least 95% according to SDS-PAGE gel electrophoresis: full-length SHP2 wild-type (SHP2-WT), SHP2 catalytic domain (SHP2cat, aa 237–529), PTP1B (aa 1–300), full-length STEP46, and full-length VHR.
Dually phosphorylated IRS-1 peptide (synthesized by PepMic, Suzhou, China) for activating SHP2-WT
Bis-Tris (Research Product International, catalog number: B75000)
Sodium chloride (NaCl, Sigma-Aldrich, catalog number: S3014)
EDTA tetrasodium salt dihydrate (BioWorld, catalog number: 40500024)
3-O-methylfluorescein phosphate cyclohexylammonium salt (OMFP; in-house synthesis)
6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP; ThermoFisher Scientific, catalog number: D22065)
Tween 20 (Fisher Bioreagents, catalog number: BP337)
DL-dithiothreitol (DTT; BioWorld, catalog number: 40400120)
Dimethyl sulfoxide (DMSO; Sigma-Aldrich, catalog number: D8418)
Milli-Q water: water purified using a Millipore Milli-Q lab water system
Bis-Tris buffer (see Recipes)
10 mM OMFP stock solution in DMSO (see Recipes)
10 mM DiFMUP stock solution in water (see Recipes)
1 M DTT stock solution (see Recipes)
Equipment
Fluorescence microplate reader with filters for 360 nm, 460 nm, 485 nm, and 535 nm (Tecan, Spark Multimode Microplate Reader)
Tabletop centrifuge with a swinging-bucket rotor (Eppendorf, 5810R equipped with A-4-62 rotor and MTP bucket)
MultidropTM Combi reagent dispenser (Thermo Fisher Scientific, catalog number: 5840300)
Small tube dispensing cassette (Thermo Fisher Scientific, catalog number: 24073290 or 24073295)
E1-ClipTipTM multichannel pipettes (Thermo Fisher Scientific, various volumes)
Echo® 555 Liquid Handler (Labcyte)
Note: To spin down plates, the Eppendorf tabletop centrifuge was used at 1,000 rpm (approximately 172 × g).
Software
Chemical and Biological Information Systems (CBIS, ChemInnovation Software, Inc.)
GraphPad PrismTM v.9 (GraphPad Software, LLC.)
MagellanTM data analysis software (Tecan)
Microsoft® Excel (Microsoft)
Note: Slopes from kinetic progression curves can be calculated using either Magellan, GraphPad Prism, or Excel. Michaelis–Menten parameters can be calculated in GraphPad Prism (or equivalent programs). Both GraphPad Prism and CBIS allow for straightforward analysis of dose-response data and calculation of IC50 values. CBIS has the advantage of providing a more automated and convenient environment for analyzing larger data sets.
Procedure
PTP Activity Assay using DiFMUP or OMFP
Note: The following procedure can be used to determine a suitable PTP concentration for the inhibition assays, and to compare or confirm the specific activity of recombinant PTP batches.
Thaw the protein stock solution on ice and mix gently.
Prepare 50 mL of substrate buffer (SB): add 5 µL of Tween-20 to 50 mL of Bis-Tris pH 6 buffer (see Recipes for details).
Note: Store SB at room temperature for no longer than one week. Alternatively, store SB at 4 °C in the dark for several months or freeze for long-term storage.
Prepare 10 mL of enzyme buffer (EB): add 50 µL of a 1 M DTT stock solution to 10 mL SB.
Note: Keep EB on ice for no longer than eight hours.
Prepare 1 mL of 500 nM enzyme intermediate solution (EIS). Use EB to dilute PTP stock solution to 500 nM and keep on ice.
Prepare PTP enzyme solution (ES) (100 µL for each) at 1.25× final concentration by serial dilutions from the EIS and using the EB (Table 1). For instance, for an OMFP assay, make enzyme solutions at 12.5 nM (ES1), 6.25 nM (ES2), 3.12 nM (ES3), 1.56 nM (ES4), and 0.781 nM (ES5) for final enzyme concentrations of 10 nM, 5 nM, 2.5 nM, 1.25 nM, and 0.625 nM. For DiFMUP assays, lower PTP concentrations are typically necessary. For instance, make enzyme solutions at 3.12 nM (ES1), 1.56 nM (ES2), 0.781 nM (ES3), 0.391 nM (ES4), and 0.195 nM (ES5) for final enzyme concentrations of 2.5 nM, 1.25 nM, 0.625 nM, 0.312 nM, and 0.156 nM (Figure 2A).
Table 1. Enzyme solutions for PTP activity assay
OMFP Assay DiFMUP Assay Concentration of PTP in the 1.25× working solution (nM) Final PTP concentration (nM) Concentration of PTP in the 1.25× working solution (nM) Final PTP concentration (nM) ES1 12.5 10 3.12 2.5 ES2 6.25 5 1.56 1.25 ES3 3.12 2.5 0.781 0.625 ES4 1.56 1.25 0.391 0.312 ES5 0.781 0.625 0.195 0.156 Using a multichannel pipette, manually dispense EB and ES into a 384-well assay plate. Add 20 µL of EB to wells A1–E1 (no-enzyme control). Add 20 µL of ES1 to wells A2–A5 (for quadruplicate measurements). Similarly, add ES2 /3 /4 /5 to wells B2–B5, C2–C5, D2–D5, and E2-E5, respectively (Figure 2B).
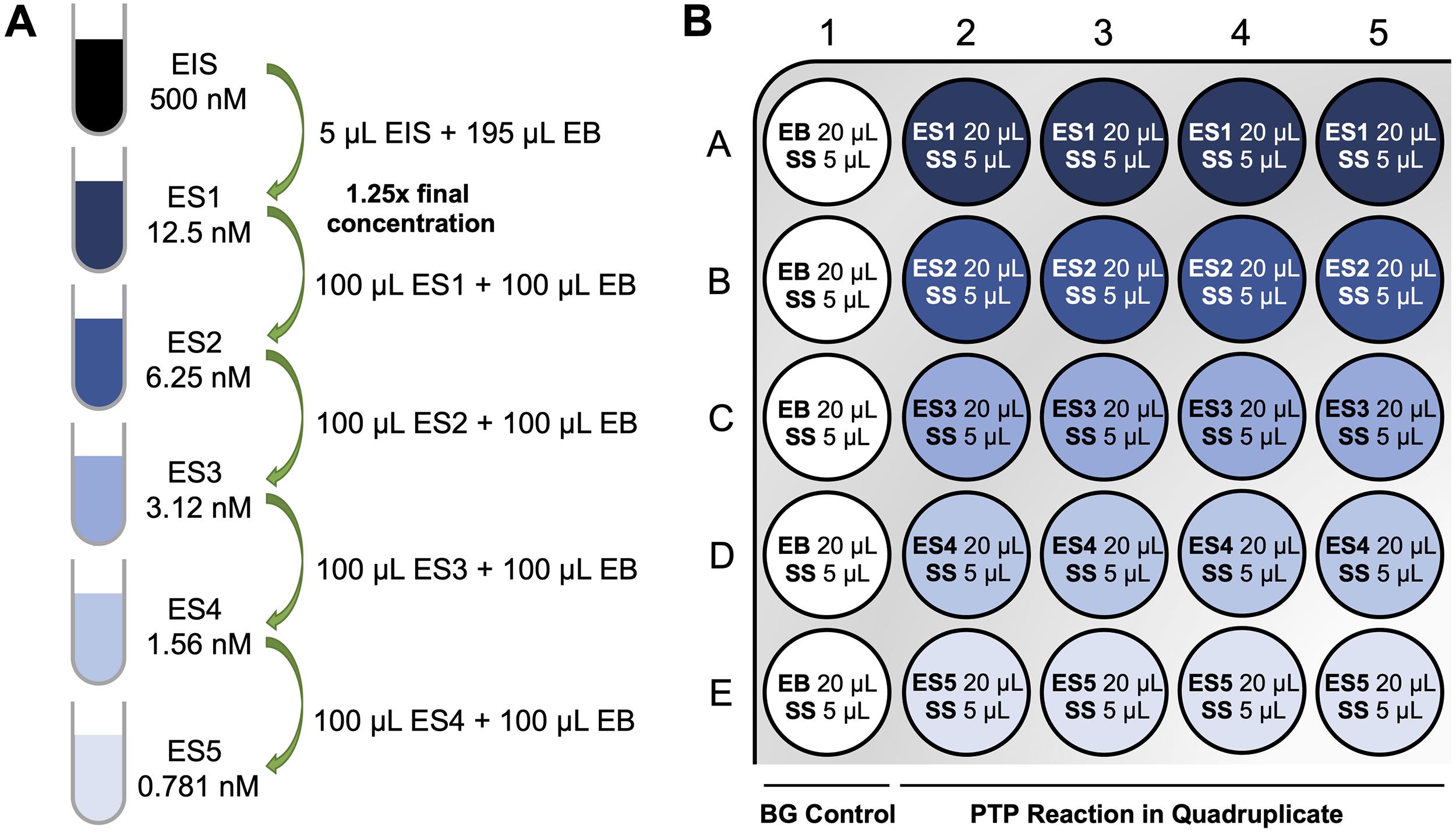
Figure 2. Enzyme serial dilution scheme (A) and assay plate layout (B) for PTP activity assay. EIS, enzyme intermediate solution; ES, enzyme solution; EB, enzyme buffer; SS, substrate solution; BG, background.Using a tabletop centrifuge with a swinging-bucket rotor, spin-down the plate for a few seconds, cover the plate with a lid (or with an additional plate), and incubate at room temperature for 20 min.
Using a 10 mM substrate stock solution and SB, prepare 1 mL of substrate solution (SS) at 5× final concentration. Use a final substrate concentration close to the Km value. If the Km of the substrate for the PTP is not known, choose 50 µM as the substrate concentration, for instance, to make a 250 µM SS. Keep SS at room temperature in the dark until use. Prepare fresh for each experiment.
Set up the microplate reader for a 30 min read in kinetic mode (see notes about microplate reader settings).
Using a multichannel pipette, add 5 µL of SS to each well (A1 through E5), immediately spin-down the plate for a few seconds, and start measurements using the microplate reader.
Analyze the fluorescence intensity data using linear regression and calculate regression coefficients (R2) and initial velocity rate (V) from the slopes, using programs such as Magellan (Tecan plate reader software), GraphPad Prism, Microsoft Excel, or similar. Representative data are shown in Figure 3.
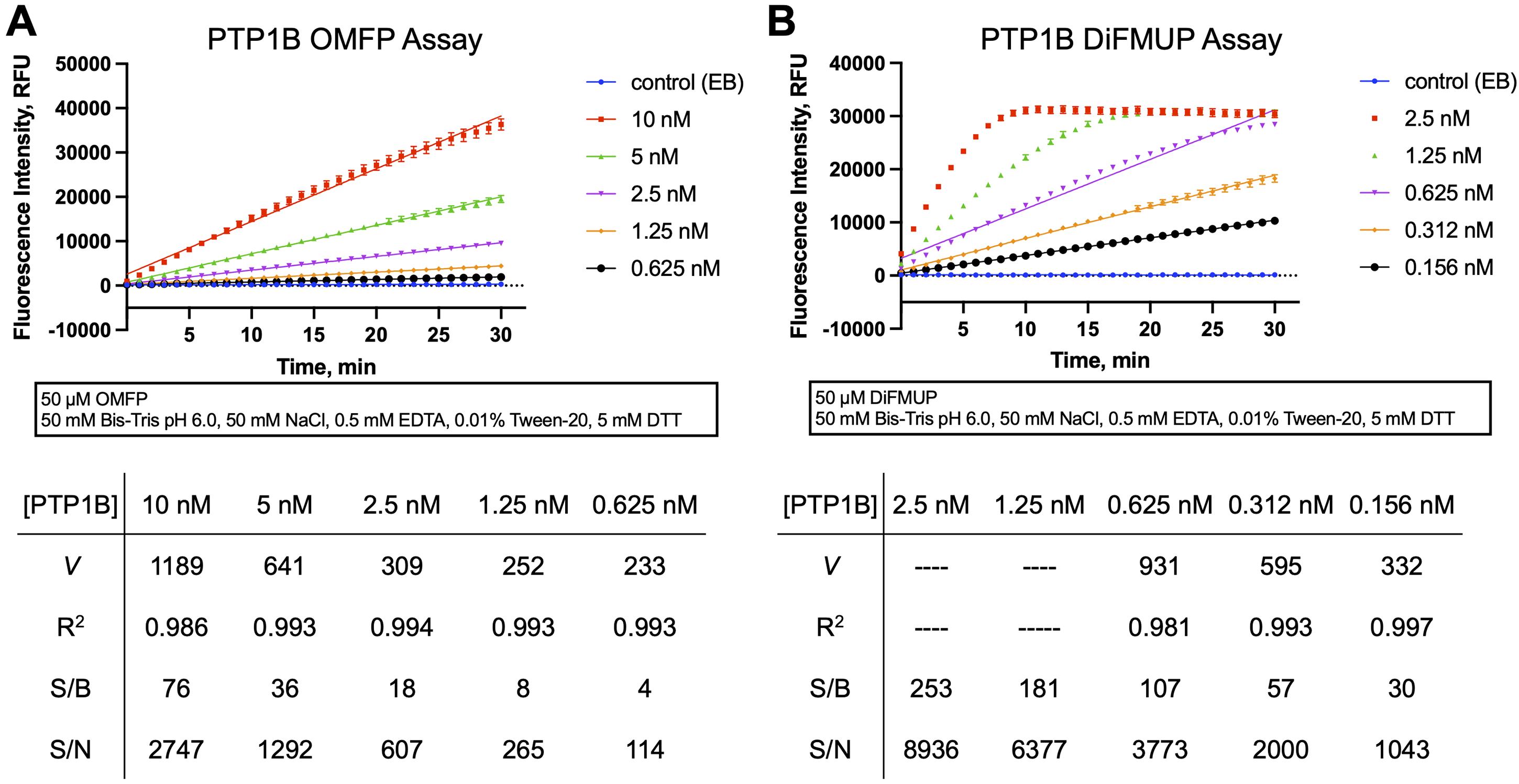
Figure 3. Continuous (kinetic) phosphatase activity assay. Representative PTP activity progression curves using OMFP (A) or DiFMUP (B) as the substrate. Plots show fluorescence intensity values in relative fluorescence units (RFU) read every minute over a 30 min period for reactions with the indicated concentrations of PTP1B or enzyme buffer (EB; no-enzyme control, background). Each data point represents the average from four reaction wells ± standard deviation (SD). Simple linear regression has been fitted to the data. Values for initial velocity rates V (slopes), regression coefficients (R2), signal to background (S/B), and signal to noise (S/N) are presented in the tables below. S/B and S/N ratios were calculated from relative fluorescence values at the 10 min time point.Note: The main criteria for determining a suitable enzyme concentration for inhibition assays are the linearity of the PTP reaction and the S/B and S/N ratios of the reaction. Linearity over the 30 min reaction can be assumed with a linear regression coefficient of R2 > 0.99. S/B and S/N ratios calculated from raw fluorescent emission values at the 10 min time point should be >10. (The 10 min time point corresponds to the length of the PTP reaction we recommend for the kinetic inhibition assays.) Judging from the progression curves shown for the PTP1B reaction with OMFP ( Figure 3A), we would recommend a PTP1B concentration of 2.5 nM, which yields acceptable linearity and S/B and S/N ratios. From the progression curves of the PTP1B reaction with DiFMUP (Figure 3B ) it is apparent that at the top two PTP1B concentrations (2.5 nM and 1.25 nM) the substrate is completely depleted within the 30 min reaction period. At 0.625 nM PTP1B, the progression curve still starts to plateau after 20 min, resulting in an R2 of 0.98. The lowest two PTP1B concentrations tested (0.312 nM and 0.156 nM) yield acceptable linearity in this experiment.
SHP2-WT Activity Assay using DiFMUP
Note: SHP2-WT adopts an autoinhibited conformation, in which one of its two SH2 domains blocks access to the active site. To assay SHP2-WT activity and test SHP2 inhibitors, a dually phosphorylated peptide derived from the insulin receptor substrate 1 (IRS-1) serves as a surrogate binding protein and is used to activate SHP2-WT (Raveendra-Panickar et al., 2022). The procedure described below determines the optimal IRS-1 concentration for SHP2-WT activation.
Thaw the protein stock solution on ice and mix gently.
Prepare SB, EB, and EIS as described in section A.
Prepare SHP2-WT ES at 2.5× final concentration (1 mL). For instance, for 0.5 nM final SHP2-WT concentration make a 1.25 nM ES: add 2.5 µL of SHP2-WT EIS to 997.5 µL EB, mix gently, and keep on ice.
Prepare serial dilutions of the IRS-1 peptide at 2.5× final concentration in EB using a 1 mM peptide stock solution. Choose dilutions in a wide range, e.g., 10 µM, 5 µM, 2.5 µM, 1.25 µM, 0.625 µM, 0.3125 µM, 0.15625 µM, and 0 µM, for final concentrations of 4 µM, 2 µM, 1 µM, 0.5 µM, 0.25 µM, 0.125 µM, 0.0625 µM, and 0 µM (Figure 4A).
Prepare eight 1.5 mL Eppendorf tubes on ice.
Dispense 198 µL of EB into tube 1.
Dispense 100 µL of EB into tubes 2–8.
Add 2 µL of the IRS-1 stock solution into tube 1, and gently mix.
Transfer 100 µL from tube 1 into tube 2 and mix, by gently pipetting up and down.
Continue the serial dilution until tube 7.
Discard 100 µL of the solution from tube 7.
Add 100 µL of SHP2-WT ES into each of the eight tubes and incubate on ice for 20 min.
Dispense solutions to 384-well assay plate for a quadruplicate experiment (Figure 4B).
Add 20 µL of EB to wells A1–A4 (no enzyme control).
Add 20 µL of tube 8 to wells B1–B4 (SHP2-WT, no peptide).
Add 20 µL of tube 7 to wells C1–C4 (SHP2-WT, lowest peptide concentration).
Continue with tubes 6 through 1, dispensing to wells D1–D4, E1–E4, F1–F4, G1–G4, H1–H4, I1–I4.
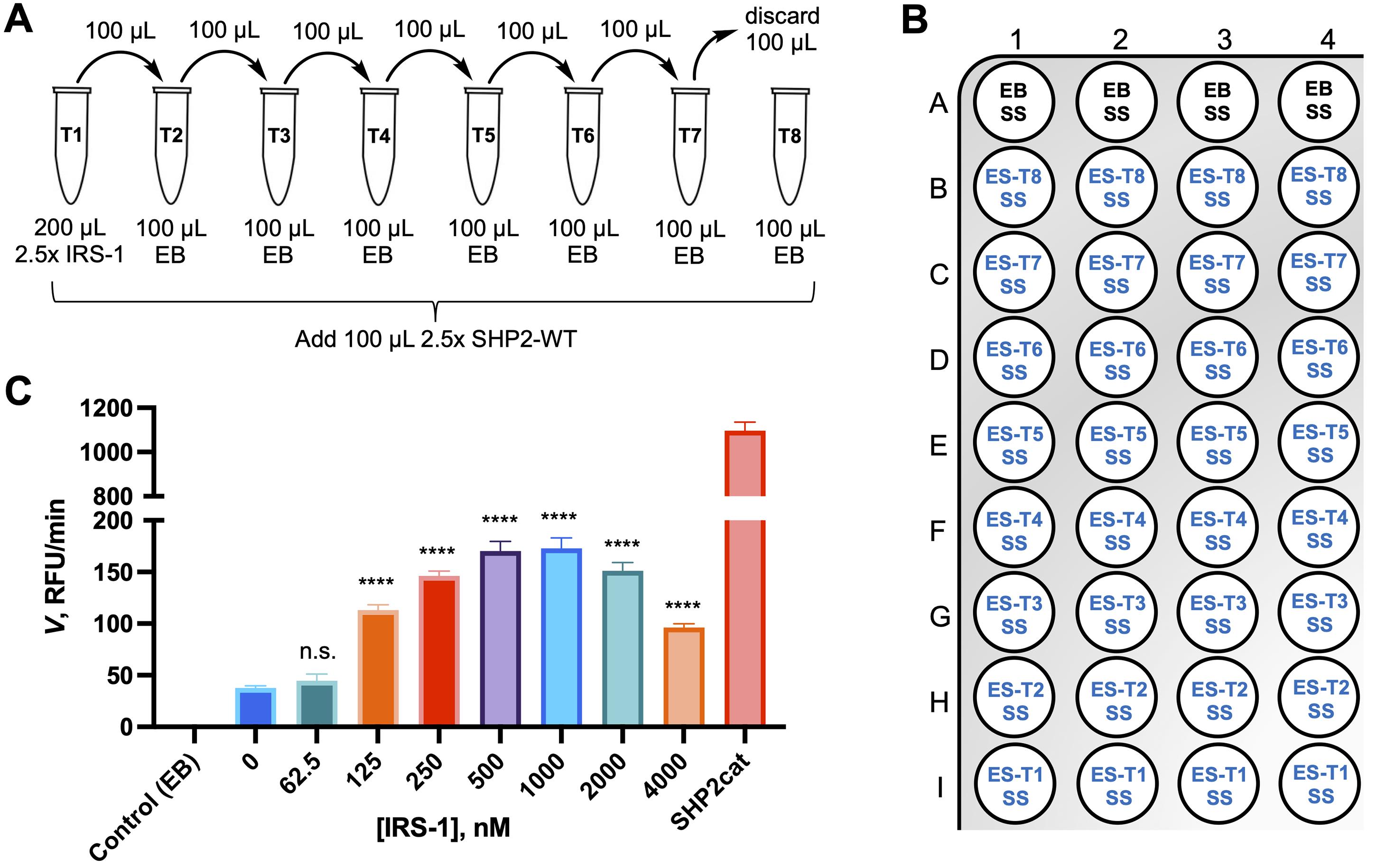
Figure 4. IRS-1 titration for SHP2-WT activity assay. (A) IRS-1 peptide serial dilution scheme. (B) Assay plate layout. Add 20 µL of enzyme buffer (EB; background control) or enzyme solutions (ES), followed by 5 µL of substrate solution (SS). (C) SHP2-WT (0.5 nM) activity (expressed as initial velocity rate V) in the presence of different IRS-1 peptide concentrations using DiFMUP (100 µM) as the substrate. For comparison, the activity of recombinant SHP2 catalytic domain (SHP2cat; 0.5 nM) without IRS-1 peptide is included. Enzyme buffer was used in the no-enzyme control experiment. The data represent the mean ± SD. Statistical significance of SHP2-WT activation by IRS-1 was determined using the unpaired t-test (n = 4; n.s.: not significant; ****p < 0.0001).Spin-down the plate for a few seconds, cover the plate, and incubate at room temperature for 20 min.
Prepare SS at 5× final concentration (1 mL). Add 50 µL of DiFMUP stock solution to 950 µL of SB to make a 500 µM DiFMUP SS for a final DiFMUP concentration of 100 µM. Keep SS at room temperature in the dark until use. Prepare fresh for each experiment.
Set up the microplate reader for a 10 min read in kinetic mode (see notes about microplate reader settings).
Using a multichannel pipette, add 5 µL of SS to each well (A1 through I4). Immediately spin-down the plate for a few seconds and start measurements using the microplate reader.
Analyze the fluorescence intensity data using linear regression and calculate R2 and V from the slopes using programs such as Magellan (Tecan plate reader software), GraphPad Prism, Microsoft Excel, or similar.
Note: As shown in Figure 4C, the maximum activation of SHP2-WT is reached at an IRS-1 peptide concentration of 500 nM, which we employed in the SHP2-WT Michaelis–Menten and inhibition assays described previously (Raveendra-Panickar et al., 2022).
Michaelis–Menten Kinetics
Note: The purpose of the Michaelis–Menten experiment is to determine the Michaelis–Menten constant (Km) of the chosen substrate for a specific PTP under specific assay conditions. The Km value is important for the inhibition assays, in which the substrate is typically used at a concentration equal to the Km. This makes inhibitor IC50 values comparable between different PTPs.
Thaw the protein stock solution on ice and mix gently.
Prepare SB, EB, and EIS as described in section A.
Prepare ES at 1.25× final concentration (2 mL) and keep on ice.
Note: A suitable final enzyme concentration for this experiment should be based on the results from the PTP activity assay (section A).
Prepare a serial dilution of substrate at 5× final concentration (Figure 5, step 1). Prepare eight to ten different SS spanning approximately three orders of magnitude in concentration. For instance, prepare the highest concentrated SS at 1 mM (for a 200 µM final concentration) and do a 1:1 serial dilution. Prepare the serial dilution in a 384-well plate for convenient transfer to reaction wells, using a multichannel pipette.
Prepare the highest concentrated SS (SS1) in an Eppendorf tube. For instance, mix 10 µL of a 10 mM substrate stock solution with 90 µL of SB for SS1 at 1 mM.
Add 30 µL of SS1 to well A6 of a 384-well assay plate.
Add 30 µL of SB to wells B6–J6.
Add 30 µL of SS1 to well B6 and mix, by gently pipetting up and down.
Transfer 30 µL from well B6 to well C6 and mix, by gently pipetting up and down.
Continue the serial dilution through well J6.
Add 20 µL of EB to wells A1–J1 (no-enzyme control used for background correction).
Add 20 µL of ES to wells A2–J4 (enzyme reaction wells in triplicate for each substrate concentration) (Figure 5, step 2).
Spin-down the plate for a few seconds, cover the plate, and incubate at room temperature for 20 min.
Set up the microplate reader for a 10 min read in kinetic mode (see notes about microplate reader settings).
Using a multichannel pipette, transfer 5 µL of SS from column 6 (rows A–J) to columns 1–4 (rows A–J) (Figure 5, step 2). Immediately spin-down the plate for a few seconds and start measurements using the microplate reader.
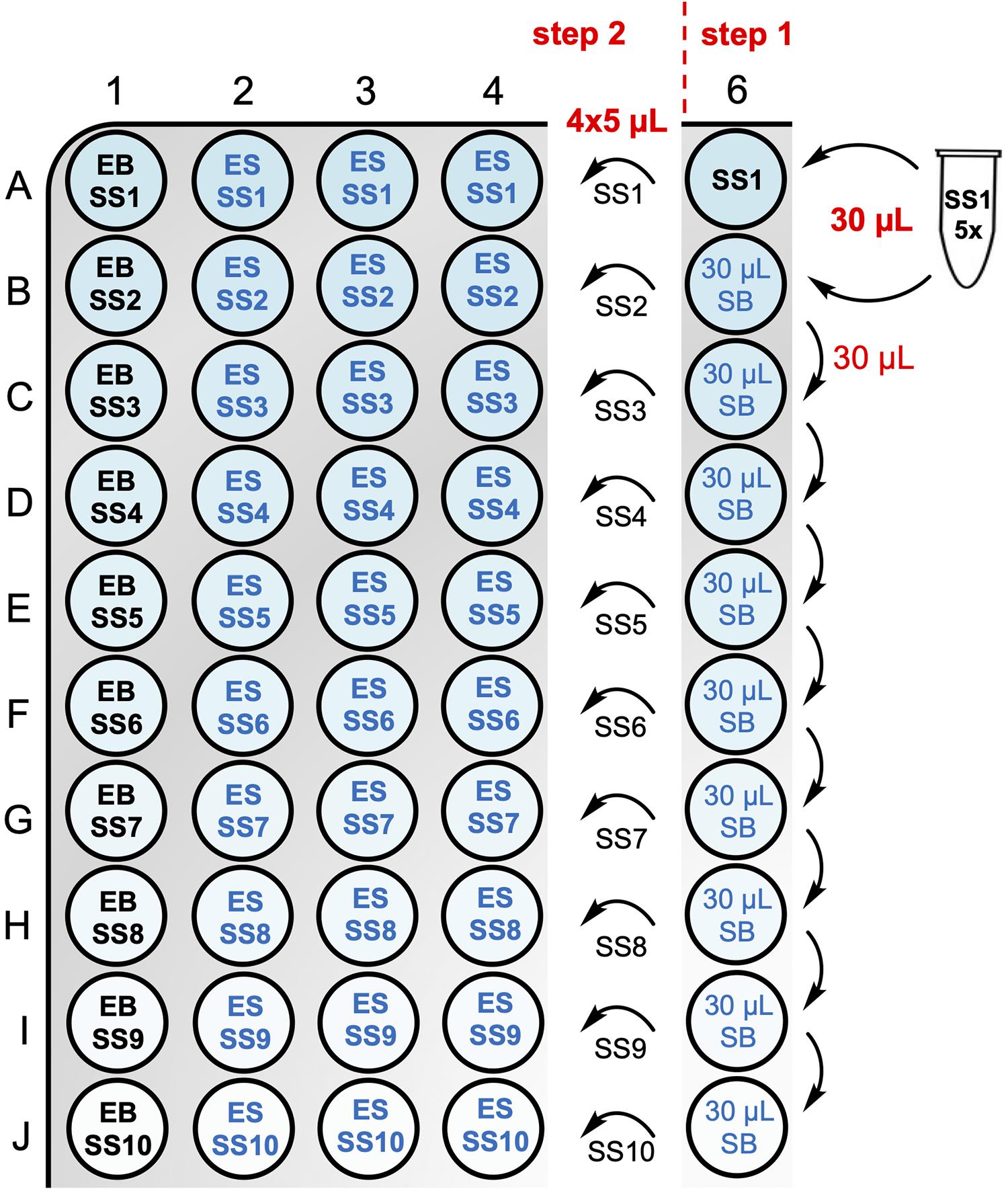
Figure 5. Substrate serial dilution scheme and assay plate layout for Michaelis–Menten kinetics assay. In step 1, a substrate serial dilution (1:1) is prepared at 5× final concentration. In step 2, 5 µL of each substrate solution (SS) is transferred into column 1, which serves as the background control and contains 20 µL of enzyme buffer (EB), and columns 2–4, which serve as the triplicate PTP reaction wells and contain 20 µL of enzyme solution (ES).Analyze the fluorescence intensity data from columns 1–4 (rows A–J) using linear regression and calculate R2 and V from the slopes using programs such as Magellan (Tecan plate reader software), GraphPad Prism, Microsoft Excel, or similar.
Analyze the background-corrected V values using the Michaelis–Menten equation and non-linear regression, and a program such as GraphPad Prism. Data from a representative Michaelis–Menten experiment using STEP46 with OMFP are shown in Figure 6.
Note: It is important to ensure that the PTP reaction is within the linear range for all concentrations included in the analysis. If necessary, adjust the enzyme concentration. Make sure the substrate concentrations cover concentration ranges below and above the Km value. For SHP2-WT, the Michaelis–Menten experiment should be conducted in the presence of IRS-1 peptide at a concentration as determined in section B. The SHP2-WT ES containing IRS-1 peptide should be prepared as described in section B.
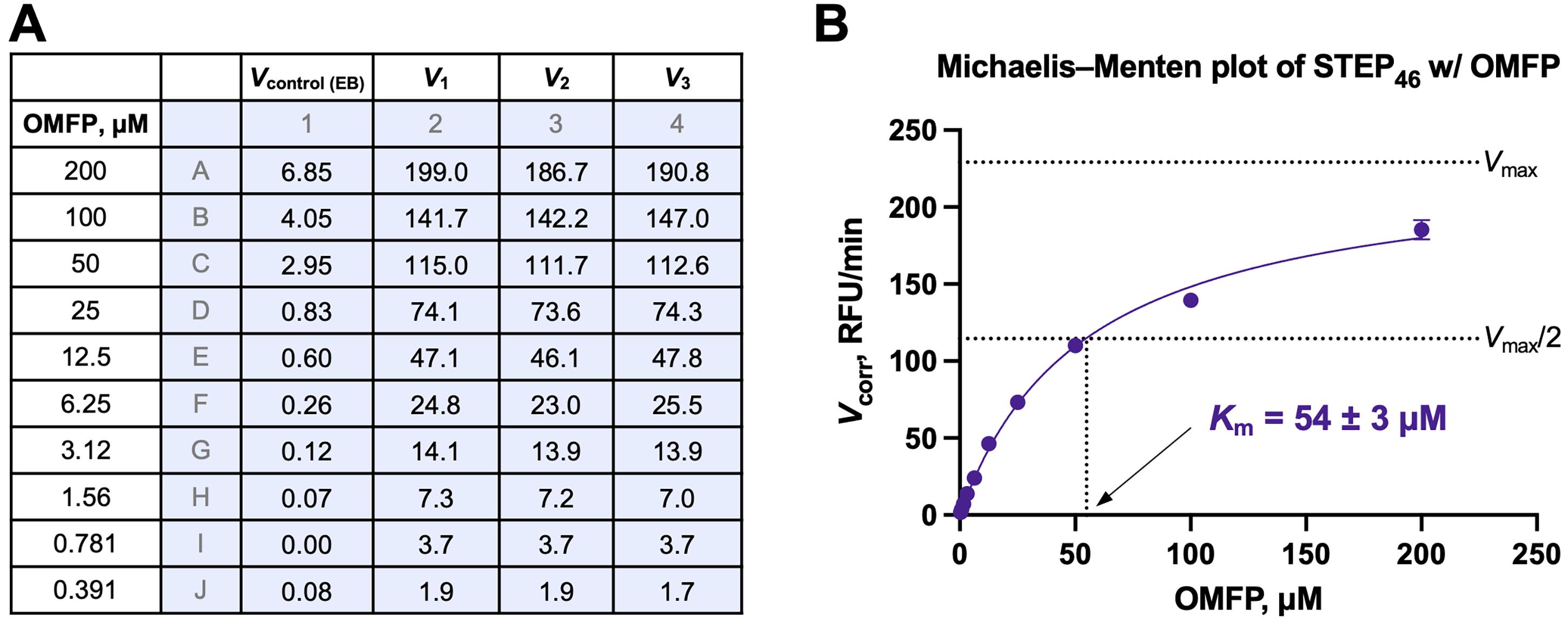
Figure 6. Michaelis–Menten kinetics. (A) Initial velocities rates (V) in relative fluorescence units per minute (RFU/min) from a Michaelis–Menten experiment for STEP46 (2.5 nM) using OMFP at the indicated concentrations. Initial rates for the enzyme buffer (EB) control (Vcontrol (EB); background) and for the STEP46 reaction in triplicate ( V1–3) were calculated from raw fluorescence emission data using the Magellan Tecan Microplate Reader software. (B) Michaelis–Menten plot using the background-corrected initial rates (V1-corr, V2-corr, V3-corr) for STEP46 from (A). The data (represented as the mean ± SD) was fitted to the Michaelis–Menten equation model (eq. 5), and the Michaelis–Menten constant (Km) was calculated using GraphPad Prism. The dashed lines indicate the STEP46 maximum velocity (Vmax) and half-maximum velocity (Vmax/2).10-Point Dose-Response PTP Inhibition Assay
Note: We used a Labcyte Echo® 555 Liquid Handler to spot DMSO (for controls) and compound solutions in DMSO (for 10-point dose-responses) via acoustic droplet dispensing. For a standard volume 384-well assay plate with a 25 μL total assay volume, we transferred 250 nL DMSO solution into each well. For manual transfer of compound stock solutions using a pipette, we recommend transferring no less than 1 μL (for accuracy reasons) and keeping the final DMSO concentration ≤5%. We recommend testing a wide range of compound concentrations of at least four log steps (e.g., 100, 33, 11, 3.7, 1.2, 0.41, 0.14, 0.045, 0.015, 0.005 μM final compound concentration). The provided volumes for ES and SS are for testing of up to four 384-well assay plates and may be adjusted according to needs.
Prepare a black, standard volume 384-well assay plate. Spot 250 nL of DMSO into negative and positive control wells (columns 1 and 2, respectively). Spot 250 nL of compound DMSO solutions in triplicate into columns 3–23.
Note: The total number of compounds that can be tested per 384-well plate is 11 (see Figure 7A for the plate map).
Thaw the protein stock solution on ice and mix gently.
Prepare SB (50 mL): add 5 µL of Tween-20 to 50 mL of Bis-Tris pH 6 buffer (see Recipes for details).
Note: Store SB at room temperature for no longer than one week.
Prepare EB (50 mL): add 250 µL of a 1 M DTT stock solution to 50 mL of SB.
Note: Keep EB on ice for no longer than eight hours.
Prepare 500 nM EIS (1 mL). Use EB to dilute PTP stock solution to 500 nM and keep on ice.
Prepare ES (approximately 45 mL) at 1.25× final concentration in a 50 mL conical tube. For instance, for 2.5 nM final PTP concentration, add 290 µL of EIS to 46.11 mL of EB, mix gently, and keep on ice.
Note: A suitable final enzyme concentration for the inhibition assay should be based on the results from the PTP activity assay (section A). For experiments with OMFP, we typically use 2.5 or 5 nM PTP final concentration. For experiments with DiFMUP, we typically use ≤0.5 nM PTP final concentration.
Prepare SS (approximately 15 mL) at 5× final substrate concentration in a 15 mL conical tube. For instance, for a 6 µM final substrate concentration, add 45 µL of the 10 mM substrate stock solution to 14.955 mL of SB, mix, and store at room temperature in the dark until use.
Note: The final substrate concentration for a specific PTP is determined by the substrate Km value obtained in the Michaelis–Menten experiment (section C).
Using a multichannel pipette, add 20 µL of EB to column 2 (positive control) of the spotted 384-well assay plate.
Using a MultidropTM Combi reagent dispenser, add 20 µL of ES to all wells, except those in column 2.
Using a tabletop centrifuge with a swinging-bucket rotor, spin-down the plate for a few seconds, cover the plate with a lid (or with an additional plate), and incubate at room temperature for 20 min.
Set up the microplate reader for a 10 min read in kinetic mode (see notes about microplate reader settings).
Using a MultidropTM Combi reagent dispenser, add 20 µL of SS to the entire plate, immediately spin-down the plate for a few seconds, and start measurements using the microplate reader.
Analyze the fluorescence intensity data using linear regression and calculate R2 and V from the slopes using programs such as Magellan (Tecan plate reader software), GraphPad Prism, Microsoft Excel, or similar. Make sure the linearity of the PTP reaction is acceptable and Z’ values are ≥0.5. For details on Z’-factor definition, please see the Data analysis section below.
Normalize the initial velocity rates using the positive (100% inhibition) and negative (0% inhibition) control values and analyze the normalized data using a nonlinear regression dose-response inhibition model (log inhibitor vs. response, variable slope, four parameters) using programs such as GraphPad Prism or CBIS, to obtain IC50 values. Data from a representative 10-point dose-response inhibition assay for VHR with OMFP are shown in Figure 7.
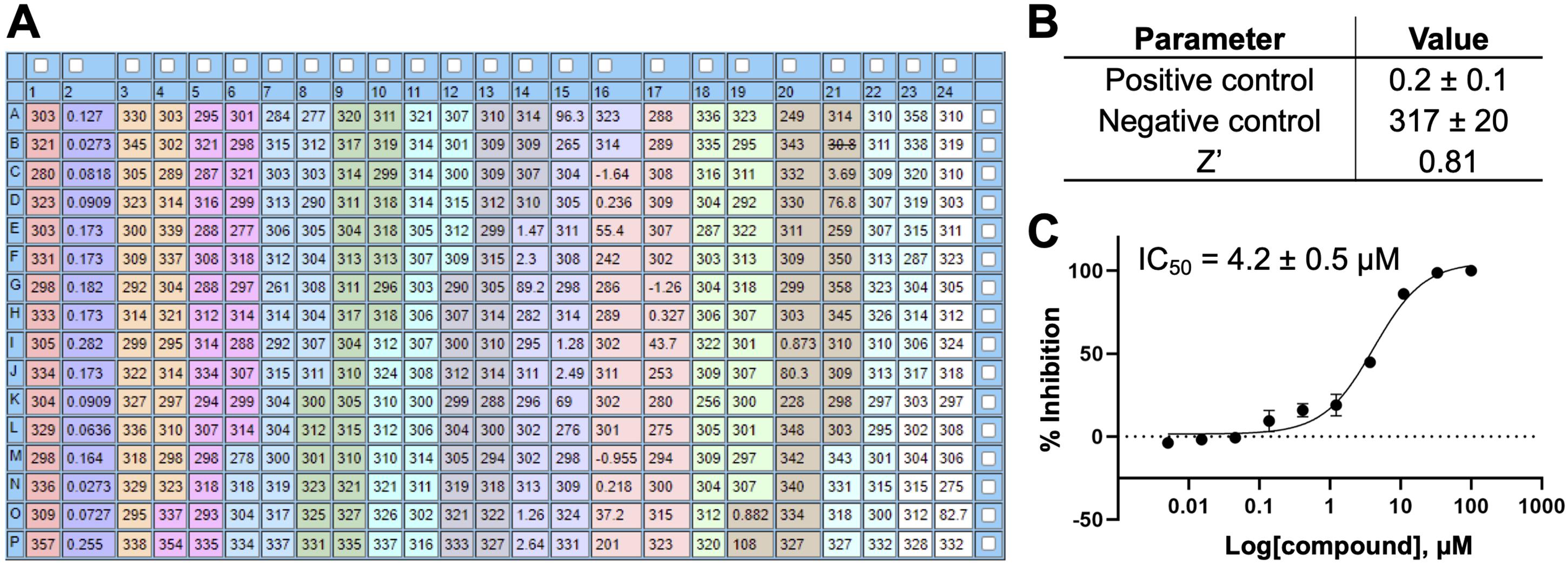
Figure 7. Representative data from a 10-point dose-response VHR inhibition assay. (A) Plate setup and initial velocity rates. Column 1 serves as the negative control (vehicle control). Column 2 serves as the positive control (contains no enzyme). Eleven candidate compounds (marked with different colors) were tested in a 10-point dose-response format in triplicate (100, 33, 11, 3.7, 1.2, 0.41, 0.14, 0.045, 0.015, and 0.005 μM final compound concentration). Wells K23 through P24 (white) do not contain DMSO or compound and are excluded from the analysis. (B) Assay plate statistical data. Initial rates for positive and negative controls are represented as mean ± SD. For Z’-factor definition and calculation see Data analysis section. (C) Example of normalized inhibition data and fitted IC50 curve (IC50 ± SE; analyzed in GraphPad Prism).
Data analysis
Statistical assay parameters
Signal to background (S/B):

where 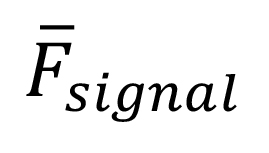 and
and ![]() are mean fluorescence values of reaction and background (no-enzyme control), respectively.
are mean fluorescence values of reaction and background (no-enzyme control), respectively.
Signal to noise (S/N):

where SDBKGD is the standard deviation of background (zero-enzyme reaction).
Z’-factor:

where SDn and SDp and ![]() and
and ![]() are the standard deviations and means of the negative and positive control initial velocity values, respectively. The Z’-factor is a statistical parameter for the quality of the assay without intervention of test compounds (Zhang, J. H. et al., 1999). Z’ values above 0.5 indicate acceptable assay performance.
are the standard deviations and means of the negative and positive control initial velocity values, respectively. The Z’-factor is a statistical parameter for the quality of the assay without intervention of test compounds (Zhang, J. H. et al., 1999). Z’ values above 0.5 indicate acceptable assay performance.
Initial velocities/slopes
All experiments were run in kinetic mode with a fluorescence intensity read every minute. The slopes of the progression curves were determined in Magellan (Tecan plate reader software), or by fitting a simple linear regression model to the kinetic data in GraphPad Prism or Excel, using the following equation:

where F represents fluorescence intensity in relative fluorescence units (RFU), x represents time (min), V represents the represents initial velocity (slope; RFU/min), and b represents the y-axis intercept (1/RFU).
Michaelis–Menten Kinetics
To determine the Michaelis–Menten constant Km (substrate concentration that yields the half-maximal velocity), the Michaelis–Menten model and GraphPad Prism were used to fit the data to the Michaelis–Menten equation:

where V represents initial velocity, Vmax the maximum enzyme velocity, and c the substrate concentration.
IC50 Determination
Initial velocity rates were normalized using the mean positive (100% inhibition) and mean negative (0% inhibition) control values to calculate the percentage of inhibition (%inhib):

where Vnc indicates the mean initial velocity rate from the negative control wells (vehicle control), Vpc indicates the mean initial velocity rate from the positive control wells (no-enzyme control), and Vinhib indicates the initial velocity rate from the reaction wells with compound at a specific concentration.
IC50 values were calculated from the %inhib by nonlinear regression, using CBIS or GraphPad Prism software. The data were fitted to a log[Inhibitor] vs. response, variable slope (four parameters) model:

where Bottom and Top are the bottom and top plateau, respectively, X is the ligand concentration, and HillSlope is the steepness of the curve.
Notes about the MultidropTM Combi reagent dispenser
It is important to fully prime the cassette tubes to avoid bubbles in the plastic tubes, which may result in dispensing errors. After each usage, flush tubes thoroughly with Milli-Q water, 70% ethanol, and again Milli-Q water.
Microplate Reader Settings – OMFP Assay
Experiment type: Kinetic Loop
Mode: Kinetic
Kinetic Cycles: 11
Interval time: 1 min
Measurement type: Fluorescence intensity
Mode: Fluorescence top reading
Excitation: Filter
Excitation wavelength: 485 nm
Excitation bandwidth: 20 nm
Emission: Filter
Emission wavelength: 535 nm
Emission bandwidth: 25 nm
Gain: 40 Manual
Mirror: automatic
Number of flashes: 10
Integration time: 40 µs
Lag time: 0 µs
Settle time: 0 ms
Total kinetic run time: 9 min 59 s
Microplate Reader Settings – DiFMUP Assay
The settings are the same as for the OMFP assay except:
Excitation wavelength: 360 nm
Excitation bandwidth: 35 nm
Emission: Filter
Emission wavelength: 465 nm
Emission bandwidth: 35 nm
Recipes
Bis-Tris buffer
50 mM Bis-Tris pH 6.0, 50 mM NaCl, 0.5 mM EDTA
In 800 mL of Milli-Q water, dissolve 10.5 g of Bis-Tris, 2.9 g of NaCl, and 208 mg of EDTA. Set pH to 6.0 with 1 M hydrochloric acid. Fill with Milli-Q water to 1 L. Store at 4 °C for up to one year.
10 mM OMFP stock solution in DMSO
Dissolve 26.3 mg of OMFP cyclohexylammonium salt in 5 mL of pure DMSO. Sonication (using a sonicator bath) may be needed to fully dissolve OMFP to a clear solution. Prepare aliquots as needed, and store at -80 °C.
10 mM DiFMUP stock solution in water
Dissolve 10 mg of DiFMUP in 3.42 mL of Milli-Q water. Prepare aliquots as needed, and store at -80 °C.
1 M DTT stock solution
Dissolve 771 mg of DTT in 5 mL of Milli-Q water. Prepare aliquots as needed, and store at -80 °C.
Acknowledgments
Research reported in this publication was supported by the National Institutes of Health under Awards Numbers R01AG065387, R21AG067155, R21AI160161, and R21CA195422 (to L.T.), and NCI Cancer Center Support Grant P30CA030199. Funding for this project has also been provided by the Polish National Science Center under award number UMO-2019/32/T/ST4/00071 (to M.B.). Additionally, this project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Chemical Biology Consortium Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The protocols described in here are partly derived from our recent research paper (Raveendra-Panickar et al., 2022).
Competing interests
The authors declare that they have no conflicts of interest with the contents of this article.
References
- Bessey, O. A., Lowry, O. H. and Brock, M. J. (1946). A method for the rapid determination of alkaline phosphates with five cubic millimeters of serum. J Biol Chem 164: 321-329.
- Chen, Y. N., LaMarche, M. J., Chan, H. M., Fekkes, P., Garcia-Fortanet, J., Acker, M. G., Antonakos, B., Chen, C. H., Chen, Z., Cooke, V. G., et al. (2016). Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535(7610): 148-152.
- Cohen, P., Cross, D. and Janne, P. A. (2021). Kinase drug discovery 20 years after imatinib: progress and future directions. Nat Rev Drug Discov 20(7): 551-569.
- Feng, B. Y. and Shoichet, B. K. (2006). A detergent-based assay for the detection of promiscuous inhibitors. Nat Protoc 1(2): 550-553.
- Goebel-Goody, S. M., Baum, M., Paspalas, C. D., Fernandez, S. M., Carty, N. C., Kurup, P. and Lombroso, P. J. (2012). Therapeutic implications for striatal-enriched protein tyrosine phosphatase (STEP) in neuropsychiatric disorders. Pharmacol Rev 64(1): 65-87.
- Groen, A., Lemeer, S., van der Wijk, T., Overvoorde, J., Heck, A. J., Ostman, A., Barford, D., Slijper, M. and den Hertog, J. (2005). Differential oxidation of protein-tyrosine phosphatases. J Biol Chem 280(11): 10298-10304.
- Huang, Z., Wang, Q., Ly, H. D., Gorvindarajan, A., Scheigetz, J., Zamboni, R., Desmarais, S. and Ramachandran, C. (1999). 3,6-Fluorescein Diphosphate: A Sensitive Fluorogenic and Chromogenic Substrate for Protein Tyrosine Phosphatases*. J Biomol Screen 4(6): 327-334.
- Hunter, T. (2009). Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol 21(2): 140-146.
- Labbe, D. P., Hardy, S. and Tremblay, M. L. (2012). Protein tyrosine phosphatases in cancer: friends and foes! Prog Mol Biol Transl Sci 106: 253-306.
- Lambert, L. J., Grotegut, S., Celeridad, M., Gosalia, P., Backer, L. J., Bobkov, A. A., Salaniwal, S., Chung, T. D., Zeng, F. Y., Pass, I., et al. (2021). Development of a Robust High-Throughput Screening Platform for Inhibitors of the Striatal-Enriched Tyrosine Phosphatase (STEP). Int J Mol Sci 22(9): 4417.
- Menegatti, A. C. O. (2022). Targeting protein tyrosine phosphatases for the development of antivirulence agents: Yersinia spp. and Mycobacterium tuberculosis as prototypes. Biochim Biophys Acta Proteins Proteom 1870(5): 140782.
- Raveendra-Panickar, D., Finlay, D., Layng, F. I., Lambert, L. J., Celeridad, M., Zhao, M., Barbosa, K., De Backer, L. J. S., Kwong, E., Gosalia, P., et al. (2022). Discovery of novel furanylbenzamide inhibitors that target oncogenic tyrosine phosphatase SHP2 in leukemia cells. J Biol Chem 298(1): 101477.
- Singh, P., Dejager, L., Amand, M., Theatre, E., Vandereyken, M., Zurashvili, T., Singh, M., Mack, M., Timmermans, S., Musumeci, L., et al. (2015). DUSP3 Genetic Deletion Confers M2-like Macrophage-Dependent Tolerance to Septic Shock. J Immunol 194(10): 4951-4962.
- Song, Y., Wang, S., Zhao, M., Yang, X. and Yu, B. (2022). Strategies Targeting Protein Tyrosine Phosphatase SHP2 for Cancer Therapy. J Med Chem 65(4): 3066-3079.
- Stanford, S. M. and Bottini, N. (2017). Targeting Tyrosine Phosphatases: Time to End the Stigma. Trends Pharmacol Sci 38(6): 524-540.
- Tautz, L., Critton, D. A. and Grotegut, S. (2013). Protein tyrosine phosphatases: structure, function, and implication in human disease. Methods Mol Biol 1053: 179-221.
- Tautz, L., Pellecchia, M. and Mustelin, T. (2006). Targeting the PTPome in human disease. Expert Opin Ther Targets 10(1): 157-177.
- Tautz, L., Senis, Y. A., Oury, C. and Rahmouni, S. (2015). Perspective: Tyrosine phosphatases as novel targets for antiplatelet therapy.Bioorg Med Chem 23(12): 2786-2797.
- Tautz, L. and Sergienko, E. A. (2013). High-throughput screening for protein tyrosine phosphatase activity modulators. Methods Mol Biol 1053: 223-240.
- Tierno, M. B., Johnston, P. A., Foster, C., Skoko, J. J., Shinde, S. N., Shun, T. Y. and Lazo, J. S. (2007). Development and optimization of high-throughput in vitro protein phosphatase screening assays. Nat Protoc 2(5): 1134-1144.
- Tonks, N. (2013). Protein tyrosine phosphatases--from housekeeping enzymes to master regulators of signal transduction.FEBS J 280(2): 346-378.
- Vainonen, J. P., Momeny, M. and Westermarck, J. (2021). Druggable cancer phosphatases. Sci Transl Med 13(588): eabe2967.
- Vang, T., Miletic, A. V., Arimura, Y., Tautz, L., Rickert, R. C. and Mustelin, T. (2008). Protein tyrosine phosphatases in autoimmunity. Annu Rev Immunol 26: 29-55.
- Welte, S., Baringhaus, K. H., Schmider, W., Muller, G., Petry, S. and Tennagels, N. (2005). 6,8-Difluoro-4-methylumbiliferyl phosphate: a fluorogenic substrate for protein tyrosine phosphatases. Anal Biochem 338(1): 32-38.
- Zhang, J. H., Chung, T. D. and Oldenburg, K. R. (1999). A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 4(2): 67-73.
- Zhang, Z. Y., Dodd, G. T. and Tiganis, T. (2015). Protein Tyrosine Phosphatases in Hypothalamic Insulin and Leptin Signaling. Trends Pharmacol Sci 36(10): 661-674.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Baranowski, M. R., Wu, J., Han, Y. N., Lambert, L. J., Cosford, N. D. P. and Tautz, L. (2022). Protein Tyrosine Phosphatase Biochemical Inhibition Assays. Bio-protocol 12(18): e4510. DOI: 10.21769/BioProtoc.4510.
- Raveendra-Panickar, D., Finlay, D., Layng, F. I., Lambert, L. J., Celeridad, M., Zhao, M., Barbosa, K., De Backer, L. J. S., Kwong, E., Gosalia, P., et al. (2022). Discovery of novel furanylbenzamide inhibitors that target oncogenic tyrosine phosphatase SHP2 in leukemia cells. J Biol Chem 298(1): 101477.
Category
Biochemistry > Protein > Activity
Drug Discovery > Drug Screening
Drug Discovery > Drug Design
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.