- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

TRACES: a Freely Accessible, Semi-automated Pipeline for Detection, Tracking, and Quantification of Fluorescently Labeled Cellular Structures
Published: Vol 12, Iss 15, Aug 5, 2022 DOI: 10.21769/BioProtoc.4479 Views: 2258
Reviewed by: Giusy TornilloVinay PanwarAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2021
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Subcellular structures exhibit diverse behaviors in different cellular processes, including changes in morphology, abundance, and relative spatial distribution. Faithfully tracking and quantifying these changes are essential to understand their functions. However, most freely accessible methods lack integrated features for tracking multiple objects in different spectral channels simultaneously. To overcome these limitations, we have developed TRACES (Tracking of Active Cellular Structures), a customizable and open-source pipeline capable of detecting, tracking, and quantifying fluorescently labeled cellular structures in up to three spectral channels simultaneously at single-cell level. Here, we detail step-by-step instructions for performing the TRACES pipeline, including image acquisition and segmentation, object identification and tracking, and data quantification and visualization. We believe that TRACES will be a valuable tool for cell biologists, enabling them to track and measure the spatiotemporal dynamics of subcellular structures in a robust and semi-automated manner.
Keywords: Particle trackingBackground
Deciphering how subcellular structures change in space and time is often key to understanding biological functions (Kholodenko et al., 2010). Thus, developing approaches to visualize, track, and measure these spatiotemporal changes has been a critical area of research in cell biology. In the past decades, advances in fluorescent protein engineering (Lippincott-Schwartz and Patterson, 2003; Giepmans et al., 2006) and optical microscopy (Tanaami et al., 2002; Stephens and Allan, 2003; Hell, 2007; Chen et al., 2014; Oreopoulos et al., 2014) have allowed scientists to observe subcellular structures with unprecedented spatiotemporal resolution. Post-imaging processing and analysis are then applied to quantify and interpret the data. For example, a popular Fiji plugin, TrackMate, can be used to visualize and analyze the motion of objects (Tinevez et al., 2017; Ershov et al., 2021), including the tracking of hundreds of centrosomes in the same spectral channel simultaneously (Aydogan et al., 2018, 2020, 2022; Alvarez-Rodrigo et al., 2019). CellProfiler (Lamprecht et al., 2007; Stirling et al., 2021), another open-source software tool, can be used to quantify data from biological images, particularly in a modular and high-throughput manner. However, TrackMate cannot track on multiple spectral channels simultaneously and is not optimal for tracking objects that are not perfectly circular. CellProfiler, while versatile and packed with extensive features, is not usually used for analyzing subcellular structures. Imaris (Bitplane, Belfast, UK) is a popular software capable of tracking objects in three dimensions with a user-friendly interface, but this and other commercially available image processing software (e.g., NIS Elements by Nikon, Tokyo, Japan) are not free to users, especially for the advanced features such as particle tracking. To overcome these limitations, we have developed TRACES (Tracking of Active Cellular Structures), a customizable and semi-automated quantification pipeline capable of simultaneously detecting, tracking, and quantifying up to three fluorescently labeled object types at single-cell resolution. TRACES is built on open-source platforms and freely accessible to users. The use of intensity- and size-based thresholding to segment objects in TRACES also makes it possible to identify and track objects that are not circular (an advantage over TrackMate). In addition, as the tracking and quantification algorithm of TRACES is written in Python and implemented using Jupyter Notebook, it can be easily modified or improved to fit users’ needs. While several available tools are optimized for tracking entire cells, TRACES is developed specifically for tracking subcellular structures at single-cell resolution, including tracking micron-sized objects, such as the centrosome. In this protocol, we use the analysis of condensation of pericentrin (PCNT) proteins and the movement of the resulting “condensates” toward the centrosome (Jiang et al., 2021) as an example, hereby demonstrating the TRACES workflow. This workflow involves image acquisition and segmentation, object identification and tracking, and data quantification and visualization.
One limitation of TRACES is its inability to track dividing cells, as the current tracking algorithm assumes one nucleus object per cell. We hope to implement object tracking features for dividing cells in the future. Moreover, while the Python algorithm is capable of tracking on multiple spectral channels simultaneously, the objects of interest in each channel need to be identified manually in the initial image segmentation step in Fiji. We hope to integrate an automated workflow of image segmentation into our future pipeline.
In sum, our TRACES method is a free and semi-automated pipeline for detecting, tracking, and measuring multiple fluorescently labeled cellular structures in up to three spectral channels simultaneously at single-cell level. If these cellular objects can be segmented and distinguished from one another, their relationships and properties (e.g., distance, size) can then be quantified using the TRACES method. Therefore, we envision that TRACES is not limited to analyzing centrosomes, their related structures, and nuclei, the three objects exemplified in this protocol. TRACES can be applied to analyzing any distinct fluorescent objects in up to three spectral channels in the cell, and will thus be a useful tool for the cell biology community, facilitating quantitative image analysis in other biological contexts.
Materials and Reagents
4-chamber 35-mm glass bottom dishes with 20-mm microwell, #1.5 cover glass (Cellvis, catalog number: D35C4-20-1.5-N)
hTERT immortalized retinal pigment epithelial (RPE-1) cells (A gift from Irina Kaverina, Vanderbilt University, Nashville, TN, catalog number: CRL-4000, RRID: CVCL_4388)
RPE-1 cells constitutively expressing mScarlet-i-H2A and miRFP670-CETN2 with stably integrated GFP-PCNT (854-1960) constructs under the control of a Doxycycline-inducible promoter (Jiang et al., 2021)
Doxycycline hyclate (MilliporeSigma, catalog number: D9891)
Dulbecco’s modified Eagle’s medium/Hams F-12 50/50 Mix (DMEM/F-12) (Corning, catalog number: 10-092-CV)
Penicillin-Streptomycin solution, 100× (Corning, catalog number: 30-002-CI)
Tetracycline negative fetal bovine serum (tet-negative FBS) (Gemini Bio, catalog number: 100-800)
Equipment
Spinning disk confocal microscope system (Dragonfly, Andor Technology, Belfast, UK)
The spinning disk confocal module is the Dragonfly 503 multimodal imaging system (Andor) with dual color TIRF, two camera ports, and 25 µm and 40 µm pinholes. It has four lines of laser launches (405 nm/100 mW, 488 nm/50 mW, 561 nm/50 mW, and 643 nm/100 mW). The base of the system is a Leica DMi8 inverted microscope (Leica, Wetzlar, Germany) with objectives spanning the range from 10× to 100× [10×/0.40 (magnification/numerical aperture) air HCX PL APO, 25×/0.95 water HC PL FLUOTAR, 40×/1.10 water HC PL APO, 63×/1.40 oil HC PL APO, 100×/1.40 oil HC PL APO, and 100×/1.47 oil HC PL APO CORR TIRF]. The images shown in this protocol were all acquired by the 63×/1.40 oil HC PL APO objective. The laser lines used in this study are 488 nm, 561 nm, and 643 nm, with the corresponding bandpass emission filters of 525–550 nm, 600–650 nm, and 725–740 nm, respectively.
iXon Ultra 888 EMCCD camera (Andor Technology)
Environmental incubator (Okolab, Pozzuoli, Italy)
Software
Anaconda (Anaconda, Inc., New York, NY)
Fiji (ImageJ) (Johannes Schindelin, Albert Cardona, Pavel Tomancak, RRID: SCR_002285)
Jupyter Notebook (Project Jupyter, RRID:SCR_018315)
Python Programming Language (Python Software Foundation, RRID: SCR_008394)
Procedure
Cell culturing and induction of protein expression
Seed approximately 6 × 104–9 × 104 cells to each chamber of a 35-mm glass bottom dish. Allow cells to attach onto the dish. Maintain cells with appropriate media in a humidified incubator supplied with 5% CO2 at 37 °C.
Note: Tet-ON-GFP-PCNT (854–1960) cells, an RPE-1 derived cell line that expresses GFP-tagged PCNT (residues 854–1960) through doxycycline induction, are used in our study (Jiang et al., 2021). Tet-ON-GFP-PCNT (854–1960) cells also stably and constitutively express mScarlet-i-H2A and miRFP670-CETN2, which label the nucleus and centrosome, respectively. Cells are maintained in DMEM/F12 media supplemented with 10% tet-negative FBS and 1× penicillin-streptomycin solution.
Allow cells to reach 40–60% confluency (12–18 h post seeding for most cultured human cells) before imaging.
Note: For best object tracking results, seed cells at a low density and distribute them evenly on the coverslip to minimize overlapping of cells.
Optional: for doxycycline-inducible cell lines, incubate cells with 1 µg/mL doxycycline hyclate (Dox) to induce transgene expression before the start of time-lapse microscopy (e.g., 3–4 h in our case).
Time-lapse image acquisition
Note: When performing live cell imaging with multiple fluorophores, it is important to select appropriate wavelength ranges, emission filters, and dichroic mirrors to minimize spectral bleed-through artifacts. Appropriate laser power, exposure time, and acquisition intervals should also be empirically determined to maximize the signal-to-noise ratio while minimizing photobleaching, especially over a long period of time-lapse imaging. We also recommend centering the cell(s) of interest in the field of view to prevent them from moving out of the field during imaging. Below are the time-lapse image acquisition settings we use in our system (Andor Dragonfly spinning disk confocal system). Using our current algorithm, TRACES can track up to three fluorescently labeled object types simultaneously, if one of the objects can be used to define the location of the cell over time (e.g., a fluorescently labeled nucleus such as mScarlet-i-H2A in Figure 1; other markers that label the cell membrane or other organelles can also be used). We use a series of images captured during the formation and movement of PCNT condensates as an example to demonstrate the TRACES workflow in this protocol (Figure 1).
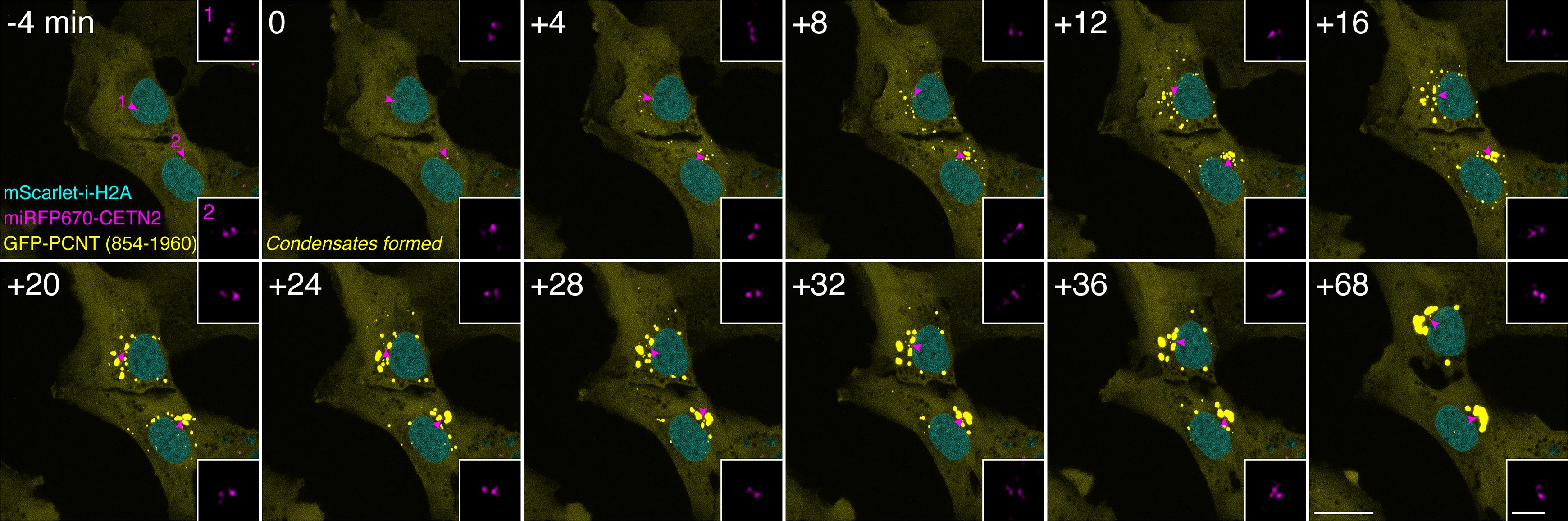
Figure 1. Examples of live-cell image acquisition. Time-lapse micrographs of Tet-ON-GFP-PCNT (854–1960) cells. Imaging started around 4 h post Dox induction. The time when the first condensates formed is marked as time 0. Note that GFP-PCNT (854–1960) forms phase-separated condensates that coalesce and move toward the centrosome (denoted by the magenta arrowheads and enlarged in insets). Scale bars: 20 µm and 2 µm (insets).Environmental control: Mount cells in a humidified chamber supplied with 5% CO2 inside a wrap-around environmental incubator with temperature set at 37 °C (for most cultured mammalian cells).
Note: Allow the microscope system to reach the desired temperature equilibrium before imaging.
Camera: iXon Ultra 888 EMCCD
Note: To minimize phototoxicity over long-term live imaging, we recommend empirically determining the lowest laser power and exposure time that can generate images with at least 2–3 signal-to-noise ratios for objects of interest. Using a highly sensitive camera, such as the one equipped with electron multiplying charge coupled device (EMCCD), is also desirable.
Objective: 63×/1.40 (magnification/numerical aperture) coupled with a 1× motorized magnification changer
Laser power: 1–10% of 50–100 mW lasers
Exposure time: 100–200 ms
Gain: 200
Pinhole size: 40 µm
Z-stack interval: 0.6 µm
For assessing colocalization or analyzing relative spatial distribution between fluorescently labeled structures, acquire all channels for each z-stack from the longest to the shortest wavelength.
Note: We also recommend setting a wide enough z-range, to account for the possibility that cells may drift out of focus during long-term imaging.
Acquisition time interval: 2–4 min
Note: Users need to optimize imaging parameters according to their specific imaging system, experimental design, and structure(s) of interest.
Data analysis
Object segmentation and identification using Fiji
Note: See Notes section A for instructions of Fiji installation.
Image import and Z-projection
Load a time-lapse image in Fiji by selecting “File” > “Open…”.
Note: Our image files are 16-bit in OME TIFF format and can be read using the Bio-Formats package (Linkert et al., 2010) that is integrated in Fiji by default. For other image formats, it might be necessary to convert the file into TIFF format or to directly activate the Bio-Formats Importer plugin to read data.
For “Bio-Formats Import options”, select “Hyperstack” for stack viewing, and “Use virtual stack” (Figure 2A).
Note: Hyperstack view displays images as multi-dimensional stacks (time, channel, Z-slice slider).
To open images with channels displayed separately, select “Default” as color mode, and “Split channel” (Figure 2A).
To perform Z-projection, select “Image” > “Stacks” > “Z Project …”. Determine the stack range by indicating the start and stop slices that will be included in the projection. Choose the appropriate projection type and select “All time frames” (Figure 2B).
Note: Maximum intensity Z-projection is used in our image analysis to select pixels from the maximum intensity of each slice to construct a 2D time-lapse image series. Users may choose other projection types that better represent the raw data.
Save the projected images as TIFF format by going to “File” > “Save As” > “Tiff…” (Figure 2C). Make sure all projected images have the same image type (e.g., 16-bit, 32-bit).
Repeat Steps A1d and 1e to process other spectral channels, if there are any.
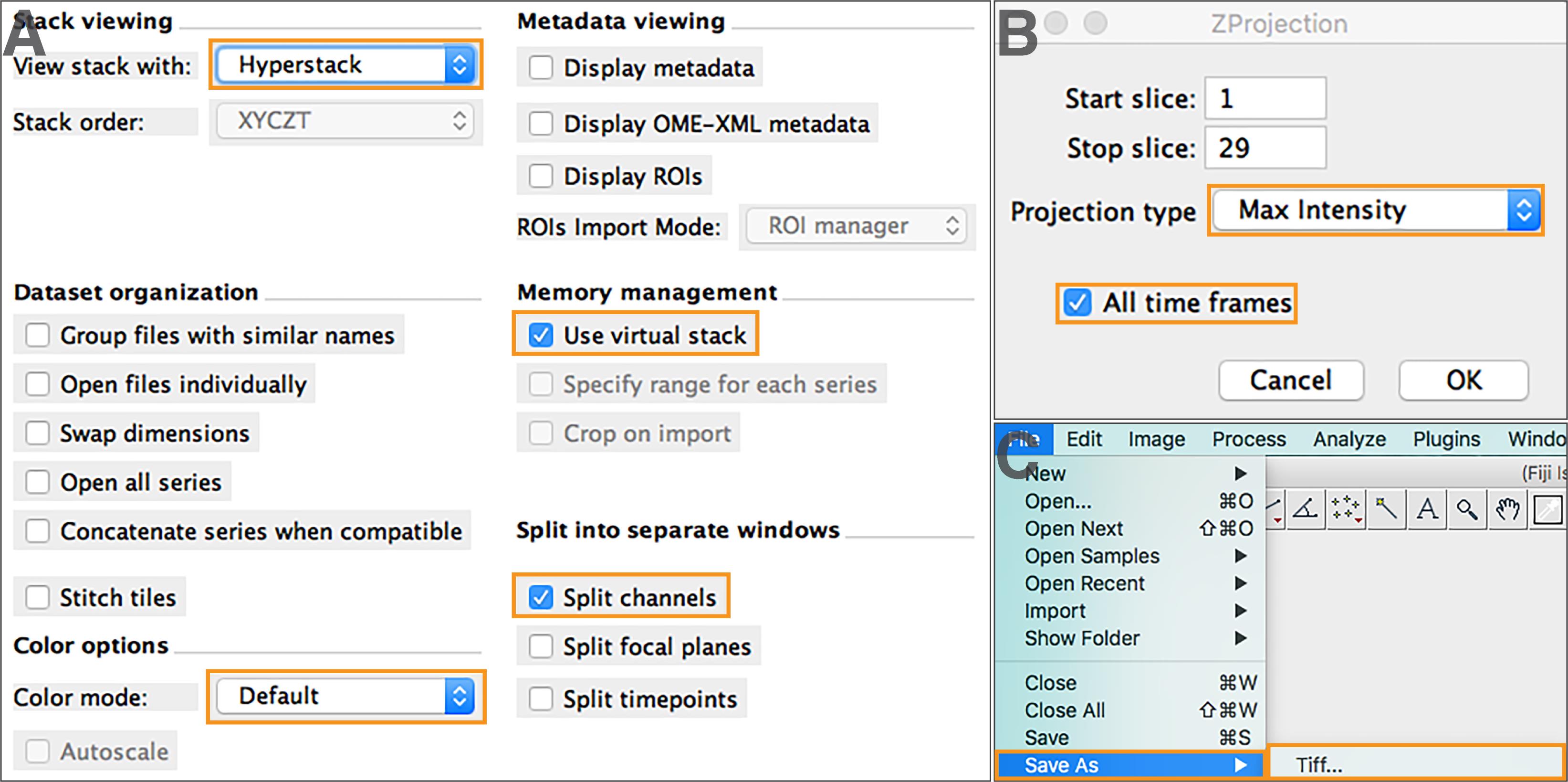
Figure 2. Image import and Z-projection. (A) Read image files using Bio-Formats. (B) Set up Z-projection parameters. (C) Save projected images as TIFF format.
Image segmentation by intensity-based thresholding
Open the projected image of a given channel in Fiji (Figure 3A). Adjust the lower and upper limits of the display range through “Image” > “Adjust” > “Brightness/Contrast…” so that objects of interest, not background signals, are clearly displayed (Figure 3B).
Note: Figures 3A and 3B show examples of before and after display adjustment of the signals for GFP-PCNT (854–1960) condensates in the +24 min time frame shown in Figure 1. The same example is also shown in Figure 5. This adjustment step only changes displayed values, not raw pixel intensity values (Ferreira and Rasband, 2012).
To apply thresholding for image segmentation, first convert image type to 8-bit through “Image” > “Type” > “8-bit”. Then, select “Image” > “Adjust” > “Threshold…”. Adjust the minimum and maximum threshold values, so that only objects of interest, not background signals, are segmented. Choose “Red” overlay, and then select “Apply” (Figure 3C). For example, threshold values of 80/255 (minimum/maximum) are used here for segmenting GFP-PCNT (854–1960) condensates.
The “Convert Stack to Binary” window will appear after selecting “Apply” from the previous step (Figure 3C). In this window, select “Default” method and “List thresholds”. Then, select “OK” (Figure 3D).
Note: Execution of Steps A2b and A2c generates a binary stack image that displays the pixels of segmented objects (or foreground) and background (e.g., Figure 5, third column). While we use the default thresholding method based on IsoData algorithm, users may choose other methods Fiji provides (e.g., Intermodes, MaxEntropy) that best segment their objects of interest. Make sure to unselect “Calculate threshold for each image”, if users want to apply the same defined threshold values across all time frames of a given time-lapse image series.
Save the resulting binary stack image with segmented objects as a TIFF file (i.e., using the same step shown in Figure 2C).
Repeat the above steps to segment other objects, if multiple objects have been acquired in different spectral channels.
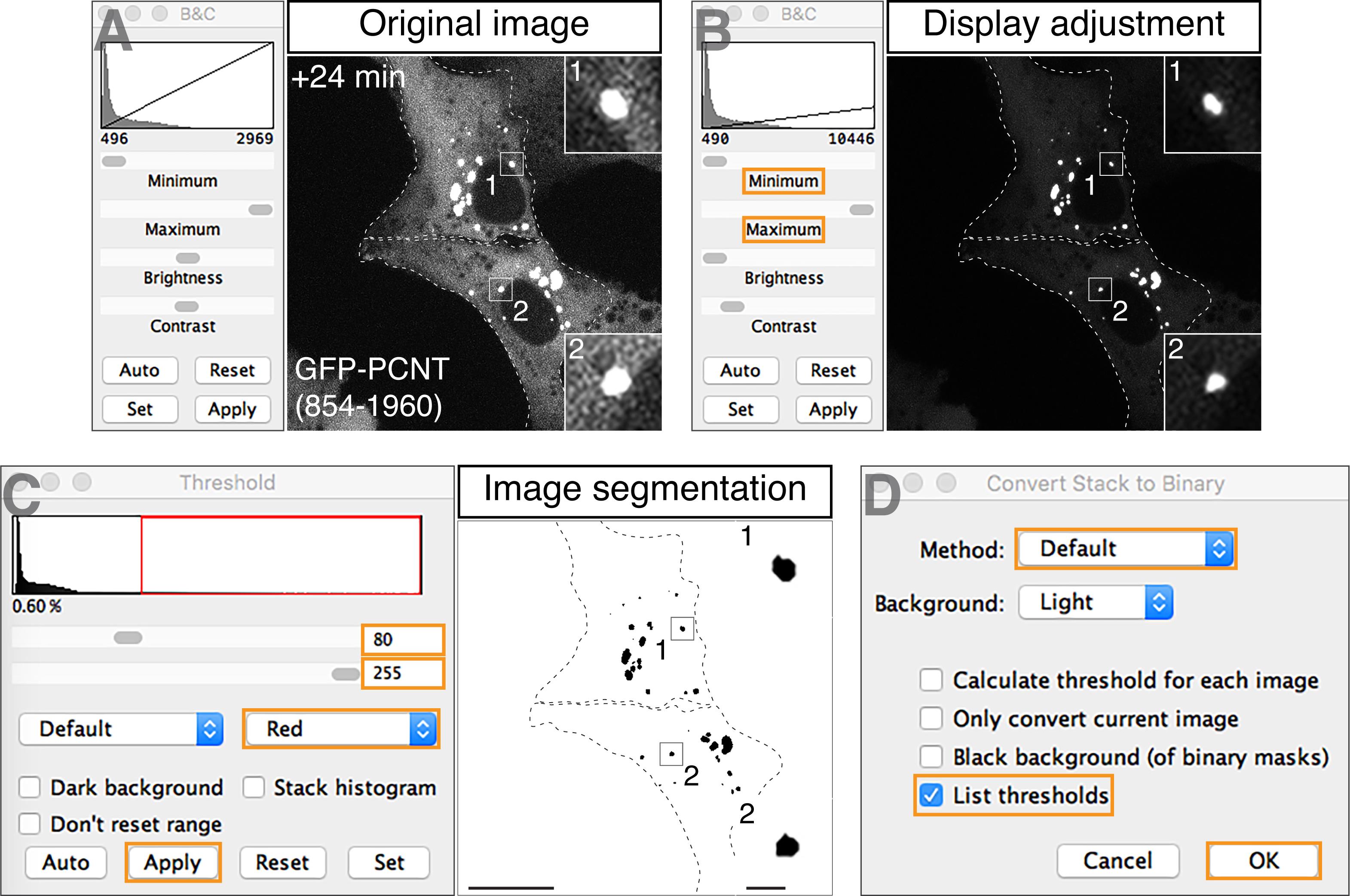
Figure 3. Examples of image segmentation by intensity-based thresholding. Using the GFP channel of the +24 min time frame in Figure 1 as an example, a step-by-step procedure to segment GFP-PCNT (854–1960) condensates is shown here, including opening the confocal z-projected image (A), adjusting display ranges (B), and applying intensity-based thresholding steps (C, D). Scale bar: 20 µm and 2 µm (insets).
Object identification and measurement
Note: This step assigns each identified object in each channel with a unique ID number and extracts measurement data, which will be used for object tracking.
Open each segmented stack image in Fiji.
Define the image scale through “Analyze” > “Set Scale…”. Then select “OK” (Figure 4A).
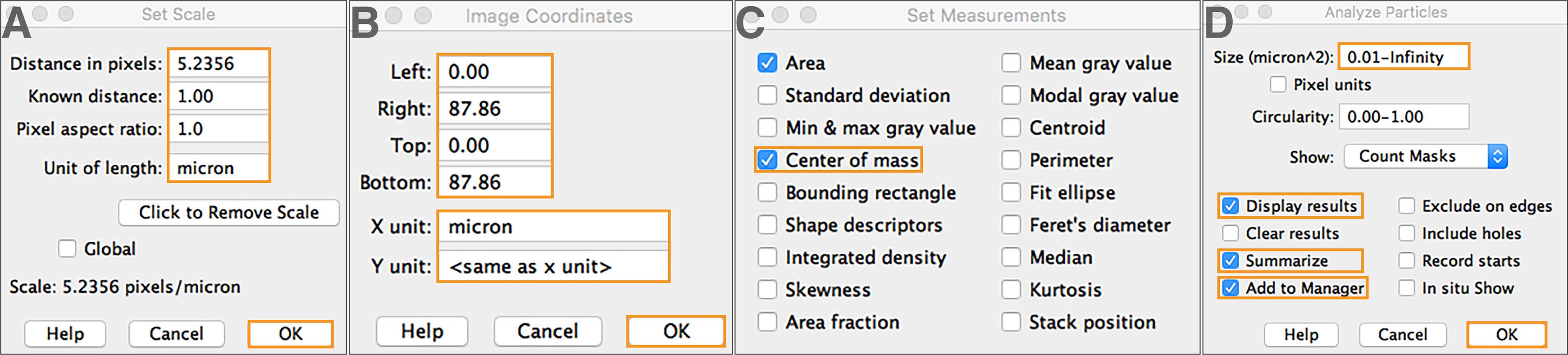
Figure 4. Object identification and measurement. (A) Define image scales. (B) Define image coordinates. (C) Specify object measurement and statistical parameters. (D) Execute the “Analyze Particles” function.Define image coordinates through “Image” > “Adjust” > “Coordinates…”. Then select “OK” (Figure 4B).
Note: It is important to set the correct image scale and coordinates prior to object measurement.
Go to “Analyze” > “Set measurement” to specify measurements and statistics to be extracted (Figure 4C).
Note: We choose to extract area and center-of-mass for our analysis. Users may select other measurements that best fit their quantification purposes. To use the Python tracking program in this pipeline, make sure to include the center-of-mass measurement, as TRACES uses (x,y) object coordinates for tracking.
Activate the “Analyze Particles” function through “Analyze” > “Analyze Particles…”. Set appropriate size thresholds to mask objects of interest and extract measurements. Check “Display results”, “Summarize”, and “Add to manager”, and then select “OK” (Figure 4D).
Note: The size thresholding in this step helps further filter out background signals that are misclassified as segmented objects from the previous intensity-based thresholding step (Figure 3). Circularity thresholding—ranging from 0.00 (elongated polygon) to 1.00 (perfect circle) (Ferreira and Rasband, 2012)—may also be applied to segment objects of interest. We set our circularity threshold as 0.00–1.00 (Figure 4D), meaning that we do not set circularity thresholds and accept all objects regardless of their circularity. “Display results” and “Summarize” options are selected so that the measurement data will be displayed and can be saved for object tracking. The “Add to manager” option is selected so that a unique ID number is automatically assigned to each identified and measured object across all time frames (e.g., Figure 5, fourth column). Because ID numbers are unique, the same object will be represented by different ID numbers across time frames.
Select “yes” to process all time frames of a time-lapse image series.
Save the resulting segmented images with objects labeled by unique ID numbers (e.g., Figure 5, fourth column).

Figure 5. Examples of display adjustment, image segmentation, and object identification for three fluorescently labeled object types. Using the +24 min time frame shown in Figure 1 as an example, z-projected confocal images with the original display settings (first column), adjusted display settings (second column), image segmentation by thresholding (third column), and object identification by executing the “Analyze Particles” function (fourth column) are shown. Note that for each object type, a unique ID number (e.g., insets, fourth column) is automatically assigned to each identified object across all time frames of the time-lapse image series. Scale bars: 20 µm and 2 µm (insets).Save the object measurement result (Figure 6A) and frame count (Figure 6B) data as .csv format for each object type.
Note: Fiji refers to each “time frame” of a time-lapse image series as “slice” (Figure 6B). To avoid confusion between “time frames” in a time series and “slices” within a z-stack, “slice” and “slice count” (initially defined by Fiji shown in Figure 6B) are subsequently re-defined as “frame” and “frame number”, respectively, in the pipeline.
Repeat the above steps for other objects if multiple objects have been acquired in different spectral channels. Figure 5 demonstrates a complete process of display adjustment, image segmentation, and object identification of three fluorescently labeled object types from a single time frame of a time-lapse image series (i.e., +24 min time frame shown in Figure 1).
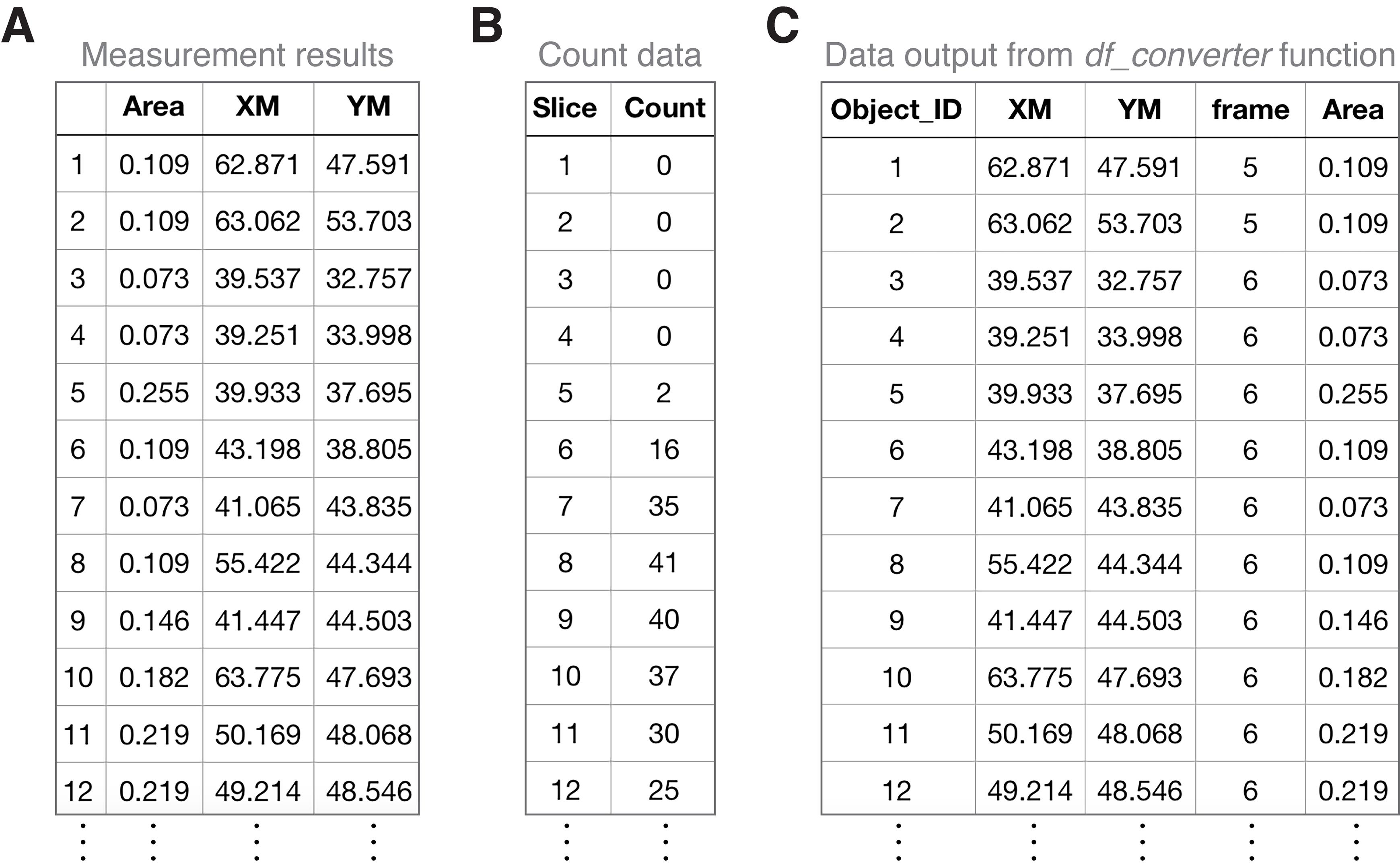
Figure 6. Examples of Fiji-exported datasets and their integration using the Python-based df_converter function. (A) Examples of the measurement results of PCNT condensate objects exported from Fiji (from the same time-lapse series shown in Figure 1). XM and YM, x and y coordinates of the object, are based on the center of mass. (B) Examples of the slice/frame count data of PCNT condensate objects exported from Fiji. “Slice” represents each time frame in a time-lapse image series. “Count” represents the number of identified condensate objects in the corresponding “slice”/time frame. (C) Examples of dataframe output of PCNT condensate objects after the execution of df_converter function. Here, “frame” indicates the frame number where each object (with a unique object ID) is located.
Object tracking using a Python-based program
Note: The TRACES object tracking program is written in Python and implemented in Jupyter Notebook. See Notes section B for installation instructions of Anaconda distribution, which conveniently includes the installation of Python, Jupyter Notebook, and packages to run TRACES. TRACES object tracking workflow includes 6 steps (Section I to VI), as detailed below (Figure 7A).

Figure 7. Object tracking, quantification, and data visualization in the TRACES pipeline. (A) Workflow for object tracking. (B) Quantification and data visualization. The highlight of TRACES script lines in each section is shown. See TRACES.ipynb for the complete script. Example plots of condensate object numbers, average condensate distances to the centrosome, and average condensate areas in a single cell are shown. See Figure 4 in Jiang et al., 2021, for examples of the final plots.Import packages and define working directory (Figure 7A, section I)
Open “TRACES.ipynb” in Jupyter Notebook.
Note: See Notes section C for instructions on launching the Jupyter Notebook application.
Import packages and define working directory.
Load files (Figure 7A, section II)
Load data files with measurement results (e.g., Figure 6A) and frame counts (e.g., Figure 6B) for each object type.
Note: For example, the filename of measurement results for the centrosome object is “centrosome_results.csv”. We thus load the file as “pd.read_csv(‘centrosome_results.csv’)” (Figure 7A, II, line 1). The data files for the measurement results and frame counts are loaded for three object types—centrosome, condensate, and nucleus—whose dataframe variables are named as “cen”, “con”, and “h2a” in the script, respectively. (Figure 7A, II). As variable names are arbitrary, users may choose their own variable names freely.
Combine datasets using the df_converter function
Note: df_converter function combines the object measurement results (e.g., Figure 6A) and frame counts (e.g., Figure 6B) from Fiji into a dataframe with specified variable orders for each object type (e.g., Figure 6C); this information will be used for object tracking.
Note: Two general steps are involved in applying a function: (a) define function and (b) specify parameters and execute function, followed by (c) data output (i.e., a dataframe is generated), as exemplified below.
In section III of the TRACES script, run scripts in the first cell to define “df_converter(df1, df2)” function.
In the second cell, specify parameters and execute function for each object type. “df1” and “df2” are the dataframe variables for the measurement results and frame counts, respectively, specified in section II of the script.
Note: For example, “cen_r” and “cen_sc” are the dataframe variables for the measurement results and frame counts of centrosome objects defined in section II, respectively. We thus specify function parameters as “df_converter(cen_r, cen_sc)” for centrosome object type (Figure 7A, III, line 2).
After execution of the function in the previous step (Step B3b), a dataframe for each object type will be generated with the following variables (from left to right): “Object_ID” (object ID number), “XM” (object x coordinate), “YM” (object y coordinate), “frame” (frame number where each object is located), and other customized measurements (e.g., “Area”) (Figure 6C).
Note: Make sure to create a unique variable name for each dataframe. For example, object dataframe variables are named “centrosome_df”, “condensate_df”, and “h2a_df” for centrosome, condensate, and nucleus object types, respectively. As variable names are arbitrary, users may choose their own variable names freely.
Group objects within cells using the object_grouping function
Note: The position of the nucleus serves as the proxy for the location of the cell. To track subcellular objects over time at single-cell resolution, we first group each object to its own cell by assigning each object to its nearest nucleus using the object_grouping function. This function first measures the distance of each object to all nuclei per time frame and then assigns each object to its nearest nucleus across time frames based on the minimal Euclidean distance.
Note: If users’ cell line of interest does not include a nucleus marker, Hoechst or other fluorescent dyes can be used to label the nucleus. Alternatively, other cytoplasmic markers that generally reflect the cell position may also be used as the reference point for grouping.
In section IV of the TRACES script, run scripts in the first cell to define the “object_grouping(channel_df, h2a_df)” function.
In the second cell, specify parameters and execute function for each object type. “channel_df” and “h2a_df” are the dataframe variables of the grouping and nucleus objects, respectively, specified in section III of the script.
Note: For example, “condensate_df” and “h2a_df” are the dataframe variables for the condensate and nucleus objects defined in section III. To group condensate objects within cells, specify function parameters as “object_grouping(condensate_df, h2a_df)” (Figure 7A, IV, line 2). After execution, both condensate and centrosome object types are then grouped within cells; the resulting dataframe variables after grouping are named “con_h2a_group” and “cen_h2a_group”, respectively.
Integrate spatiotemporal information of different objects using the dist_between_channels function
Note: The dist_between_channels function integrates measurements (e.g., distance, area) between specified object types (e.g., condensate and centrosome) within each cell across time frames. The previous object grouping step only yields a pairwise relationship of the specified two objects (e.g., condensate to nucleus, or centrosome to nucleus). This step is to integrate these two separate pairwise datasets into a single dataset. Therefore, a three-object relationship can be obtained (e.g., the relationship among condensate, nucleus, and centrosome objects).
In section V of the TRACES script, run scripts in the first cell to define the “dist_between_channels(channel1_h2a_df, channel2_h2a_df)” function.
In the second cell, specify parameters and execute function. “channel1_h2a_df” and “channel2_h2a_df” are the dataframe variables of the object grouping specified in section IV of the script.
Note: For example, “con_h2a_group” and “cen_h2a_group” are the dataframe variables for object grouping of condensates and centrosomes specified in section IV, respectively. To integrate different measured values between condensate and centrosome objects within each cell, we specify function parameters as “dist_between_channels(con_h2a_group, cen_h2a_group)” (Figure 7A, V, line 2). After execution, an integrated object dataframe variable, “con_cen_df”, is generated (Figure 7A, V, line 2), in which the relationship among condensates, centrosomes, and nuclei is defined. We will use this dataframe for object tracking in the next step.
Track objects in a target cell using the object_tracker function
Note: The object_tracker function simultaneously tracks all three object types (e.g., centrosome, condensate, and nucleus) of a cell specified by the user. To use this function, users need to specify a target cell for tracking. We use the nucleus object ID to define a target cell. Users may find the nucleus object ID information by viewing the binary segmented image with objects labeled by unique ID numbers generated from the object identification and measurement step in Fiji (e.g., Figure 5, fourth column).
Note: The current TRACES tracking algorithm only tracks objects in non-dividing cells, as the program assumes one nucleus object per cell. We hope to implement object tracking features for dividing cells in the future.
In section VI of the TRACES script, run scripts in the first cell to define the “object_tracker(start_index, end_frame, dataframe)” function.
In the second cell, specify parameters and execute function. The “start_index” parameter is the nucleus object ID of a target cell at the starting frame. The “end_frame” is the ending frame number for tracking. The “dataframe” is the dataframe variable of the integrated object specified in section V of the script.
Note: For example, we want to track objects in a target cell from frame number 5 to 54. The nucleus object ID of the target cell is “21” at frame number 5, and “con_cen_df” is the integrated object dataframe variable specified in section V. Thus, we specify function parameters as “object_tracker(21, 54, con_cen_df)” (Figure 7A, VI, line 2).
After execution, a dataframe named “track_result” is generated, and automatically exported to the working directory as a .csv file named “track_result.csv” (Figure 7A, VI, line 5). This csv file contains the following columns (from left to right): “frame_num” (frame number), “orig_c1_index” (condensate object ID), “orig_c2_index” (centrosome object ID), “orig_h2a_index” (nucleus object ID), “H2A_X”, “H2A_Y” (x and y coordinates of nucleus objects), “c1_XM”, “c1_YM” (x and y coordinates of condensate objects), “c2_XM”, “c2_YM” (x and y coordinates of centrosome objects), and “distance_c1_to_c2” (distance between condensate to centrosome objects). We will use this dataframe for quantification in the next steps.
Note: If users want to track multiple cells in a time-lapse image series, specify a new target cell and repeat the above steps.
Quantification and data visualization using a Python-based program
Quantify object numbers using the count_by_frame function
Note: The count_by_frame function calculates the number of objects per time frame in the target cell and generates a plot for data visualization (Figure 7B, VII). Users need to specify an object type of interest for quantification.
In section VII of the TRACES script, run scripts in the first cell to define the “count_by_frame(track_table, track_table_object_colname) function.
In the second cell, specify parameters and execute function. The “track_table” parameter is the dataframe variable of tracking results specified in section VI. The “track_table_object_colname” is the object ID column of interest in the tracking result dataframe.
Note: For example, the dataframe variable for tracking results is “track_result”. The object ID column name of condensate objects is “orig_c1_index”. To quantify the condensate object number, we specify function parameters as “count_by_frame(track_result, ‘orig_c1_index’)” (Figure 7B, VII, line 2).
After execution, the quantification data and plot (Figure 7B, VII) are automatically exported to the working directory as .csv and .png files, respectively.
Note: The graph shows condensate object numbers as a function of frame number in a target cell (Figure 7B, VII). Users can also convert the x-axis from frame number to time by specifying the time interval between frames. See Figure 4B in Jiang et al., 2021 for examples of the final plots.
Quantify object distances using the distance_plot function
Note: The distance_plot function calculates the average distance between two object types (e.g., the condensate-to-centrosome distance) per time frame in the target cell and generates a plot for data visualization (Figure 7B, VIII).
In section VIII of the TRACES script, run scripts in the first cell to define the “distance_plot(track_table)” function.
In the second cell, specify parameters and execute function. The “track_table” parameter is defined as above in Step C1b.
Note: For example, the dataframe variable for tracking results is “track_result”. To quantify the average condensate-to-centrosome distance per time frame, we specify function parameters as “distance_plot(track_result)” (Figure 7B, VIII, line 2).
After execution, the quantification data and plot (Figure 7B, VIII) are automatically exported to the working directory as .csv and .png files, respectively.
Note: The graph shows the average condensate-to-centrosome distance as a function of frame number in a target cell (Figure 7B, VIII). Users can also convert the x-axis to time, and combine the quantification result from multiple cells. See Figure 4C in Jiang et al., 2021 for examples of the final plots.
Measure the user-specified object properties using the measurement_plot function
Note: Users may choose to measure other properties of objects (e.g., area) during the object identification and measurement step in Fiji. Users can apply the measurement_plot function to measure user-specified properties. Figure 7B, IX shows an example of measuring the area of condensate objects.
In section IX of the TRACES script, run scripts in the first cell to define the “measurement_plot(track_table, track_table_object_colname, object_df, m_type)” function.
In the second cell, specify parameters and execute function. The “track_table” and “track_table_object_colname” parameters are defined as above in Step C1b. The “object_df” is the dataframe variable for the object of interest specified in section III of the script. The “m_type” is the column name of measurement type of interest in the “object_df” dataframe (Figure 6C).
Note: For example, the dataframe variable for tracking results is “track_result”. The object ID column name of the condensate object is “orig_c1_index”. The condensate object dataframe is “condensate_df”, as specified in section III of the script. “Area” is the column name of the area measurement in “condensate_df” dataframe (Figure 6C). To quantify the average condensate area per time frame, we specify function parameters as “measurement_plot(track_result, ‘orig_c1_index’, condensate_df, ‘Area’)” (Figure 7B, IX, line 2).
After execution, the quantification data and plot (Figure 7B, IX) are automatically exported to the working directory as .csv and .png files, respectively.
Note: The graph shows the average condensate area as a function of frame number in a target cell (Figure 7B, IX). Users can also convert the x-axis to time and combine the quantification result from multiple cells. See Figure 4A in Jiang et al., 2021 for examples of the final plots.
Notes
Installation of Fiji
Follow instructions in https://imagej.net/software/fiji/downloads to install Fiji.
Installation of Anaconda
Anaconda is a distribution platform of Python/R programming languages and data-science packages. Downloaded Anaconda also includes Jupyter Notebook, a convenient web-based application for generating and editing Python/R language scripts.
Follow instructions in https://docs.anaconda.com/anaconda/install/ to install Anaconda.
Launching Jupyter Notebook
Note: For detailed instructions, follow this link to open the Jupyter Notebook application.
Download and save “TRACES.ipynb” in a local folder.
To launch Jupyter Notebook, first open the computer terminal.
For Mac users, type “jupyter notebook” in the terminal, and then press the “Enter” button.
The Jupyter Notebook dashboard will appear in a new browser window.
Select “TRACES.ipynb” to open in Jupyter Notebook.
Line numbers on Jupyter Notebook
If line numbers do not show up on Jupyter Notebook, go to “View” > “Toggle Line Numbers”.
Manual correction of misgrouped objects in the target cell using the manual_correct function
Note: As post-analysis quality control, users should confirm the accuracy of the tracking process, by manually evaluating the behaviors of objects in the original time-lapse images with those of the segmented objects and the exported quantification results. Occasionally, certain background signals might be missegmented as objects during image processing steps, which are then incorrectly grouped into the target cell. To solve this issue, we have developed the manual_correct function for users to remove misgrouped objects from the target cell. This step is optional and should only be applied to remove misgrouped objects prior to the workflow of quantification and data visualization.
In the “Optional step” section of the TRACES script, run scripts in the first cell to define the “manual_correct(track_table, track_table_object_colname, index_list)” function.
In the second cell, specify parameters and execute function (Figure 8). The “track_table” parameter is the dataframe variable of tracking results specified in section VI. The “track_table_object_colname” is the object ID column of interest in the tracking result dataframe. The “index_list” is a list of ID numbers of misgrouped objects to be removed from the target cell.
Note: For example, if we want to remove two misgrouped condensate objects with ID number 366 and 375, first specify the “index_list” as “rm_index_ls = [366,375]” (figure 8, line 2). The dataframe variable for tracking results is “track_result”. The column name of condensate object ID in the dataframe is “orig_c1_index”. Thus, we specify function parameters as “manual_correct(track_result, ‘orig_c1_index’, rm_index_ls)” (Figure 8, line 3). The dataframe of the corrected tracking results is automatically exported to the working directory as a .csv file (Figure 8, line 6).

Figure 8. Remove misgrouped objects using the manual_correct function. A highlight of the TRACES script lines. See TRACES.ipynb for the complete script.
- Alvarez-Rodrigo, I., Steinacker, T. L., Saurya, S., Conduit, P. T., Baumbach, J., Novak, Z. A., Aydogan, M. G., Wainman, A. and Raff, J. W. (2019). Evidence that a positive feedback loop drives centrosome maturation in fly embryos. eLife 8: e50130.
- Aydogan, M. G., Hankins, L. E., Steinacker, T. L., Mofatteh, M., Saurya, S., Wainman, A., Wong, S. S., Lu, X., Zhou, F. Y. and Raff, J. W. (2022). Centriole distal-end proteins CP110 and Cep97 influence centriole cartwheel growth at the proximal-end. J Cell Sci. doi: 10.1242/jcs.260015.
- Aydogan, M. G., Steinacker, T. L., Mofatteh, M., Wilmott, Z. M., Zhou, F. Y., Gartenmann, L., Wainman, A., Saurya, S., Novak, Z. A., Wong, S. S., et al. (2020). An Autonomous Oscillation Times and Executes Centriole Biogenesis. Cell 181(7): 1566-1581 e1527.
- Aydogan, M. G., Wainman, A., Saurya, S., Steinacker, T. L., Caballe, A., Novak, Z. A., Baumbach, J., Muschalik, N. and Raff, J. W. (2018). A homeostatic clock sets daughter centriole size in flies. J Cell Biol 217(4): 1233-1248.
- Chen, B. C., Legant, W. R., Wang, K., Shao, L., Milkie, D. E., Davidson, M. W., Janetopoulos, C., Wu, X. S., Hammer, J. A., 3rd, Liu, Z., et al. (2014). Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science 346(6208): 1257998.
- Ershov, D., Phan, M. S., Pylvänäinen, J. W., Rigaud, S. U., Le Blanc, L., Charles-Orszag, A., Conway, J. R. W., Laine, R. F., Roy, N. H., Bonazzi, D., et al. (2021). Bringing TrackMate into the era of machine-learning and deep-learning. bioRxiv: 2021.2009.2003.458852.
- Giepmans, B. N., Adams, S. R., Ellisman, M. H. and Tsien, R. Y. (2006). The fluorescent toolbox for assessing protein location and function. Science 312(5771): 217-224.
- Hell, S. W. (2007). Far-field optical nanoscopy. Science 316(5828): 1153-1158.
- Jiang, X., Ho, D. B. T., Mahe, K., Mia, J., Sepulveda, G., Antkowiak, M., Jiang, L., Yamada, S. and Jao, L. E. (2021). Condensation of pericentrin proteins in human cells illuminates phase separation in centrosome assembly. J Cell Sci 134(14): jcs258897.
- Kholodenko, B. N., Hancock, J. F. and Kolch, W. (2010). Signalling ballet in space and time. Nat Rev Mol Cell Biol 11(6): 414-426.
- Lamprecht, M. R., Sabatini, D. M. and Carpenter, A. E. (2007). CellProfiler: free, versatile software for automated biological image analysis. Biotechniques 42(1): 71-75.
- Linkert, M., Rueden, C. T., Allan, C., Burel, J. M., Moore, W., Patterson, A., Loranger, B., Moore, J., Neves, C., Macdonald, D., et al. (2010). Metadata matters: access to image data in the real world. J Cell Biol 189(5): 777-782.
- Lippincott-Schwartz, J. and Patterson, G. H. (2003). Development and use of fluorescent protein markers in living cells. Science 300(5616): 87-91.
- Oreopoulos, J., Berman, R. and Browne, M. (2014). Spinning-disk confocal microscopy: present technology and future trends. Waters, J. C. and Wittman, T. (Eds.). In: Methods in Cell Biology. Academic Press, 153-175.
- Stephens, D. J. and Allan, V. J. (2003). Light microscopy techniques for live cell imaging. Science 300(5616): 82-86.
- Stirling, D. R., Swain-Bowden, M. J., Lucas, A. M., Carpenter, A. E., Cimini, B. A. and Goodman, A. (2021). CellProfiler 4: improvements in speed, utility and usability. BMC Bioinformatics 22(1): 433.
- Tanaami, T., Otsuki, S., Tomosada, N., Kosugi, Y., Shimizu, M. and Ishida, H. (2002). High-speed 1-frame/ms scanning confocal microscope with a microlens and Nipkow disks. Appl Opt 41(22): 4704-4708.
- Tinevez, J. Y., Perry, N., Schindelin, J., Hoopes, G. M., Reynolds, G. D., Laplantine, E., Bednarek, S. Y., Shorte, S. L. and Eliceiri, K. W. (2017). TrackMate: An open and extensible platform for single-particle tracking. Methods 115: 80-90.
Acknowledgments
We thank Dr. Sean R. Collins and Dr. Mark Winey for suggestions on optimizing the TRACES pipeline, and all members of the Jao Lab for discussion. This protocol was adapted from a previous work (Jiang et al., 2021). This work was supported by the National Institutes of Health (1R01GM144435-01 to L.-E.J.) and UC Davis Predoctoral Fellowships (Emmy Werner and Stanley Jacobsen Fellowship and Floyd and Mary Schwall Dissertation Year Fellowship to X.J.).
Competing interests
The authors declare no competing or financial interests.
References
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Jiang, X., Jiang, L. and Jao, L. E. (2022). TRACES: a Freely Accessible, Semi-automated Pipeline for Detection, Tracking, and Quantification of Fluorescently Labeled Cellular Structures. Bio-protocol 12(15): e4479. DOI: 10.21769/BioProtoc.4479.
Category
Bioinformatics and Computational Biology
Cell Biology > Cell imaging > Confocal microscopy
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.