- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Visualization, Quantification, and Modeling of Endogenous RNA Polymerase II Phosphorylation at a Single-copy Gene in Living Cells
Published: Vol 12, Iss 15, Aug 5, 2022 DOI: 10.21769/BioProtoc.4482 Views: 2960
Reviewed by: Zinan ZhouPooja VermaRama Reddy Goluguri
Original research article
The authors used this protocol in:

May 2021

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
In eukaryotic cells, RNA Polymerase II (RNAP2) is the enzyme in charge of transcribing mRNA from DNA. RNAP2 possesses an extended carboxy-terminal domain (CTD) that gets dynamically phosphorylated as RNAP2 progresses through the transcription cycle, therefore regulating each step of transcription from recruitment to termination. Although RNAP2 residue-specific phosphorylation has been characterized in fixed cells by immunoprecipitation-based assays, or in live cells by using tandem gene arrays, these assays can mask heterogeneity and limit temporal and spatial resolution. Our protocol employs multi-colored complementary fluorescent antibody-based (Fab) probes to specifically detect the CTD of the RNAP2 (CTD-RNAP2), and its phosphorylated form at the serine 5 residue (Ser5ph-RNAP2) at a single-copy HIV-1 reporter gene. Together with high-resolution fluorescence microscopy, single-molecule tracking analysis, and rigorous computational modeling, our system allows us to visualize, quantify, and predict endogenous RNAP2 phosphorylation dynamics and mRNA synthesis at a single-copy gene, in living cells, and throughout the transcription cycle.
Graphical abstract:
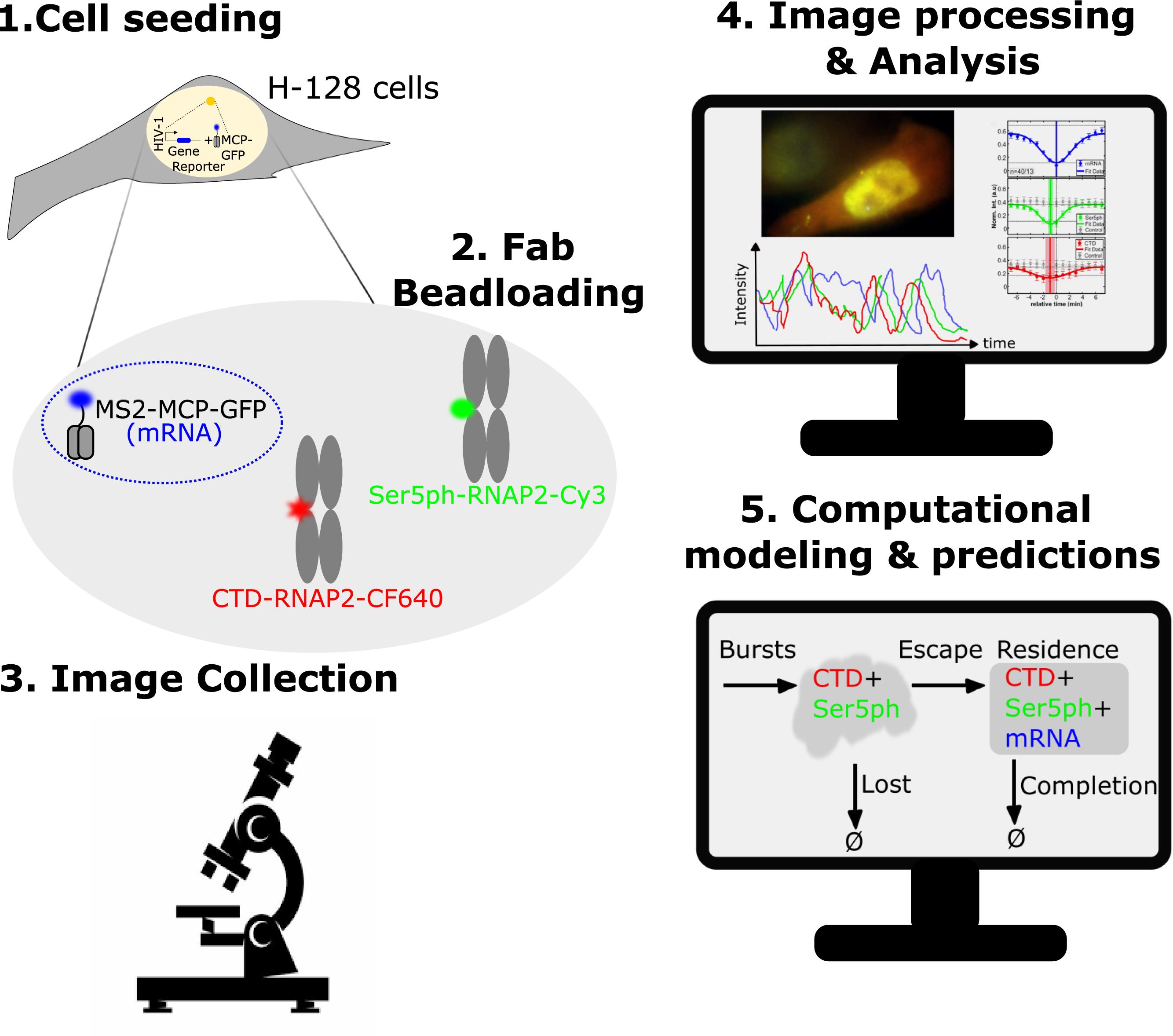
Schematic of the steps for visualizing, quantifying, and predicting RNAP2 phosphorylation at a single-copy gene.
Background
Interest in the phosphorylation status of the CTD increased due to recent studies showing its correlation with RNAP2 clustering (Cissé et al., 2013; Cho et al., 2016; Boehning et al., 2018; Pancholi et al., 2021). These findings suggest that RNAP2 clusters form around gene promoters, and early in the transcription cycle, they are enriched in unphosphorylated-RNAP2 and Ser5ph-RNAP2 (Nagashima et al., 2019). RNAP2 phosphorylation distribution along the 1D genome has been extensively studied using immunoprecipitation-based assays (Heidemann et al., 2013; Harlen and Churchman, 2017). However, these techniques are performed using fixed cells and require averaging over a population of cells, limiting temporal resolution and masking heterogeneity (Coulon et al., 2013). Recent advances in fluorescent microscopy and single-molecule tracking (Tokunaga et al., 2008; Chen et al., 2014; Li et al., 2019) have overcome these limitations. Now, it is possible to monitor single RNAP2 dynamics at different locations throughout the genome (Cissé et al., 2013; Steurer et al., 2018) and specific single-copy genes (Cho et al., 2016; Li et al., 2019) pre-marked with MS2 (Tantale et al., 2016; Pichon et al., 2018) or PP7 (Larson et al., 2011; Coulon et al., 2014) RNA stem-loops. When transcribed, MS2 and PP7 tags form hairpin-like secondary structures, which are then bound by their fluorescence-labeled coating proteins, MCP and PCP, respectively. Unfortunately, previous protocols use permanent fluorescent fusion tags to track RNAP2, which cannot distinguish between the crucial RNAP2 phosphorylation states that control transcription.
Fluorescent antibody-based (Fab) probes can detect post-translational modifications to RNAP2 by binding and lighting up specific modifications to the CTD of RNAP2 in vivo (Hayashi-Takanaka et al., 2009; Stasevich et al., 2014; Kimura et al., 2015; Lyon and Stasevich, 2017). However, freely diffusing and unbound Fab probes lead to a high background; hence, this technique was previously limited to large tandem gene arrays (Stasevich et al., 2014) that include multiple copies of the same gene to enhance the signal-to-noise ratio. However, using such a protocol averages over many gene copies, and masks heterogeneity from one gene copy to another. Recently, our lab developed a protocol to measure and predict the spatiotemporal dynamics of RNAP2 phosphorylation and mRNA production throughout the transcription cycle of a single-copy gene. This protocol, which we describe here, combines multicolor single-molecule microscopy, complementary Fabs, and rigorous computational modeling (Forero-Quintero et al., 2021). For our system (Forero-Quintero et al., 2021), we utilized an established HeLa cell line (H-128) expressing a single-copy reporter gene controlled by an HIV-1 promoter, comprising an MS2-tag and its MS2 coating protein tagged with GFP (MCP, blue) (Tantale et al., 2016). Our reporter gene is predominantly active due to persistent stimulation by Tat, leading to a bright MCP-GFP signal, that indicates the location of the transcription site within the nucleus, allowing us to observe fluctuations in mRNA synthesis in real-time (Figure 1a). Our loaded Fabs recognize the CTD of RNAP2 (CTD-RNAP2, conjugated with CF640, red) without or with residue-specific phosphorylation, and specific phosphorylation at the Serine 5 within the CTD of RNAP2 (Ser5ph-RNAP2, conjugated with Cy3, green). This combination of imaging probes makes it possible to observe RNAP2 regions enriched or depleted with Ser5ph within the nucleus (Figure 1a, b). Fab binding and unbinding from their targets occurs rapidly, making it a valuable tool to monitor temporal changes in the phosphorylation status of RNAP2 (Hayashi-Takanaka et al., 2011; Stasevich et al., 2014; Kimura et al., 2015). At the HIV-1 reporter transcription site, we typically observe both Fabs present, but on occasion, the Ser5ph-RNAP2 signal becomes dim or absent despite the presence of CTD-RNAP2 and mRNA signals. Furthermore, all signals occasionally reduce to background levels, evidencing correlated fluctuations of the signals, which are caused by natural bursts and pauses in the transcriptional activity of the reporter gene (Figure 1b). By combining these multicolor elements, we can distinguish three distinct steps of the transcription cycle at the HIV-1 reporter gene: (1) RNAP2 recruitment (marked by Fab targeting CTD-RNAP2), (2) initiation (marked by Fab targeting CTD-RNAP2 and Fab targeting Ser5ph-RNAP2), and (3) elongation (marked by both Fabs and MCP binding to mRNA) (Figure 1a). To add a quantitative interpretation to these observations, we explored many possibilities and identified a simple computational model that matches all data. The optimal model that fits all data, but which avoided overfitting, consists of five basic rate parameters that describe four reactions: (1) the recruitment of RNAP2 in geometrically distributed bursts of average size (β) and frequency (ω), (2) the departure of unsuccessful or lost RNAP2 (kab), (3) promoter escape of RNAP2 (kesc), and (4) a combined rate of transcription completion and RNAP2 release (kc). Our simple mechanistic mathematical model allows us to estimate each of the above rates with excellent precision, as well as to predict the number of unphosphorylated and/or phosphorylated RNAP2 at each state throughout the transcription cycle at the HIV-1 reporter gene, and to simulate additional experimental features, such as RNAP2 positional densities or response to different transcriptional inhibition mechanisms.
Our integrated technique possesses several advantages: (1) Fab binds endogenous RNAP2, so all RNAP2 present in the cell at a given time has a high probability to be labeled; (2) The fluorescence of the Fabs is naturally amplified by the 52 heptad repeats contained in the CTD of RNAP2; (3) Fab continually binds and unbinds RNAP2, reducing the loss of fluorescence due to photobleaching; (4) Our technology can be employed to measure the RNAP2 dynamics and phosphorylation status at other single-copy genes, provided the location of the gene can be identified without (Gu et al., 2018) or with labeling techniques [e.g., using MS2 or PP7 tags (Larson et al., 2011; Coulon et al., 2014; Tantale et al., 2016; Pichon et al., 2018), or other labeling systems such as ROLEX (Ochiai et al., 2015), ANCHOR (Mariamé et al., 2018), DNA FISH (Takei et al., 2017) or CasFISH (Deng et al., 2015)]; and (5) our modeling approach directly considers the temporal and statistical fluctuations of individual transcription sites, and applies information criteria to identify the simplest model that matches all experimental data.
In this protocol, we describe step-by-step how to fragment and fluorescently label antibodies targeting the CTD of RNAP2, how we put them into human cells using bead-loading (McNeil and Warder, 1987; Hayashi-Takanaka et al., 2009; Stasevich et al., 2014; Cialek et al., 2021), the imaging conditions we employed and how to tune them to get a good signal-to-noise ratio based on the experimental needs, how to quantify signal intensities at the transcription site from 3D imaging movies, and how to infer an appropriate mathematical model to reproduce these data without overfitting.

Figure 1. Schematic of our multicolor system to visualize RNAP2 phosphorylation at the transcription site of a reporter gene. A. The reporter gene contains a 128x MS2 insert (blue) that, when transcribed, produces stem-loops which are then recognized and bound by MCP genetically fused to GFP, thus lighting up the location of the gene within the nucleus, as well as the synthesis of nascent and mature mRNAs. Our Fabs are capable of identifying the CTD of RNAP2, as well as its phosphorylation at serine 5, which were fluorescently labeled with CF640 (red) and Cy3 (green), respectively. By combining these three elements, it is possible to visualize the recruitment (by CTD-RNAP2-CF640), initiation (by Ser5ph-RNAP2-Cy3), and elongation (by MS2-MCP-GFP) steps at the transcription site of a reporter gene. B. Crops from an example cell displaying, from top to bottom, the mRNA, Ser5ph-RNAP2, CTD-RNAP2, and merge signals, at three characteristic time points, in which (left) all three signals are present, (middle) mRNA and CTD-RNAP2 are present, but Ser5ph-RNAP2 is dim or absent, and (right) all signals are absent. These data demonstrate transcription fluctuations and the occurrence of multiple transcription cycles (example cell image adapted from Forero-Quintero et al., 2021).
Materials and Reagents
CTD-RNAP2 & Ser5ph-RNAP2 Fab generation and dye-conjugation
15 mL conical collection tubes (Fisher Scientific, catalog number: 14-959-49D)
500 mL Steritop Threaded Bottle Top Filter, 0.22 µm, Polyethersulfone (PES), Sterile, 33 mm fitting, (Millipore Sigma, catalog number: SCGPS05RE)
Amicon Ultracel-4 (10 kDa-cutoff) centrifugal filter unit with cellulose membrane, 4 mL sample volume (Millipore Sigma, catalog number: UFC801024)
Amicon Ultracel-10 (10 kDa-cutoff) 0.5 centrifugal filter unit (Fisher Scientific, catalog number: UFC501024)
0.6 mL and 1.7 mL low retention microcentrifuge tubes (Thomas Scientific, catalog numbers: 1149J99 and 1149K01, respectively)
PD-mini G-25 desalting column (VWR, GE Healthcare, catalog number: 95055-984)
Rack/stand for filtering using 15 mL conical tubes (we use the rack provided with the NucleoBond Xtra Midi Kit, from Macherey-Nagel, catalog number: 740420.50)
Pierce Mouse IgG1 Fab and F(ab’)2 Preparation Kit (Thermo Fisher Scientific, catalog number: PI44980), store at 4 °C
Anti-CTD-RNAP2 and anti-Ser5ph-RNAP2 monoclonal antibodies used were kindly provided by Dr. Hiroshi Kimura, but now they are commercially available (Cosmo Bio USA, catalog numbers: MABI 0601 and MABI 0603, respectively), store at 4 °C
Sodium Azide (Millipore Sigma, catalog Number: 71289-5G), store at RT
Phosphate-buffered saline (10× PBS) (Fisher Scientific, catalog number: BP661-10), store at RT
Sodium Bicarbonate (NaHCO3) (Millipore Sigma, catalog number: S5761-500G), store at RT, and 4 °C when in solution
Dimethyl sulfoxide (DMSO) (Millipore Sigma, catalog number: D8418), stored at RT
Cy3 N-hydroxysuccinimide ester mono-reactive dye pack (VWR, catalog number: 95017-373), store at -20 °C
CF640R Succinimidyl Ester (Biotium, catalog number: 92108), store at -20 °C
Sodium Bicarbonate (NaHCO3) (Millipore Sigma, catalog number: S5761-500G), store at RT, and 4 °C when in solution
1× PBS (see Recipes)
H-128 cell culture
100 mm tissue culture dishes (VWR, CELLSTAR, catalog number: 82050-546)
Serological pipettes Polystyrene 10 mL (VWR, catalog number: 82050-482 (CS))
1 L glass graduated cylinder (Chemistry stock room CSU)
HeLa Flp-in H9 cells (H-128) (Kindly provided by Dr. Edouard Bertrand (Tantale et al., 2016), store at -80 °C, and maintain at 37 °C in culture)
Dulbecco’s modified Eagle medium (DMEM), high glucose, no glutamine (Thermo Fisher Scientific, catalog number: 11960-044), store at 4 °C
L-glutamine (L-glut) (200 mM)-100× (Thermo Fisher Scientific, catalog number: 25030081), store at -20 °C
Penicillin Streptomycin (P/S) (10,000 U/mL) (Thermo Fisher Scientific, catalog number: 15140122), store at -20 °C
Fetal Bovine Serum (FBS), 100% US Origin (Atlas Biologicals, catalog number: F-0050-A), store at -20 °C
Hygromycin B (Gold Biotechnology, catalog number: H-270-1), store at -20 °C
Trypsin-EDTA (0.05%), phenol red (Thermo Fisher Scientific, catalog number: 25300062, store at -20 °C for long term, and at 4 °C when in regular use)
DMEM to maintain H-128 cells (see Recipes)
DMEM to image H-128 cells (see Recipes)
Transcription Inhibitors recipes (see Recipes)
Loading CTD-RNAP2-CF640 & Ser5ph-RNAP2-Cy3 Fabs into living H-128 cells
Glass bottom dishes (35 mm, 14 mm glass) (MatTek Corporation, catalog number: P35G-1.5-14-C).
Dye-conjugated Fabs “CTD-RNAP2-CF640 & Ser5ph-RNAP2-Cy3” (see details in Materials & Procedure sections A), store at 4 °C
Custom-made bead-loader with 106 μm glass beads [see details in our bead-loading protocol (Cialek et al., 2021)].
DMEM medium-high glucose, no glutamine, phenol red-free (Thermo Fisher Scientific, catalog number: 31053-028), store at 4 °C
Imaging
Triptolide, Tripterygium wilfordii (Millipore Sigma, catalog number: 645900), store at -20 °C
Flavopiridol, Alvocidib (Selleck Chemicals, catalog number: S1230), store at -20 °C
THZ1 2HCl, CDK7 Inhibitor (Selleck Chemicals, catalog number: S7549), store at -20 °C
Note: The inhibitors above were reconstituted in DMSO before storage.
Equipment
Rocker/rotator (Thermo Fisher Scientific, model: HulaMixer PRS-5/12, catalog number: 15920D)
Arduino Uno-R3 (SparkFun Electronics, catalog number: DEV-11021), and mini rotor (SparkFun Electronics, Servo-Generic Sub-Micro Size, catalog number: ROB-09065)
Tissue culture CO2 Incubator for cells (Heraeus, model: Heracell 150)
Tabletop centrifuge (Beckman Coulter, model: Microfuge 20)
Tabletop centrifuge capable of cooling (Thermo Fisher Scientific, model: Accusping Micro 17R, and/or Sorvall Legend XFR with F14 6x250LE)
UV-vis Spectrophotometer (Thermo Fisher Scientific, model: NanoDrop OneC, Catalog number: ND-ONE-W)
Biological safety cabinet (Nuaire, model: Class II type A/B3, NU-425-400)
TC20 Automated Cell Counter (Bio-Rad, catalog number: 145-0102)
Pure water filtration (Thermo Fisher Scientific, model: Barnstead NANOpure II)
Digital water bath (Fisher Scientific, model: ISOTEMP 210)
Fluorescence microscope with highly inclined illumination and a stage top incubator [we employed our custom-made microscope (Forero-Quintero et al., 2021)]
Laptop or desktop computer with Mathworks Matlab
Software
Preprocess the images with Fiji ImageJ (Schindelin et al., 2012) (https://fiji.sc/)
Analyze the preprocessed images using custom-made code written in Wolfram Mathematica 11.1.1 (https://www.wolfram.com/mathematica/), and available at https://github.com/MunskyGroup/Forero_2020/tree/master/Bioprotocol_Codes.
Model identification scripts and model-exploration Graphical User Interface (GUI) were created using MATLAB R2019b (https://www.mathworks.com/products/matlab.html), and are available at https://github.com/MunskyGroup/Forero_2020/tree/master/Bioprotocol_Codes.
Procedure
CTD-RNAP2 & Ser5ph-RNAP2 antibodies fragmentation & dye-conjugation (see Figure 2 for visualization of the major steps in this preparation)
Use the Pierce Mouse IgG1 Fab and F(ab’)2 Preparation Kit to fragment the Fabs.
Follow the instructions from the manufacturer available at: https://assets.fishersci.com/TFS-Assets/LSG/manuals/MAN0011653_Pierce_Mouse_IgG1_Fab_Fab2_Prep_UG.pdf.
From the manufacturer’s protocol above, we used the following conditions for the CTD-RNAP2 and/or Ser5ph-RNAP2 antibodies fragmentation process, which resulted in good quality/concentrated Fabs.
Begin with the IgG sample preparation. Repeat the centrifugation step of the Zeba Spin Desalting Column [this column is included in the Pierce Mouse IgG1 Fab and F(ab’)2 Preparation Kit, see details above in step A.1.a.] with 1 mL of Digestion buffer four times.
Add 0.5 mL of CTD-RNAP2 or Ser5ph-RNAP2 full-length antibody in 1× PBS at the maximum concentration recommended by the manufacturer (4 mg).
Important notes:
(1) The preparation of the CTD-RNAP2 and Ser5ph-RNAP2 Fabs should be done on separate days, since the kit only provides a single NAb protein column. This column can be reused up to ten times for different antibodies, as long as it is properly regenerated after each use.
(2) We used highly concentrated CTD-RNAP2 & Ser5ph-RNAP2 antibodies, but if you are purchasing commercial antibodies (usually provided at 1 mg/mL), you should concentrate the antibodies to a higher concentration before generating and purifying Fabs (see details on how to concentrate antibodies at Koch et al., 2021).
After equilibrating the immobilized Ficin in a spin column tube, add the IgG sample obtained in the step above (A.1.b.ii.), and incubate the digestion reaction for four hours (or five hours for better digestion) in constant mixing of the resin at 37 °C.
Note: The manufacturer recommends an end-over-end mixer or tabletop rocker, but sometimes these do not fit in a 37 °C incubator. To be able to keep the resin constantly mixing during the incubation, we used a mini rotor and programmed it to rotate 360° using an Arduino, and place it into our 37 °C tissue culture incubator.
Place the NAb Protein A Column to equilibrate at RT for approximately 30 min before finishing the digestion incubation. Then, perform the purification process as indicated by the manufacturer. This process involves a couple of washing and elution steps, in which the antibody can be lost. Thus, we recommend keeping the flow-throughs resulting from the washing and elution steps separately.
Measure the concentrations of the Fab, as well as the flow-throughs using the absorbance at 280 nm. We use a nanodrop, but any other spectrophotometer or a BCA protein assay would work.
Note: The Fabs generated with this protocol produce 50 kDa Fab, which corresponds to a third of the full-length antibody. Thus, if the starting mass of the antibody is 4 mg, it will produce 2.67 mg Fab. However, during digestion and purification processes, some of the antibodies are lost, resulting in 1–2 mg Fab mass. In many cases mass of Fab above 0.8 mg after digestion and purification could still result in good quality Fab (enough to concentrate, label, and later on, visualize transcriptional processes).
Do not forget to regenerate the NAb protein A column after finishing the purification step to be able to reuse it. Place the column back at 4 °C in 1× PBS with 0.02% sodium azide).
Fabs concentration.
Add the Fab and the flow-through Fab from washes 1 and 2 up to 3 mL into the Amicon Ultracel-4 centrifugal filter unit with a cellulose membrane, and centrifuge at 7,500 × g and 4 °C for 20 min.
Note: Mixing the Fab and flow-through Fab from washes 1 and 2 after the purification process helps to increase the concentration of Fab, if the concentration measured after the purification was between 0.8 and 1.5 mg/mL.
Add the 1 mL left to the filter unit and wash the 15 mL conical tube that contained the Fab using 2 mL of 1× PBS to complete 3 mL in the filter unit. Centrifuge one more time at 7,500 × g and 4 °C for 20 min.
Retrieve the solution retained by the filter unit. This is the concentrated Fab.
Measure the concentration of the concentrated Fab in the Nanodrop.
Another way to concentrate Fab is by using an Amicon Ultracel-10 filter 0.5 centrifugal unit:
Mix the Fab and the flow-through Fab from washes, and spin down 0.5 mL at a time, at 12,000 × g and 4 °C for 5 min, until all the volume (~3 mL) flows through.
Some volume containing the Fab will remain in the filter unit, add the adequate amount of 1x PBS up to 0.5 mL, centrifuge at 12,000 × g and 4 °C for 5 min. Repeat this step once.
Repeat step (A.2.e.ii.) one more time, but now for 20 min.
Carefully, flip the filter unit into a 1.7-mL low binding tube, and centrifuge at 300 × g and 4 °C for 1 min.
Add 30 μL of 1× PBS to wash the filter unit, and centrifuge once again as in step (A.2.E.iv.).
Measure the concentration using a nanodrop.
Purified unconjugated Fab can be stored at 4 °C until use. It can last up to a year or two without degrading, if properly stored.
Fabs dye-conjugation.
Get purified and concentrated Fab protein.
Use one low-binding tube per protein.
In one 0.6-mL low-binding tube, mix: 10 μL of 1 M NaHCO3 (for better results, prepare fresh) with 100 μg of the purified Fab protein, and add 1× PBS to make a total volume of 100 μL.
Note: e.g., if the purified Fab protein concentration is 1.8 mg/mL, 100 μg corresponds to 55.5 μL. Thus, the mixture will consist of 55.5 μL of purified Fab protein, 10 μL of 1 M NaHCO3, and 34.5 μL of 1× PBS, for a total volume of 100 μL.Add the correct amount of dye. Use 2 μL of 1 μM CF640, and 2.66 μL of 1 μM Cy3 (both dyes were previously diluted in DMSO). After adding the dyes to the respective mixtures (CF640 to CTD-RNAP2, and Cy3 to Ser5ph-RNAP2), pipette up and down, and tap the tube to distribute the mixture evenly. Then, incubate at RT and protected from light for 2 h, while constantly rotating the mixture in a rotator.
Purify dye-conjugated Fabs using a G-25 mini trap desalting column (use one per conjugated Fab protein).
Discard the buffer in the mini trap column and replace it with 1× PBS (repeat this step three times for equilibration).
After equilibrating the column, take the incubated mixture (containing the labeled Fab), and add it to the top of the mini trap column (make sure it is centered and straight). Add 450 μL of 1× PBS for circulation (add it from above, do not touch the sides of the mini trap column).
Note: The Fab protein conjugated with the dye is heavier than the unconjugated one; thus, it would pass through the mini trap column faster. You will be able to see two separate strips forming. The lower one contains your conjugated Fab.
If the desired band is close to the bottom of the mini trap column, add 50 μL of 1× PBS; if further from the bottom, add up to 100 μL of 1× PBS. This will help to bring down the conjugated Fab.
Note: Keep a close look at the mini trap column, since you want to make sure you catch the conjugated Fab.
Start cooling down the centrifuge to 4 °C.
Get one 1.7-mL low-binding tube for each conjugated Fab, place it underneath the respective mini trap column, and add 500 μL of 1× PBS.
Note: Watch carefully, since this step will bring down all the conjugated Fab drop by drop.
Add the conjugated Fab resulting from the step above (A.3.e.v.) to the top of the Amicon Ultracel-10 (10 kDa-cutoff) 0.5 centrifugal filter unit (this filter allows proteins smaller than 10 kDa). Centrifuge at 12,000 × g and 4 °C for 5 min, and discard the flow-through.
Note: Align the filter perpendicularly to the centrifuge, so the protein is not centrifuged directly into the filter.
Add 500 μL of 1× PBS from the top of the filter, centrifuge one more time at 12,000 × g and 4 °C for 5 min, and discard the flow-through.
Repeat the step above (A.3.e.vii.), but now centrifuge for 10 min.
Note: If your flow-through is colored, it means the filter may be defective. The flow-through should be clear, meaning your conjugated Fab remains in the filter, and the flow-through is disposable.
Take the filter containing the conjugated Fab, and tightly place it in a 1.7-mL low-binding tube on top of it, and flip it. Centrifuge at 300 × g and RT for 2 min.
Measure the concentration and absorbance spectrum for each protein to calculate the degree of labeling.
Use a nanodrop or other spectrophotometer that measures the absorbance spectrum. Choose IgG as sample type, and the corresponding dye (CF640 or Cy3). Ideally, you should get a concentration higher than 1 mg/mL for your protein, and a 1.2 for the ratio of the absorbances.
Calculate the degree of labeling (DOL) of the Fab using the following equation:
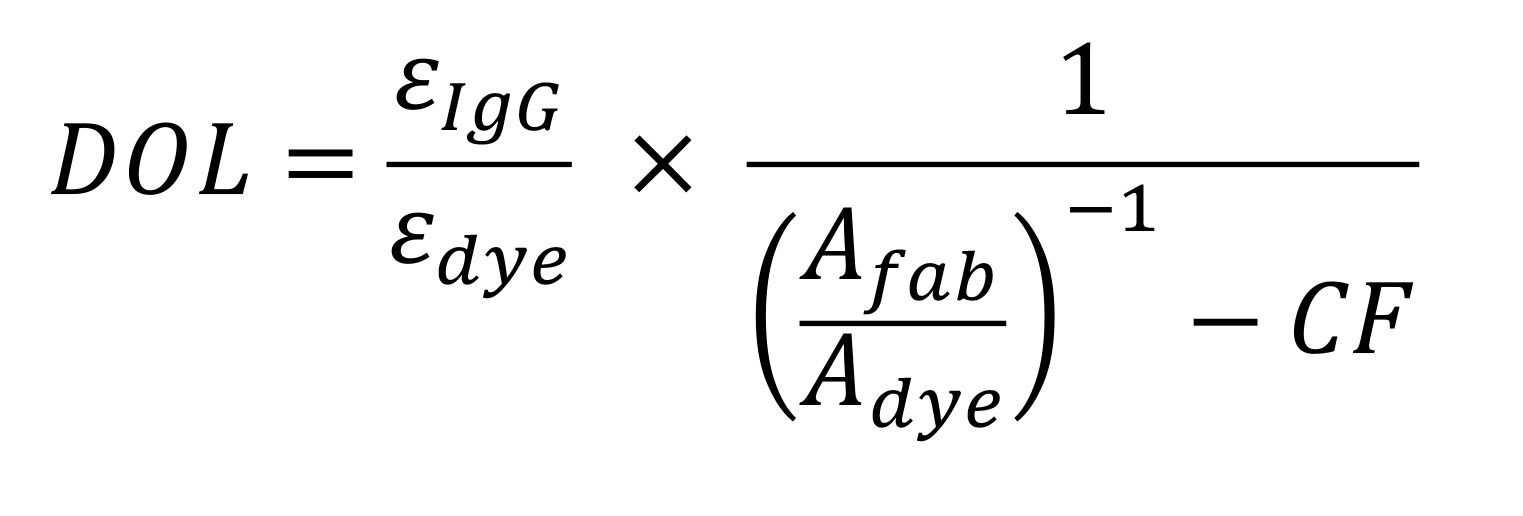
Where ϵIgG and ϵdye are the extinction coefficients of the Fab protein (IgG) at 280 nm (83,000 M-1cm-1) and the dyes (150,000 M-1cm-1 for Cy3, and 105,000 M-1cm-1 for CF640, respectively). These numbers are provided by the manufacturer. AFab and ADye are the absorbances determined at 280 nm for the IgG protein, and for the dyes at 550 nm for Cy3 and 650 nm for CF640, and CF is the correction factor for the dye at 280 nm (0.08, and 0.37 for Cy3, and CF640, respectively, provided by the manufacturer). Store the labeled Fabs at 4 °C for up to a year or so.
Note: In our study (Forero-Quintero et al., 2021), Fabs with a DOL between 0.75 and 1 were used successfully for live-imaging experiments.
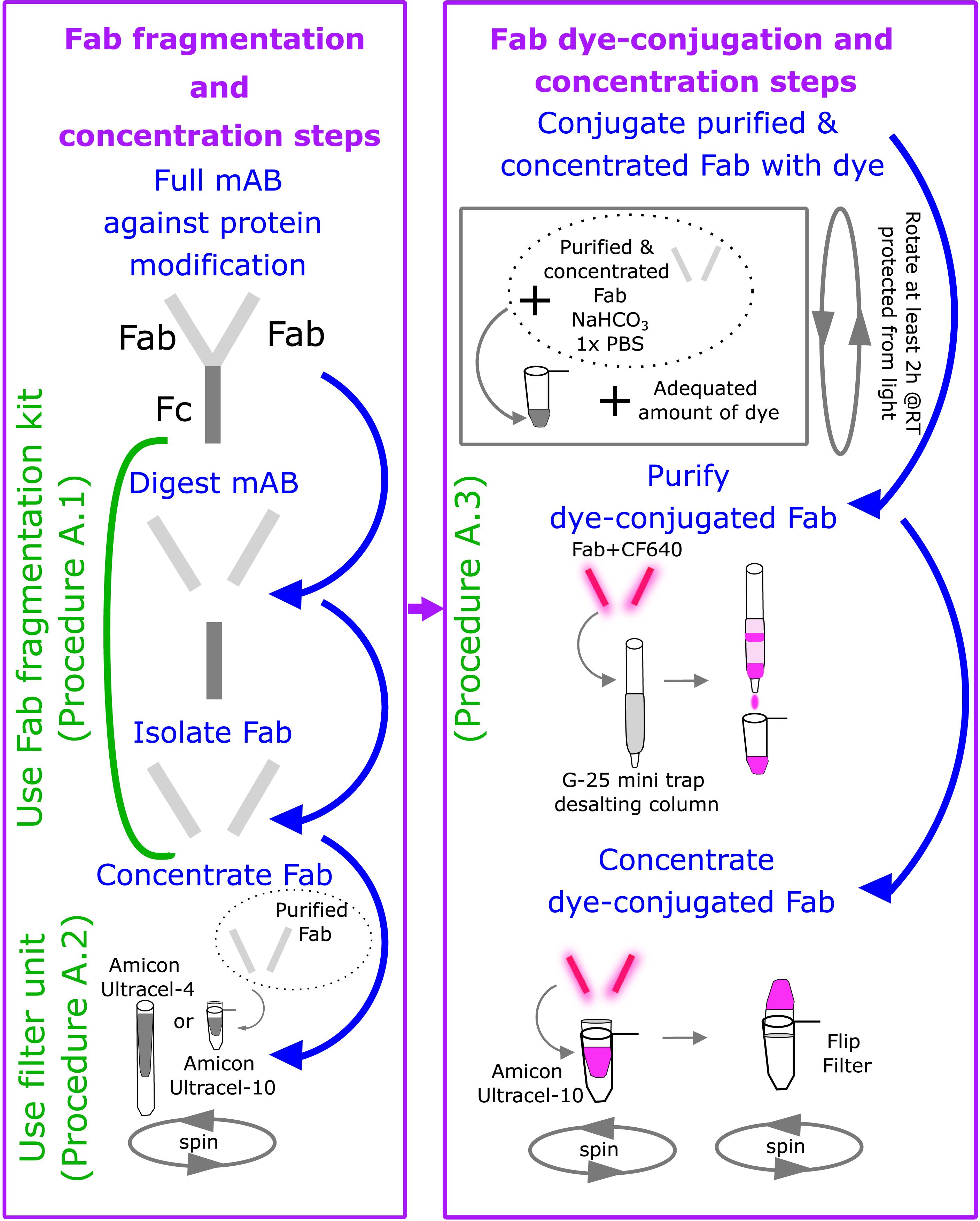
Figure 2. Major steps in fragmenting, labeling, and concentrating antibodies. (Left), Digest the full monoclonal antibody (mAB) against the desired protein modification, and then isolate Fab by using the Pierce Mouse IgG1 Fab and F(ab’)2 Preparation Kit to fragment the Fabs (Procedure A.1). Concentrate the isolated Fab using either an Amicon Ultracel-4 or -10 filter unit, as described in procedure A.2. (Right), Conjugate the purified and concentrated Fab with the desired fluorescent dye by assembling a reaction containing purified Fab, NaHCO3, PBS, and the dye. Purify and concentrate the dye-conjugated Fab, as described in procedure A.3.
H-128 cell culture
Prepare DMEM to maintain H-128 cells (see details in the Recipes section).
Cells are maintained to a confluency not greater than 95% in supplemented DMEM medium (see Recipe 2 below) at 37 °C, 5% CO2 incubator.
Note: The cells were passed a maximum of 25 times before bringing up a new batch of cells. Splitting of the cells was performed twice a week at most, to avoid stressing the cells due to the trypsinization procedure.
The day before performing an imaging experiment, plate the cells into MatTek dishes as follows:
Warm up the 0.05% trypsin stock, and supplemented DMEM to maintain H-128 cells.
Transfer the 100-mm dish from the incubator to the tissue culture hood.
Remove all the medium.
Wash the cells with 5 mL of 1× PBS three times.
Add 4 mL of 0.05% trypsin to cover the surface of the 100-mm dish. Let it stand at RT for approximately 40 s, and remove the trypsin with an aspirator. A thin layer will remain.
Place the cells back in the incubator for 5 min. After incubation, tap the bottom and the edges of the dish, to detach and singularize the cells from the bottom of the dish.
Observe the cells using a light microscope, to confirm the cells are detached and singularized.
Add 10 mL of supplemented DMEM medium to the dish. Using a 10-mL serological pipette, pipette the cells in the suspension up and down, placing the tip of the pipette against an edge of the dish, to break up cell clusters or groups of cells that did not singularize when tapping the cells. To make sure all the cells at the bottom of the dish are in the suspension, tilt the dish, and let the mixture of cells and media run from different angles in the dish while pipetting up and down.
Take a volume of 10 μL of the suspension, and count the cells using a cell counter.
Considering the number of cells measured, plate enough cells on the glass bottom region of the MatTek to reach a final concentration of 1.5 × 105 cells/mL. Let the resuspended cells stand on the glass region for a few seconds, before adding the supplemented DMEM medium. Approximately 0.8 mL of cells in suspension (3 × 105 cells/mL) and 1.2 mL of supplemented DMEM medium are required to maintain H-128 cells. This ratio results in good confluency of cells, to bead-load conjugated Fabs by bead-loading.
Place the cells in the MatTek chambers back in the 37 °C, 5% CO2 incubator, and let them sit for at least 24 h before any further procedure is done.
Loading CTD-RNAP2-CF640 & Ser5ph-RNAP2-Cy3 Fabs into living H-128 cells
Build your own bead loader consisting of 106 μm glass beads, a 100 μm nylon mesh [Spectramesh Woven Filters Polypropylene Opening: 105-µm (Spectrum Labs, catalog number: 148496)], and a MatTek dish with the glass bottom part removed [seeCialek et al. (2021) andKoch et al. (2021) for details on the construction].
On your bench, place a 0.6-mL low-binding tube, and mix:
~0.75 μg of CTD-RNAP2-CF640 protein Fab (~1 μL of the conjugated Fab obtained in procedure step A).
~0.5 μg of Ser5ph-RNAP2-Cy3 protein Fab (~1 μL of the conjugated Fab obtained in procedure step A).
Add 1× PBS to a total volume of 4 μL.
Note: Spin down the tubes containing the conjugated Fabs, and mix well by pipetting up and down before taking the volumes out for the mixture. Depending on the concentration of the conjugated Fabs, you can use up to 10 μL of the mixture described above, as long as the concentrations are maintained.
Mix well by pipetting up and down, but avoid creating bubbles in the mixture. Spin down.
Start warming 50-mL aliquots of the DMEM medium to image H-128 cells (see Recipe 3, below), without (DMEM-) and with (DMEM+) supplementation.
Note: This DMEM is red phenol-free, to facilitate imaging.
Place the following items in the cell culture hood, and perform the following steps in there:
A custom-made bead loader [to see a detailed explanation of the bead loader construction and procedure, see our recent publication (Cialek et al., 2021)].
The mixture of conjugated Fabs (described above).
A 15-mL conical tube per chamber to be bead-loaded.
A MatTek chamber with H-128 cells (plated at least 24 h before).
Load the 4 μL of conjugated Fab in the pipette tip, and set it aside. Remove all the medium (including the remnants around the rim in the glass bottom region, to ensure cells are not scrapped with the pipette tip) from the MatTek chamber, and place it aside in a 15-mL conical tube.
Note: It is important to remove all medium, especially around the glass region, since even the smallest remanent might (1) affect the concentration of conjugated Fabs added, and (2) the beads might float on that medium, and therefore not come in direct contact with the cells.
Add the 4 μL mixture of conjugated Fabs directly on top of the cells, right on the center of the glass bottom area.
Immediately after, place the custom-made bead loader on top of the MatTek chamber and gently sprinkle the glass beads on top of cells, by taping both chambers against the bench once.
Set aside the custom-made bead loader.
Tap the MatTek chamber against the bench 7–10 times, using enough strength for the glass beads to exert force against the cell membranes and let the Fabs in, but gently enough so the cells do not peel off the glass.
Notes:
(1) Similar to electroporation, the bead loading technique induces tiny tears in the cell membranes by a mechanical force, which allows the diffusion of conjugated Fabs into the cells through these tiny tears. This technique does not affect cell viability, and cells recover within hours post-procedure.
(2) Some cell types are more susceptible than others to peeling, especially if they do not attach directly to the glass and require some pre-treatment of the glass, like laminin, or polylysine, in which case bead loading might work, but the strength of tapping must be experimentally determined.
Gently pour the medium removed in step (C.5.) back into cells through a side of the chamber, not directly on top of the cells, ensuring not to disturb the cells.
Place the cells back in the incubator at 37 °C, 5% CO2, and let the cells recover for at least 1 h.
Bring the cells back to the hood and remove the medium. Wash out the glass beads by adding 1 mL of DMEM- at a time and pouring the solution into a liquid waste container. Repeat this as many times as needed, until most of the glass beads are gone. Use a light microscope to visually confirm relatively few beads remain.
Note: Tilting the MatTek chamber will help to bring down the beads, to the rim of the cover glass portion, where they can be aspirated with a pipette tip.
Remove the last addition of DMEM- and add 2 mL of DMEM+. Place the cells back in the incubator at 37 °C, 5% CO2. Let them fully recover for another 4 h before beginning imaging.
Note: In our experience, we observe cells recover fully and display active transcription sites co-localizing with RNAP2 5 h post-bead loading. To see a detailed explanation of the bead loading protocol and its applications, see our recent publication (Cialek et al., 2021).
Imaging
Imaging can be performed on a microscope equipped with 488, 561, and 637 nm lasers with appropriate filters, a stage top incubator to maintain the cells at 37 °C, 5% CO2, and sensitive EM-CCD cameras. We recommend using a widefield fluorescence microscope with highly inclined illumination (HILO) (Tokunaga et al., 2008) for better visualization of active and inactive transcription sites above the background.
Place the cells onto the stage-top incubator (37 °C, 5% CO2).
Define the imaging conditions based on the activation rate (measured or expected) of the gene studied.
Set up the imaging experiment to cover the entire nucleus of the cells. For H-128 (HeLa) cells, 13 z-stacks with 0.5 μm spacing are enough.
Notes:
(1) Capturing the whole volume of the nucleus is important to study transcription dynamics more precisely, and to guarantee that when the transcription sites disappear, this is not caused by it going out of the recorded region, but instead due to inactivation periods.
(2) The Z range we use covers from top to bottom HeLa, U-2 OS, HEK293, RPE1, and fibroblast cells.
Set up the temporal intervals according to the activation rate of the gene of interest and/or the type of event you desire to observe.
To visualize transcription fluctuations, you can set up your imaging experiment on the scale of minutes, depending on your gene of interest. The transcription site of our HIV-1 reporter gene exhibits fluctuations within the minute range. Thus, we did a short live-cell imaging set, in which each cell was imaged every minute, for 30 min. In this type of experiment, we were able to observe that mRNA, CTD-RNAP2, and Ser5ph-RNAP2 transcription fluctuations were nicely correlated; however, we were not able to observe multiple transcription cycles (multiple active and inactive periods) in a single cell record.
To visualize multiple transcriptional active and inactive periods of a gene, you should set up your imaging experiment in the order of hours, and depending on the total length of the measurement, set the scan of each cell every 1 or few minute(s) apart. You can also reduce the laser power if your movie lasts for several hours. Altering these two conditions would help to prevent photobleaching of the fluorescent probes and cell damage over the course of a movie. In our system, we recorded cells for a longer period of time; each cell was imaged every 1 min for 200-time points (~3 h 20 min), covering the entire cell (a representative recording is shown in Video 1). In our HIV-1 reporter gene, the mRNA signal rarely disappeared completely, however, longer movies allowed us to visualize these rare events in some of our cells. In some cases, the mRNA, Ser5ph-RNAP2, and CTD-RNAP2 signals turned on and off up to four times in a single movie, with RNAP2 signals appearing slightly earlier than the mRNA, indicating bursts of transcription and multiple complete transcription cycles.
To determine short time delays between signals, you should set up your experiment to a faster imaging rate. In our system, we observed a time delay between the CTD-RNAP2 and Ser5ph-RNAP2 signals at the HIV-1 transcription site. We expected serine 5 phosphorylation of the CTD tail of RNAP2 to occur in the order of seconds; therefore, we were not able to resolve the time lag between our RNAP2 signals using our 1-min rate movies. Thus, we imaged faster, 1 frame every 150 ms, for a total of 10,000-time points (150 s) in a single plane. Note that faster imaging is limited to a single plane, which is not problematic in this case, since transcription sites do not move much between z planes in the order of seconds.
Add triptolide to test for active transcription.
Set up your imaging experiment to capture the entire cell (13 z-stacks with 0.5 μm spacing), scanning each cell every 1 min for 35 min.
Acquire five time points.
After the first five time points, add triptolide at a final concentration of 5 μM directly to the top of the chamber.
Notes:
(1) For drug experiments, we usually withdraw 1 mL of the DMEM+ medium from the MatTek chamber before mounting it onto the microscope incubator and keep it at 37 °C. A few seconds before adding the stimuli to the cells, add triptolide to the reserved DMEM+, mix well, and add the mixture to cells.
(2) When adding the mixture to cells, make sure not to touch the chamber; otherwise, your field of view will go out of focus and be changed.
Continue to image the cells for 30 time points (30 min) after adding triptolide.
Note: Active transcription sites together with RNAP2 Fab signals disappear within 5–10 min upon addition of triptolide at the transcription site of our HIV-1 reporter gene. Degradation of mature mRNAs occurs in the order of hours. Triptolide experiments are good to determine whether nuclear spots of the right size and brightness are active transcription sites.
Add THZ1 and flavopiridol to inhibit later steps in the transcription cycle (initiation, and elongation, respectively).
Set up your imaging experiment to capture the entire cell (13 z-stacks with 0.5-μm spacing), scanning each cell every 1 min for 35 or 55 min, for flavopiridol and THZ1, respectively.
Note: It takes 20–25 min for THZ1 to completely inhibit transcription initiation, and therefore mRNA synthesis. For this reason, we imaged for an extended time, so we could visualize complete transcriptional initiation inhibition at the transcription site of our HIV-1 reporter gene.Acquire five time points.
After the first five time points, add THZ1 at a final concentration of 15 μM or flavopiridol at a final concentration of 1 μM directly to the top of the chamber, as described above.
Continue to image the cells for 55 time points (55 min) after adding THZ1, and for 30 time points (30 min) after adding flavopiridol.
 Video 1. Representative 3-color movie from a long-time imaging course after processing.
Video 1. Representative 3-color movie from a long-time imaging course after processing.
Maximum projection of a 13 z-stack three-color movie, representing an exemplary H-128 cell. The dashed white circle shows the transcription site of the HIV-1 reporter, in which the mRNA, CTD-RNAP2, and Ser5ph-RNAP2 signals are co-localized. The images were acquired every 1 min for a total of 200 min. The field of view is 512 by 512 pixels = 66.56 μm × 66.56 μm. Scale bar, 10 μm.
Data analysis
Image processing and signal quantification:
Correct the 3D images for photobleaching and laser fluctuation in each z-stack, by dividing the movie by the mean intensity of the whole cell or the nucleus in each channel to create a new corrected 3D movie. We used a script in Mathematica to perform this task. The code is available at https://github.com/MunskyGroup/Forero_2020/tree/master/Bioprotocol_Codes, saved as “BleachCorrectionZ_bioprotocol”.
Note: A max of the max image is required to construct a mask in this step (create a maximum projection through z, and then a maximum projection of the z-maximum projection in time). Creating the maximum projections is straight-forward using Fiji ImageJ max-projection (see Figure 3 for further details).
Using Fiji ImageJ, preprocess the corrected 3D movie from step E.1.a. (see Figure 3 for further explanation in how to create the following files), and:
Subtract the background intensity from each channel.
Create a 2D maximum projection through z.
Create a maximum projection in time of the 2D maximum projection in z (from step E.1.b.ii).
Save the 3D image sequences in a folder named with the cell number.

Figure 3. Image pre-processing pipeline. The flow chart displays how to pre-process a 3D movie to generate the files needed to quantify the intensity signals at the transcription site, using our custom-made script in Mathematica.Note: We performed the tasks described in the following steps (E.1.c–E.1.l) using a custom code written in Mathematica, saved as “TranscriptionSiteTrackingCode_Bioprotocol” and available on Github athttps://github.com/MunskyGroup/Forero_2020/tree/master/Bioprotocol_Codes.
Create a mask delineating the nucleus of the cell.
Note: Use the maximum projection in time of the 2D maximum projection in z (“max of the max image”).
Optional: If the signal-to-noise ratio is poor, it is a good idea to create a running average projection over time (using a few frames, ~3).
Select the thresholds in each channel to visualize spots at the transcription site, and a bandpass filter to highlight just the transcription site in the mRNA channel.
Binarize the image generated in step E.1.e.
Track the transcription site over time. You can use the Trackmate (Tinevez et al., 2017) plugin in Fiji, or create your own tracking routine. We tracked transcription sites over time by using the ComponentMeasurements-IntensityCentroid built-in Mathematica routine, together with our click and track function, which allowed us to find the XY coordinates through time.
Create two masks for each time point: one marking the transcription site (TS) and one marking the background (BG).
Using the 3D image sequences (folder created in step E.1.b.iv.), determine the Z coordinate or z-stack at which the transcription site in XY has its maximum (“best z”).
Note: If the transcription site disappears due to transcription inactivation or inhibition, the Z or Zs where there is no signal remaining are replaced by the Z coordinate of the last visible position.
Calculate a new 2D maximum projection from the XYZ coordinates considering the “best z” in step E.1.i. at each time point.
Calculate the mean intensity values over time for the transcription site and background, using the TS and BG masks created as described in step E.1.h.
Note: We calculated the raw and normalized intensity vectors per channel using a moving average of three points to display the raw intensity as a function of time, and we calculated the normalized intensity by dividing the raw intensity by the average 95% intensity from all transcription sites.
To display the transcription sites over time, generate cropped images of the 3-time point moving average trims from the “best z” in each channel. Center each trim with the intensity centroid of the transcription site in the mRNA channel. We displayed CTD-RNAP2, Ser5ph-RNAP2, mRNA, and a merge.
Notes:
(1) Our Mathematica script “TranscriptionSiteTrackingCode_Bioprotocol” can be tested using the original raw movie (without processing except for the photobleaching and laser fluctuations correction) corresponding to Video 1. The movie is saved as “SupFig3a_PBC_Cell14_woProj_ExemplaryCell.tif”, and available at https://doi.org/10.6084/m9.figshare.14187011 [repository created for our original publication (Forero-Quintero et al., 2021) from which this protocol is derived]. We also provided an image of beads saved as “Beads02.tif” in the same repository, to correct for camera offset.
(2) We calculated the raw mean intensity for each channel at the transcription site over time, including background subtraction for each cell, without running average, and saved a file collecting all the data for all cells analyzed as “Raw_Intensities Analysis-BLC_W_BG_WO_RunAve_Date.XLS”. This file was then used in step E.2.
Biophysical parameters that can be extracted from the data analyzed in the steps above:
XYZ transcription site position through time without generalized maximum projections in Z [see Sup. Figure 3d in the original paper (Forero-Quintero et al., 2021), from which this protocol is derived].
Transcription dynamics throughout the transcription cycle (by CTD-RNAP2, Ser5ph-RNAP2, mRNA intensity signals at the transcription site, Figure 4).
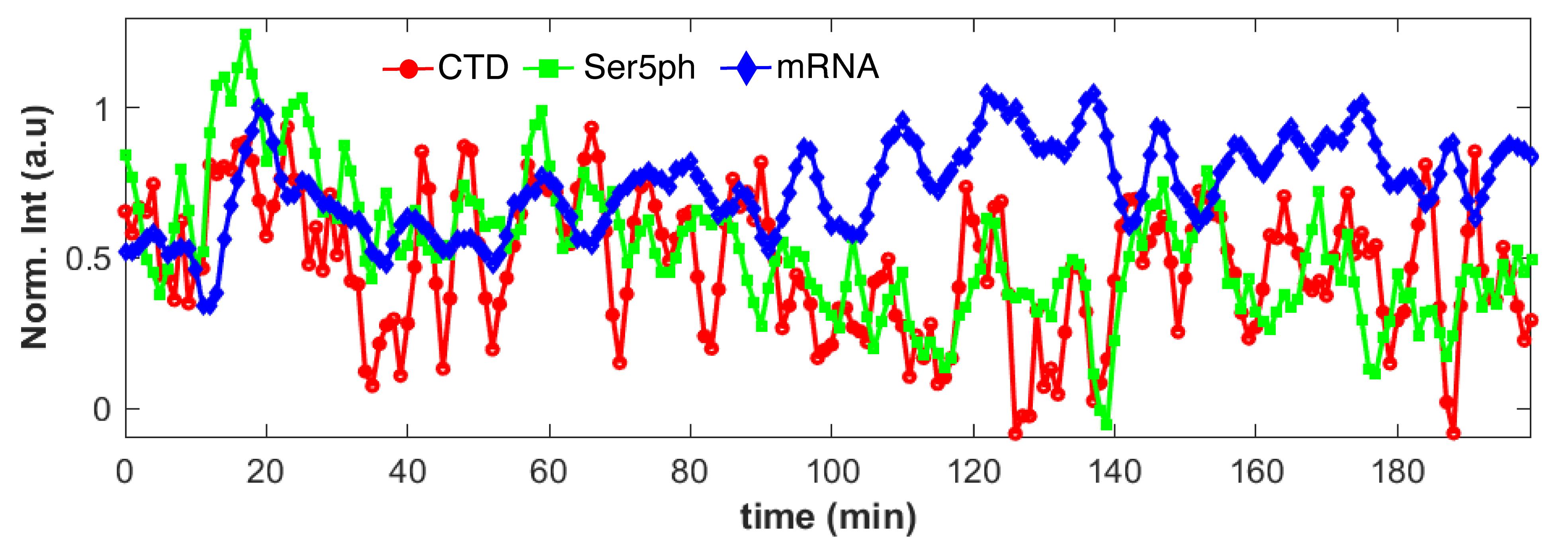
Figure 4. Representative data after signal quantification. Normalized signal intensities over time for the exemplary 3D movie shown in Video 1. The signals co-localize at the transcription site, and represent CTD-RNAP2 (red circles), Ser5ph-RNAP2 (green squares), and mRNA (blue diamonds). Figure adapted fromForero-Quintero et al. (2021).Analysis of minima signals when transcription is inactive [see Figure 2d in the original paper (Forero-Quintero et al., 2021)].
Analysis of transcription site spatial organization through time and the transcription cycle [see Figure 2e–h, Sup. Figure 3h, and Sup. Figure 4 in the original paper (Forero-Quintero et al., 2021)].
Note: Our “TranscriptionSiteTrackingCode_Bioprotocol” includes a Distance Analysis tab that can be adjusted for this quantification according to your purpose.
Quantification of the number of mRNAs per transcription site [see Figure 3d in the original paper (Forero-Quintero et al., 2021)].
Probability distributions for CTD-RNAP2, Ser5ph-RNAP2 intensity signals, and mRNA counts at the transcription site [see Figure 3d in the original paper (Forero-Quintero et al., 2021)].
Auto- and cross-correlations at the transcription site for CTD-RNAP2, Ser5ph-RNAP2, and mRNA signals [see Figure 3a–b in the original paper (Forero-Quintero et al., 2021)].
Transcription response upon transcriptional inhibition at different steps in the transcription cycle [see Figure 4 in the original paper (Forero-Quintero et al., 2021)].
Please refer to our original paper (Forero-Quintero et al., 2021) for details on criteria for data inclusion/exclusion, details on the number of replicates in each experiment, and statistical tests.
Quantitative model of transcription
Download Forero2020/Bioprotocol_Codes/ from: https://github.com/MunskyGroup/Forero_2020/tree/master/Bioprotocol_Codes.
Load trajectory data from XLS file (named as “Raw_Intensities Analysis-BLC_W_BG_WO_RunAve_Date.XLS” in our example) and normalize intensities and correlations (Section 1 in “ComputationalProtocol.m”).
Intensity data should be formatted such that it contains one column for each channel and (number of time points) × (number of cells) rows, where each subsequent cell is appended vertically,
For example, 3 channels with 20 cells of 200-min trajectories should have a shape of 4000 rows × 3 columns.
Change intensity normalization options if desired:
Default settings are minimum 0, and maximum 1.5, normalization to the 95th percentile of each intensity trajectory.
Change correlation lengths and normalization options:
Default settings linearly interpolate to find the zero-lag autocorrelation G(τ=0) and then average over all cells to find
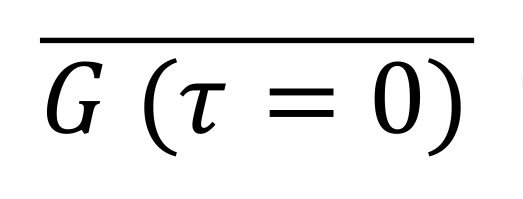 for each signal. This provides an estimate of the variance after removal of shot noise. Cross-correlations are computed separately for each cell, and then divided by the average correlation at zero lag time. By default, correlations are calculated for delays of -10 to +10 min for cross-correlations, and for 0 to 30 min for auto-correlations.
for each signal. This provides an estimate of the variance after removal of shot noise. Cross-correlations are computed separately for each cell, and then divided by the average correlation at zero lag time. By default, correlations are calculated for delays of -10 to +10 min for cross-correlations, and for 0 to 30 min for auto-correlations.
Define the model (Section 2 in “ComputationalProtocol.m”).
Specify the number of states.
Specify the number and initial guess for parameters and noise.
Specify stoichiometry matrix, S, and affine linear propensity functions W = W1*x + W0.
Specify intensity transformation matrix, c.
Additional details on formulating bursting models are found in Methods. Alternative models are described in Forero-Quintero et al. (2021, Sup. Figure 6).
Solve the model to compute steady state means, variances, auto-, and cross- correlations at specified time points (Section 3 in “ComputationalProtocol.m”).
Details for solution methods are provided inForero-Quintero et al. (2021), Section 9, Methods, “A quantitative model of transcription.”
Calculate log-likelihood of the data given the model (Section 4 in “ComputationalProtocol.m”).
Provide measured mean expression values (‘DataMeans’) for each channel, standard error of the mean for the molecules quantified (‘DataSEMs’), and the number of transcription sites (‘Nmolecules’) for each species. These values can be left as zero (0) in the code for channels that are not quantified. In our original paper (Forero-Quintero et al., 2021), we determined the number of nascent mRNAs at the transcription site (‘Ntranscripts’) by counting mature mRNAs and comparing their average intensity to the transcription site intensity for the reporter gene.
Specify which parameters are fixed “par_fixed” and which are allowed to change “par_changed” as needed for parameter searching.
Conduct parameter search to identify maximum likelihood estimate (MLE) (Section 5 in “ComputationalProtocol.m”).
We recommend using 20 or more iterative combinations of genetic algorithm (GA) and fminsearch analyses with multiple initial guesses and then select the best fit over these iterations.
Calculate Bayes Information Criterion (BIC) or Akaike Information Criterion (AIC) for model selection (Section 6 in “ComputationalProtocol.m”).
Specify the number of free parameters (k=5 in our example).
If using BIC, specify an estimated number of degrees of freedom in the data. We recommend a conservative estimate as (e.g., n=8). If using AIC, it is not necessary to estimate the number of degrees of freedom.
Run Metropolis Hastings search for parameter uncertainty quantification (Section 7 in “ComputationalProtocol.m”).
We recommend settings of 5,000 samples over 50 segments with a thinning rate of 20 for at least 20 chains (10 million total samples). For a proposal function, we recommend an N-dimensional Gaussian with variances selected to be 3%–5% of each parameter.
Plot correlation fit (Section 8 in “ComputationalProtocol.m”).
Additional Analyses (Sections 9–12 in “ComputationalProtocol.m”).
Sample model intensities.
Compare model and data intensity histograms.
Predict ChIP from the model.
Predict perturbation analyses.
Recipes
1× PBS
Bring into the cell culture hood:
10× PBS [add the whole content of PBS powder concentrate to 1 L of ultrapure water from a Milli Q, and mix properly. Make sure the pH is ~7.4, and then filter the solution using a Steritop Threaded Bottle Top Filter (0.22 µm)].
Four autoclaved bottles (500 mL).
900 mL of ultrapure water from a Nanopure.
Parafilm.
Steritop Threaded Bottle Top Filter (for 500 mL).
Add 100 mL of 10× PBS to 900 mL of ultrapure water from a Milli Q in a 1-L graduated cylinder, cover with parafilm, and mix it properly. Open a new Steritop Threaded Bottle Top Filter, and place it in the first bottle, add 500 mL of the solution, turn on the vacuum, and plug the hose.
Note: To make good use of the filter, make 3 L of 1× PBS at a time.
Store at RT until use. It lasts several months on the shelf.
DMEM to maintain H-128 cells
Thaw 50 mL of FBS, 5 mL of P/S, and 5 mL of L-glut. Add them to 500 mL of DMEM, high glucose, no glutamine, with red phenol, and mix well. Store at 4°C until use.
Make a 50-mL aliquot of the supplemented medium described above and add 150 μL of hygromycin (previously diluted in ultrapure water from Milli-Q, at a concentration of 50 mg/mL) to obtain a final concentration of 150 μg/mL of hygromycin in the medium.
Note: Hygromycin is necessary to maintain the expression of the HIV-1 reporter in the H-128 stable cell line. When the medium is supplemented with hygromycin, use it within two weeks and store at 4 °C when not in use.
DMEM to image H-128 cells
Follow the same instructions as above (Recipe 2), but now use DMEM, high glucose, no glutamine, without red phenol.
Note: A clear medium is necessary to guarantee crisp fluorescent images. Store at 4 °C. It is stable for several weeks when properly stored.
Transcription Inhibitors
Add 555 μL of DMSO to the whole content of Triptolide (TPL, 1 mg), and mix well, to obtain a stock concentration of 5 mM.
Add 6.2214 mL of DMSO to the whole content of Flavopiridol (Flav, 5 mg), and mix well, to obtain a stock concentration of 2 mM.
Add 3.9125 mL of DMSO to the whole content of THZ1 (5 mg), and mix well, to obtain a stock concentration of 2 mM.
Note: Store all the inhibitor stocks at -20 °C when not in use.
Acknowledgments
We thank all the members of the Stasevich and Munsky labs for their support and helpful discussion and suggestions. T.J.S was supported by the NIH (grant no. R35GM119728) and the NSF (grant no. MCB-1845761). L.S.F.Q., B.M., and W.R. were supported by the NIH (grant no. R35GM124747), and L.S.F.Q. was also supported by the W.M. Keck Foundation. This protocol was derived from the work published in Forero-Quintero et al. (2021).
Competing interests
The authors declare no competing interests.
References
- Boehning, M., Dugast-Darzacq, C., Rankovic, M., Hansen, A. S., Yu, T., Marie-Nelly, H., McSwiggen, D. T., Kokic, G., Dailey, G. M., Cramer, P., et al. (2018). RNA polymerase II clustering through carboxy-terminal domain phase separation. Nat Struct Mol Biol 25(9): 833-840.
- Chen, B. C., Legant, W. R., Wang, K., Shao, L., Milkie, D. E., Davidson, M. W., Janetopoulos, C., Wu, X. S., Hammer, J. A., 3rd, Liu, Z., et al. (2014). Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science 346(6208): 1257998.
- Cho, W. K., Jayanth, N., English, B. P., Inoue, T., Andrews, J. O., Conway, W., Grimm, J. B., Spille, J. H., Lavis, L. D., Lionnet, T., et al. (2016). RNA Polymerase II cluster dynamics predict mRNA output in living cells. Elife 5: e13617.
- Cialek, C. A., Galindo, G., Koch, A. L., Saxton, M. N. and Stasevich, T. J. (2021). Bead Loading Proteins and Nucleic Acids into Adherent Human Cells. J Vis Exp (172).
- Cissé, II, Izeddin, I., Causse, S. Z., Boudarene, L., Senecal, A., Muresan, L., Dugast-Darzacq, C., Hajj, B., Dahan, M. and Darzacq, X. (2013). Real-time dynamics of RNA polymerase II clustering in live human cells. Science 341(6146): 664-667.
- Coulon, A., Chow, C. C., Singer, R. H. and Larson, D. R. (2013). Eukaryotic transcriptional dynamics: from single molecules to cell populations. Nat Rev Genet 14(8): 572-584.
- Coulon, A., Ferguson, M. L., de Turris, V., Palangat, M., Chow, C. C. and Larson, D. R. (2014). Kinetic competition during the transcription cycle results in stochastic RNA processing. Elife 3: e03939.
- Deng, W., Shi, X., Tjian, R., Lionnet, T. and Singer, R. H. (2015). CASFISH: CRISPR/Cas9-mediated in situ labeling of genomic loci in fixed cells. Proc Natl Acad Sci U S A 112(38): 11870-11875.
- Forero-Quintero, L. S., Raymond, W., Handa, T., Saxton, M. N., Morisaki, T., Kimura, H., Bertrand, E., Munsky, B. and Stasevich, T. J. (2021). Live-cell imaging reveals the spatiotemporal organization of endogenous RNA polymerase II phosphorylation at a single gene. Nat Commun 12(1): 3158.
- Gu, B., Swigut, T., Spencley, A., Bauer, M. R., Chung, M., Meyer, T. and Wysocka, J. (2018). Transcription-coupled changes in nuclear mobility of mammalian cis-regulatory elements. Science 359(6379): 1050-1055.
- Harlen, K. M. and Churchman, L. S. (2017). The code and beyond: transcription regulation by the RNA polymerase II carboxy-terminal domain. Nat Rev Mol Cell Biol 18(4): 263-273.
- Hayashi-Takanaka, Y., Yamagata, K., Wakayama, T., Stasevich, T. J., Kainuma, T., Tsurimoto, T., Tachibana, M., Shinkai, Y., Kurumizaka, H., Nozaki, N., et al. (2011). Tracking epigenetic histone modifications in single cells using Fab-based live endogenous modification labeling. Nucleic Acids Res 39(15): 6475-6488.
- Hayashi-Takanaka, Y., Yamagata, K., Nozaki, N. and Kimura, H. (2009). Visualizing histone modifications in living cells: spatiotemporal dynamics of H3 phosphorylation during interphase. J Cell Biol 187(6): 781-790.
- Heidemann, M., Hintermair, C., Voss, K. and Eick, D. (2013). Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim Biophys Acta 1829(1): 55-62.
- Kimura, H., Hayashi-Takanaka, Y., Stasevich, T. J. and Sato, Y. (2015). Visualizing posttranslational and epigenetic modifications of endogenous proteins in vivo. Histochem Cell Biol 144(2): 101-109.
- Koch, A. L., Morisaki, T. and Stasevich, T. J. (2021). A Multi-color Bicistronic Biosensor to Compare the Translation Dynamics of Different Open Reading Frames at Single-molecule Resolution in Live Cells. Bio-protocol 11(14): e4096.
- Larson, D. R., Zenklusen, D., Wu, B., Chao, J. A. and Singer, R. H. (2011). Real-time observation of transcription initiation and elongation on an endogenous yeast gene. Science 332(6028): 475-478.
- Li, J., Dong, A., Saydaminova, K., Chang, H., Wang, G., Ochiai, H., Yamamoto, T. and Pertsinidis, A. (2019). Single-Molecule Nanoscopy Elucidates RNA Polymerase II Transcription at Single Genes in Live Cells. Cell 178(2): 491-506 e428.
- Lyon, K. and Stasevich, T. J. (2017). Imaging Translational and Post-Translational Gene Regulatory Dynamics in Living Cells with Antibody-Based Probes. Trends Genet 33(5): 322-335.
- Mariamé, B., Kappler-Gratias, S., Kappler, M., Balor, S., Gallardo, F. and Bystricky, K. (2018). Real-time visualization and quantification of human Cytomegalovirus replication in living cells using the ANCHOR DNA labeling technology. J Virol 92(18), e00571-18.
- McNeil, P. L. and Warder, E. (1987). Glass beads load macromolecules into living cells. J Cell Sci 88 ( Pt 5): 669-678.
- Nagashima, R., Hibino, K., Ashwin, S. S., Babokhov, M., Fujishiro, S., Imai, R., Nozaki, T., Tamura, S., Tani, T., Kimura, H., et al. (2019). Single nucleosome imaging reveals loose genome chromatin networks via active RNA polymerase II. J Cell Biol 218(5): 1511-1530.
- Ochiai, H., Sugawara, T. and Yamamoto, T. (2015). Simultaneous live imaging of the transcription and nuclear position of specific genes. Nucleic Acids Res 43(19): e127.
- Pancholi, A., Klingberg, T., Zhang, W., Prizak, R., Mamontova, I., Noa, A., Sobucki, M., Kobitski, A. Y., Nienhaus, G. U., Zaburdaev, V., et al. (2021). RNA polymerase II clusters form in line with surface condensation on regulatory chromatin. Mol Syst Biol 17(9): e10272.
- Pichon, X., Lagha, M., Mueller, F. and Bertrand, E. (2018). A Growing Toolbox to Image Gene Expression in Single Cells: Sensitive Approaches for Demanding Challenges. Mol Cell 71(3): 468-480.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nature Methods 9(7): 676-682.
- Stasevich, T. J., Hayashi-Takanaka, Y., Sato, Y., Maehara, K., Ohkawa, Y., Sakata-Sogawa, K., Tokunaga, M., Nagase, T., Nozaki, N., McNally, J. G., et al. (2014). Regulation of RNA polymerase II activation by histone acetylation in single living cells. Nature 516(7530): 272-275.
- Steurer, B., Janssens, R. C., Geverts, B., Geijer, M. E., Wienholz, F., Theil, A. F., Chang, J., Dealy, S., Pothof, J., van Cappellen, W. A., et al. (2018). Live-cell analysis of endogenous GFP-RPB1 uncovers rapid turnover of initiating and promoter-paused RNA Polymerase II. PNAS 115(19), E4368–E4376.
- Takei, Y., Shah, S., Harvey, S., Qi, L. S. and Cai, L. (2017). Multiplexed Dynamic Imaging of Genomic Loci by Combined CRISPR Imaging and DNA Sequential FISH. Biophys J 112(9): 1773-1776.
- Tantale, K., Mueller, F., Kozulic-Pirher, A., Lesne, A., Victor, J.-M., Robert, M.-C., Capozi, S., Chouaib, R., Bäcker, V., Mateos-Langerak, J., et al. (2016). A single-molecule view of transcription reveals convoys of RNA polymerases and multi-scale bursting. Nature Communications 7(1): 12248.
- Tinevez, J. Y., Perry, N., Schindelin, J., Hoopes, G. M., Reynolds, G. D., Laplantine, E., Bednarek, S. Y., Shorte, S. L. and Eliceiri, K. W. (2017). TrackMate: An open and extensible platform for single-particle tracking. Methods 115: 80-90.
- Tokunaga, M., Imamoto, N. and Sakata-Sogawa, K. (2008). Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat Methods 5(2): 159-161.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Forero-Quintero, L. S., Raymond, W., Munsky, B. and Stasevich, T. J. (2022). Visualization, Quantification, and Modeling of Endogenous RNA Polymerase II Phosphorylation at a Single-copy Gene in Living Cells. Bio-protocol 12(15): e4482. DOI: 10.21769/BioProtoc.4482.
Category
Biophysics > Microscopy
Bioinformatics and Computational Biology
Biochemistry > RNA > Single-molecule Activity > Imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.