- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

DSP-crosslinking and Immunoprecipitation to Isolate Weak Protein Complex
Published: Vol 12, Iss 15, Aug 5, 2022 DOI: 10.21769/BioProtoc.4478 Views: 9608
Reviewed by: Chiara AmbrogioSoumya MoonjelyAdriano Bolondi
Original research article
The authors used this protocol in:

Oct 2021
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Detecting protein-protein interactions (PPIs) is one of the most used approaches to reveal the molecular regulation of protein of interests (POIs). Immunoprecipitation of POIs followed by mass spectrometry or western blot analysis enables us to detect co-precipitated POI-binding proteins. However, some binding proteins are lost during cell lysis or immunoprecipitation if the protein binding affinity is weak. Crosslinking POI and its binding proteins stabilizes the PPI and increases the chance of detecting the interacting proteins. Here, we introduce the method of DSP (dithiobis(succinimidyl propionate))-mediated crosslinking, followed by tandem immunoprecipitation (FLAG and HA tags). The eluted proteins interacting with POI can be analyzed by mass spectrometry or western blotting. This method has the potential to be applied to various cytoplasmic proteins.
Graphical abstract:

Background
Detection of proteins interacting with POIs is one of the effective ways to explore how the POIs are regulated by other proteins in the cells. Immunoprecipitation of the POI is a widely used method to co-precipitate the associating proteins, which can be detected by following mass spectrometry or western blot analysis. When the interaction between a protein and the POI is stable and strong enough, crosslinking is not necessary to isolate the binding protein. However, the simple immunoprecipitation method may not be suitable for the detection of binding proteins that bind to the POI weakly or transiently. DSP is a cell-permeable chemical crosslinker that reacts with amino groups such as lysine residue. Therefore, treatment of cells with DSP can strengthen the PPI in the cells (Lomant and Fairbanks, 1976; Zlatic et al., 2010) (Figure 1). Furthermore, since DSP has a disulfide bond in its spacer arm, this crosslinker can be cleaved by a reducing agent after the isolation of the protein complex to obtain linear proteins.
By performing DSP crosslinking and following immunoprecipitation and mass spectrometry analysis, we have previously investigated IL-1β-dependent PPI of Regnase-1, an RNase which degrades mRNAs coding inflammatory genes. We discovered that SKP1, CUL1, F-box (SCF) proteins, and 14-3-3 proteins bind to Regnase-1 in an IL-1β stimulation-dependent manner (Akaki et al., 2021).
Here, we describe the method of DSP crosslinking and immunoprecipitation using HeLa cells expressing POIs. Whereas previous methods of DSP-crosslinking and protein purification were performed with single immunoprecipitation (Zlatic et al., 2010; Wang et al., 2019), we precipitated FLAG- and HA-tagged POI by tandem affinity purification after crosslinking to isolate binding proteins with low background. As the lysis buffer used in this method is sufficient to dissolve Regnase-1, which predominantly localizes in the cytoplasm, this method is suggested to be applicable to investigate PPIs of other cytoplasmic proteins.
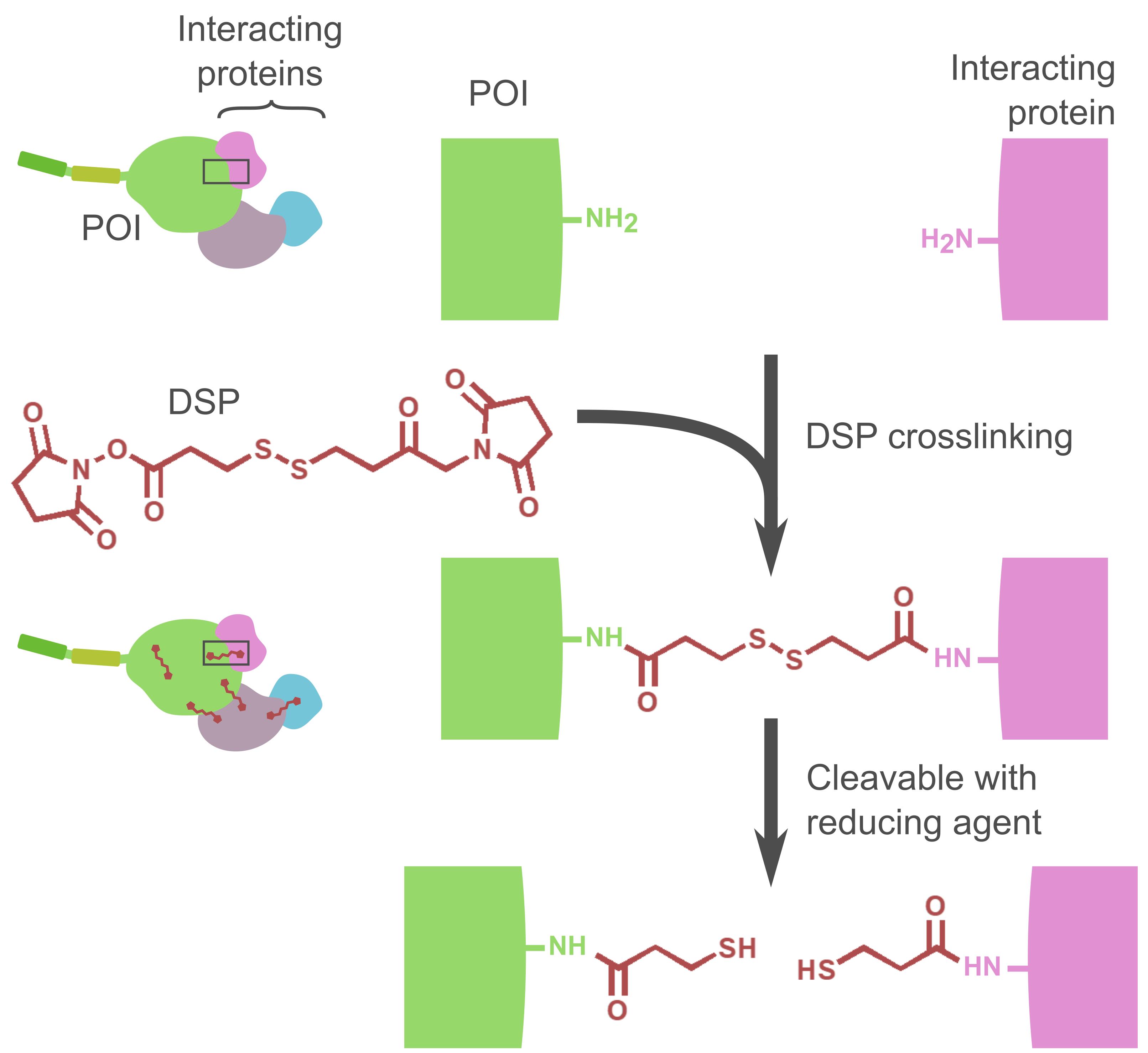
Figure 1. Schematic illustration of the DSP reaction. One DSP molecule reacts with two proximal amino groups. After the purification of the crosslinked POI and proteins, the linkers can be cleaved by reducing agents such as 2-mercaptoethanol or dithiothreitol (DTT).
Materials and Reagents
1.5 mL tube (Eppendorf, Eppendorf Safe-Lock Tubes, 1.5 mL, Eppendorf QualityTM, catalog number: 0030120086)
50 mL tube (Thermo Scientific, 50 mL Conical Sterile Polypropylene Centrifuge Tubes, catalog number: 339652)
10 cm dish (Corning, Falcon® 100 mm TC-treated Cell Culture Dish, catalog number: 353003)
Scraper (VIOLAMO, Violamo Cell Lifter, catalog number: 1-2249-01)
20–200 μL pipette tips (Labcon, catalog number: 1093-260-000-9)
1,000 μL pipette tips (Labcon, catalog number: 1045-260-000-9)
5 mL pipettes (Thermo Scientific, NuncTM 5 mL Serological Pipette, catalog number: 170355N)
10 mL pipettes (Thermo Scientific, NuncTM 10 mL Serological Pipette, catalog number: 170356N)
25 mL pipettes (Thermo Scientific, NuncTM 25 mL Serological Pipette, catalog number: 170357N)
50 mL pipettes (Thermo Scientific, NuncTM 50 mL Serological Pipette, catalog number: 170358N)
HeLa cells
DSP (dithiobis(succinimidyl propionate)) (Tokyo Chemical Industry Co., Di(N-succinimidyl) 3,3'-Dithiodipropionate [Cross-linking Reagent], catalog number: D2473)
Dynabeads Protein G (Invitrogen, DynabeadsTM Protein G for Immunoprecipitation, catalog number: 10004D)
FLAG Peptide (Millipore, FLAG® Peptide, catalog number: F3290, Dissolve in TBS as described in “FLAG peptide stock solution”. Aliquot the solution in 20 μL and store at -30 °C.)
Anti-FLAG antibody (Merck, Monoclonal ANTI-FLAG® M2 antibody produced in mouse, catalog number: F3165)
Anti-HA antibody (Merck, Anti-HA antibody Mouse monoclonal, catalog number: H3663)
Lipofectamine 2000 (Invitrogen, Lipofectamine® 2000, catalog number: 11668500; any transfection reagent can be used if it works)
1 M Tris HCl pH7 (Invitrogen, Tris (1 M), pH 7.0, RNase-free, catalog number: AM9850G)
1 M Tris HCl pH8 (Invitrogen, Tris (1 M), pH 8.0, RNase-free, catalog number: AM9855G)
5 M NaCl (Invitrogen, NaCl (5 M), RNase-free, catalog number: AM9760G)
NP-40 (Nacalai Tesque, Nonidet(R) P-40, catalog number: 23640-94)
cOmplete Mini EDTA-free (Roche, cOmpleteTM, Mini, EDTA-free Protease Inhibitor Cocktail, catalog number: 11836170001)
PhosSTOP (Roche, PhosSTOPTM, catalog number: 4906837001)
H2O (Invitrogen, UltraPureTM DNase/RNase-Free Distilled Water, catalog number: 10977-023)
DMSO (Merck, Dimethyl sulfoxide, catalog number: D2650)
Urea (Nacalai Tesque, catalog number: 35940-65)
Tris (Nacalai Tesque, Tris(hydroxymethyl)aminomethane, catalog number: 35434-21)
HCl (Nacalai Tesque, Hydrochloric Acid(35%), catalog number: 18321-05)
SDS (Nacalai Tesque, Sodium Lauryl Sulfate, catalog number: 08933-05)
Glycerol (Nacalai Tesque, catalog number: 17045-65)
Bromophenol blue (Nacalai Tesque, catalog number: 05808-61)
DMEM (Nacalai Tesque, DMEM (4.5 g/L Glucose) with L-Gln, without Sodium Pyruvate, liquid, catalog number: 08459-64)
FBS (Gibco, catalog number: 10270-106, LOT: 42G9391K)
PBS (Nacalai Tesque, D-PBS(-) without Ca and Mg, liquid, catalog number: 14249-24)
Penicillin/Streptomycin (Nacalai Tesque, catalog number: 09367-34)
100 mM DSP (see Recipes)
Tris-HCl (1 M, pH 7.4) (see Recipes)
STOP solution (see Recipes)
Wash buffer (see Recipes)
IP buffer (see Recipes)
TBS (Tris Buffered Saline) (see Recipes)
FLAG peptide stock solution (see Recipes)
FLAG-elution buffer (see Recipes)
Urea elution buffer (see Recipes)
3× SDS sample buffer (see Recipes)
1× SDS elution buffer (see Recipes)
Equipment
CO2 incubator (SANYO, model: MCO-19AIC)
Cell counter (Beckman Coulter, model: Z1 Coulter Particle Counter)
Water bath (TAITEC, model: 0068750-000)
Magnetic stand (Invitrogen, DynaMagTM-2 Magnet, model: 12321D)
Rotating incubator (TAITEC, model: RT-50)
Centrifuge (TOMY, model: MX-307)
Heat block (astec, Block Incubator, model: BI-516S)
Micropipette (up to 20 μL) (Gilson, PIPETMAN P20)
Micropipette (up to 200 μL) (Gilson, PIPETMAN P200)
Micropipette (up to 1,000 μL) (Gilson, PIPETMAN P1000)
Pipette controller (Drummond Scientific Company, Pipet-Aid XPress, model: 4-040-135)
Procedure
Preparing cells expressing POI
Notes:
Before the transfection, we culture HeLa cells in DMEM with 10% (FBS), 1% Penicillin/Streptomycin, and 100 µM 2-Mercaptoethanol. Do not use Penicillin/Streptomycin-containing media at the time of transfection.
As a negative control, always prepare sample(s) not expressing FLAG-HA tagged POI.
Alternatively, one can utilize the doxycycline-inducible system. In this case, one should establish a cell line expressing POI in a doxycycline-dependent manner. For the negative control, prepare sample(s) not treated with doxycycline.
Plate 8.0 × 105 HeLa cells in 10 cm dish per sample (10 mL of DMEM containing 10% FBS per dish).
Note: The number of cells can be scaled up or down depending on the purpose of the experiment. The size of dishes and how much plasmid DNA, buffers, beads, antibodies, etc., used should also be scaled up or down along with the amount of the cells.
Incubate the cells at 37 °C, 5% CO2 for 24 h.
Transfect plasmids for FLAG-HA-tagged POI expression using Lipofectamine 2000 according to manufacturer's instructions.
Note: We transfected HeLa cells in 10 cm dish with 4.0 μg of plasmids using 12 μL of Lipofectamine 2000 (each of them is diluted in 200 μL of serum-free DMEM). As too much overexpression of Regnase-1 (our POI) causes cell toxicity, we adjusted the amount of plasmid DNA used to avoid this. In addition, since the efficient amount of POI for IP and following analysis varies depending on POI, one might have to optimize the amount of plasmid DNA used for the transfection.
Incubate the cells at 37 °C, 5% CO2 overnight.
Preparing Wash buffer and IP buffer
On the day of DSP-crosslinking and immunoprecipitation, prepare Wash buffer and IP buffer (see Recipes) before the crosslinking. Keep them on ice.
DSP-crosslinking
Prepare 100 mM DSP just prior to DSP-crosslinking.
Dilute 100 mM DSP to 0.1 mM DSP in pre-warmed (37 °C) PBS.
Rinse the dishes twice with 5 mL of pre-warmed (37 °C) PBS/dish.
Discard the PBS from the dishes.
Add 5 mL of 0.1 mM DSP (prepared in step C2) in each dish.
Note: The higher the concentration of DSP is, the larger the crosslinked protein complex becomes. The concentration of DSP can be optimized depending on the PPI you aim to detect (Figure 2). Note that too much crosslinking makes the protein complex too large, which may result in non-specific crosslinking of non-relevant proteins.
Incubate the dishes at 37 °C for 30 min in a CO2 incubator.
Note: Alternatively, dishes can be incubated at 4 °C for ~2 h. In this case, crystals may appear on the dishes, and it is difficult to remove them. We found that these crystals do not interrupt immunoprecipitation, but we do not know the exact effect of these crystals on the result.
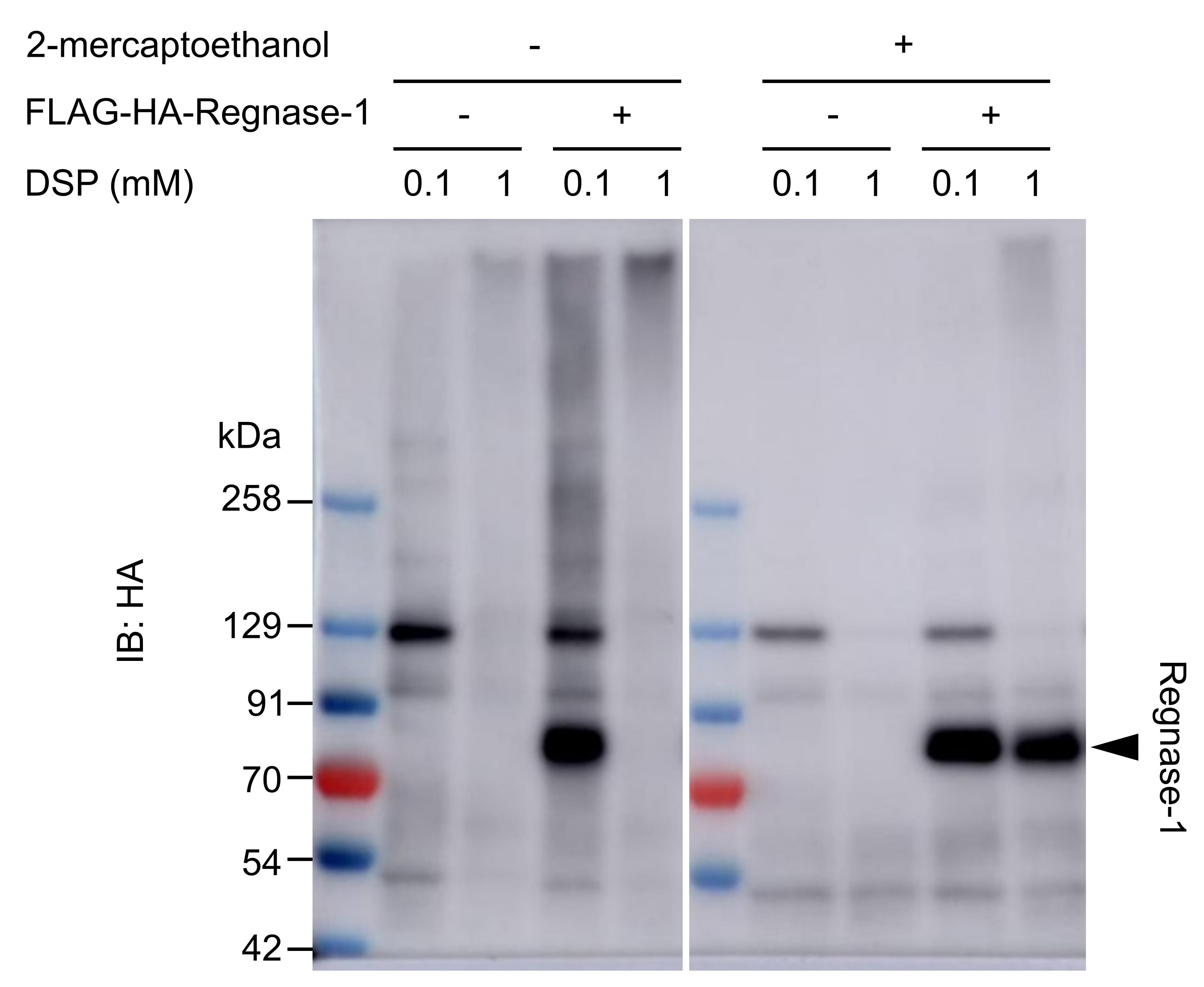
Figure 2. Optimization of DSP concentration. HeLa cells transiently expressing FLAG-HA-Regnase-1 (POI) were crosslinked with different concentrations of DSP (37 °C for 30 min), and the cell lysates were analyzed by western blotting. The smear band in 0.1 mM-DSP-treated sample indicates Regnase-1 crosslinked with other proteins; 1 mM DSP crosslinking resulted in huge protein complexes that could not migrate into the polyacrylamide gel. The effect of crosslinking can be abolished by reducing agents such as 2-mercaptoethanol.Preparing beads for the first IP
During the DSP-crosslinking (step C6), prepare 40 μL of Dynabeads Protein G/sample in one 1.5 mL tube (e.g., 40 μL × 5* = 200 μL in one 1.5 mL tube for 4 samples; *5 = 4 + 1 for dead volume. This ratio should be used hereafter.)
Wash the Dynabeads Protein G with the same amount of Wash buffer three times.
Set the tube containing Dynabeads Protein G on a magnetic stand and let the beads accumulate onto the magnet.
Discard the supernatant with a pipette.
Add ice-cold Wash buffer (e.g., 40 μL × 5 = 200 μL for 4 samples).
Remove the tube from the magnetic stand.
Resuspend the beads by pipetting up and down.
Repeat step D2 twice more.
Set the tube containing the washed beads on a magnetic stand and discard the supernatant.
Resuspend the beads with ice-cold IP buffer (e.g., 40 μL × 5 = 200 μL for 4 samples).
Add 1 μL of anti-FLAG antibody per one sample (e.g., 1 μL × 5 = 5 μL of the antibody into 200 μL of the washed beads).
Incubate the beads on a rotating incubator at 4 °C for 1 h. (The speed of the rotator is set around the middle between the lowest and the fastest speed. This setting should be used hereafter.)
Note: We usually rotate samples at the middle speed (approximately 24 rotations/min; the radius of the rotating incubator is approximately 10 cm).
Stopping DSP-crosslinking
After the 30-min incubation at step C6, discard the 0.1 mM DSP and rinse the dishes once with 5 mL of pre-warmed (37 °C) PBS/dish.
Discard the PBS and add 5 mL of STOP solution (room temperature)/dish.
Incubate the dishes at room temperature for 15 min.
First IP
Discard the STOP solution from the dishes.
Rinse the dishes twice with 5 mL of ice-cold PBS/dish.
Discard the PBS from the dishes.
Add 500 μL of ice-cold IP buffer/dish.
Scrape off the cells with a scraper and transfer them with all the IP buffer on the dish into a 1.5 mL tube (one tube for one sample).
Note: Collect the cells as much as possible. We usually scrape all the edges of a dish first and then scrape together the cells and IP buffer to one side of the dish. Tilt the dish to the side of the gathered cells and collect them using a pipette.
Resuspend the cells by pipetting up and down.
Incubate the tubes on ice for 10 min to complete cell lysis.
Centrifuge the tubes containing the lysates at 20,000 × g at 4 °C for 5 min.
Transfer 500 μL of the supernatant to a new 1.5 mL tube for IP. (The remaining supernatant can be used as an input sample to check the expression of POI by western blotting.)
Add 40 μL of anti-FLAG-antibody-bound beads (prepared at step D6) into the tube containing the supernatant.
Incubate the tubes on a rotating incubator at 4 °C for 2 h.
Preparing beads for the second IP
Approximately 30 min before finishing the 2-h incubation at step F11, prepare and wash new Dynabeads Protein G, as in steps D1 to D4.
Add 1 μL of anti-HA antibody per one sample (e.g., 1 μL × 5 = 5 μL of the antibody into 200 μL of the washed beads).
Incubate the beads on a rotating incubator at 4 °C for 1 h.
Protein elution with FLAG peptides
Prepare FLAG-elution buffer by diluting FLAG peptide stock solution with TBS and keep it on ice.
After the 2-h incubation at step F11, wash the beads with ice-cold 700 μL of Wash buffer three times. (Discard the supernatant first. See step D2 for the way of beads washing.)
Discard the Wash buffer from the beads.
Resuspend the beads with 100 μL of FLAG-elution buffer (prepared at step H1) gently by pipetting up and down.
Incubate the beads with FLAG-elution buffer on a rotating incubator at 4 °C for 10 min.
Set the tubes on a magnetic stand and transfer the supernatant (containing POI and POI-bound proteins) to new 1.5 mL tubes.
Repeat steps H4 to H5 once more.
Set the tubes on a magnetic stand and transfer the supernatant to the tubes containing first eluted proteins at step H6. (The total volume is 200 μL/sample.)
Add 300 μL of ice-cold IP buffer to the collected supernatant. (The final volume is 500 μL/sample.)
Second IP
Mix the 500 μL of supernatant at step H9 with 40 μL of anti-HA-antibody-bound beads (prepared at step G3).
Incubate the beads on a rotating incubator at 4 °C for 2 h.
Final elution
After the 2-h incubation at step I2, wash the beads with 700 μL of ice-cold Wash buffer three times. (Discard the supernatant first. See step D2 for the way of beads washing.)
Elute the proteins by desired methods.
For western blotting
Add 75 μL of 1× SDS elution buffer and mix by pipetting up and down.
Note: If desired, add 2-mercaptoethanol (IP buffer:3× SDS sample buffer:2-mercaptoethanol = 40:17:3) to cleave the disulfide bond in DSP (Figure 2).
Incubate the samples at 95 °C for 5 min on a heat block.
Cool down the samples on ice.
Note: It takes approximately 5 minutes to cool down. The cooled samples can be stored at -80 °C.
Centrifuge the tubes at 20,000 × g at room temperature for 1 min.
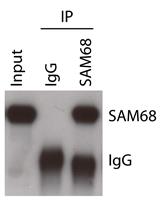
Use the supernatant for western blotting (Figure 3).
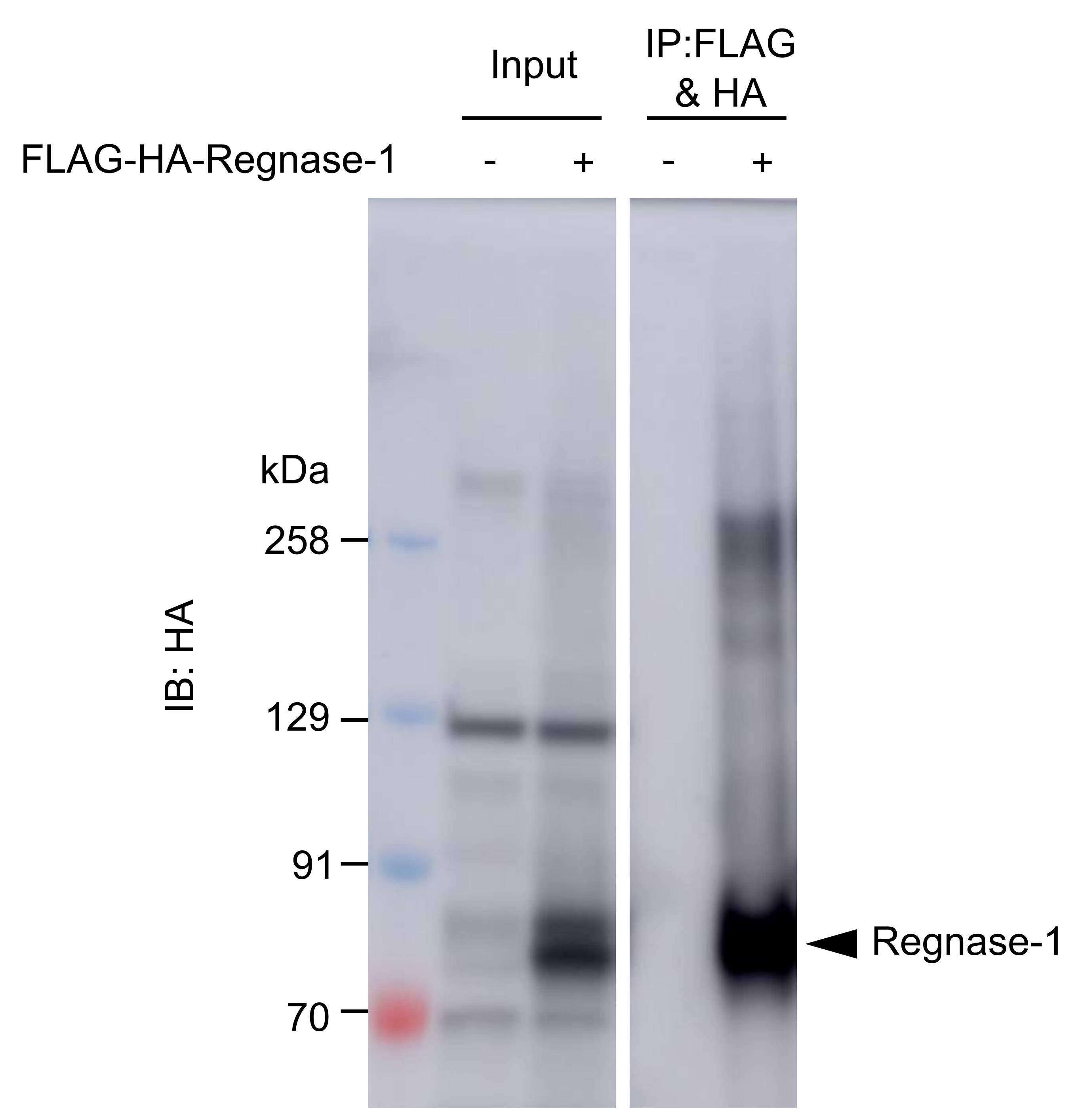
Figure 3. A result of western blotting after protein elution. FLAG-HA-Regnase-1 (POI) transiently expressed in HeLa cells was crosslinked, immunoprecipitated, and eluted as above.For mass spectrometry analysis
Note: Depending on efficiency or specificity of the elution of POI, optimization of elution method might be needed.
Add 100 μL of Urea elution buffer and mix by pipetting up and down.
Incubate the samples on ice for 5 min.
Set the tubes on a magnetic stand and collect the supernatant into new 1.5 mL tubes.
Some of the supernatant can be used for western blotting to check successful crosslinking and IP before mass spectrometry analysis. Mix 10 μL of the supernatant with 5 μL of 3× SDS sample buffer and follow the steps J2a-ii to J2a-iii.
Store the rest of the supernatant at -80 °C until sample preparation for mass spectrometry analysis.
Recipes
100 mM DSP
Reagent Final concentration Amount DSP 100 mM 10 mg DMSO n/a 247 μL Total n/a n/a Tris-HCl (1 M, pH 7.4)
Reagent Final concentration Amount Tris-HCl (1 M, pH 7) n/a 16 mL Tris-HCl (1 M, pH 8) n/a 4 mL Total n/a 20 mL STOP solution
Reagent Final concentration Amount Tris-HCl (1 M, pH 7.4) 20 mM 600 μL PBS n/a 29.4 mL Total n/a 30 mL Wash buffer
Reagent Final concentration Amount Tris-HCl (1 M, pH 7.4) 20 mM 800 μL NaCl (5 M) 150 mM 1.2 mL NP-40 (10%) 0.5% 2 mL H2O n/a 36 mL Total n/a 40 mL IP buffer
Reagent Final concentration Amount Wash buffer n/a 10 mL cOmplete Mini EDTA-free n/a 1 tablet PhosSTOP n/a 1 tablet Total n/a 10 mL TBS (Tris Buffered Saline)
Reagent Final concentration Amount Tris-HCl (1 M, pH 7.4) 50 mM 500 μL NaCl (5 M) 150 mM 300 μL H2O n/a 9.2 mL Total n/a 10 mL FLAG peptide stock solution
Reagent Final concentration Amount FLAG peptide 5 mg/mL 4 mg TBS n/a 800 μL Total n/a 800 μL FLAG-elution buffer
Reagent Final concentration Amount FLAG peptide stock solution 100 μg/mL 20 μL TBS n/a 980 μL Total n/a 1 mL Urea elution buffer
Reagent Final concentration Amount Urea 8 M 4.8 g Tris-HCl (1 M, pH 8.0) 50 mM 500 μL H2O n/a Up to 10 mL Total n/a 10 mL 3× SDS sample buffer
Reagent Final concentration Amount Tris-HCl (1 M, pH 6.8) 150 mM 7.5 mL SDS 6% 3 g Glycerol 30% 15 mL Bromophenol blue 0.25% 125 mg H2O n/a Up to 50 mL Total n/a 50 mL 1× SDS elution buffer
Reagent Final concentration Amount 3× SDS sample buffer 1/3 200 μL IP buffer 2/3 400 μL Total n/a 600 μL
Acknowledgments
We thank Y Ishihama and K Ogata at Kyoto university for the great advice about optimizing the method suitable for mass spectrometry analysis. This work was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI [18H05278] to OT and [19H03488] to TM; Japan Agency for Medical Research and Development (AMED) [JP20gm4010002, JP21ae0121030 and JP20fk0108454] to OT. KA was supported by ‘Kibou Projects’ Scholarship for doctoral Students in Immunology.
The original research paper for this protocol was published in eLife, DOI: 10.7554/eLife.71966.
Competing interests
We have no competing interests to declare.
References
- Akaki, K., Ogata, K., Yamauchi, Y., Iwai, N., Tse, K. M., Hia, F., Mochizuki, A., Ishihama, Y., Mino, T. and Takeuchi, O. (2021). IRAK1-dependent Regnase-1-14-3-3 complex formation controls Regnase-1-mediated mRNA decay. Elife 10: e71966.
- Lomant, A. J. and Fairbanks, G. (1976). Chemical probes of extended biological structures: synthesis and properties of the cleavable protein cross-linking reagent [35S]dithiobis(succinimidyl propionate). J Mol Biol 104(1): 243-261.
- Zlatic, S. A., Ryder, P. V., Salazar, G. and Faundez, V. (2010). Isolation of labile multi-protein complexes by in vivo controlled cellular cross-linking and immuno-magnetic affinity chromatography. J Vis Exp (37): 1855.
- Wang, H., He, M., Willard, B. and Wu, Q. (2019). Cross-linking, immunoprecipitation and proteomic analysis to identify interacting proteins in cultured cells. Bio-protocol 9(11): 3258.
Article Information
Copyright
![]() Akaki et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Akaki et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Akaki, K., Mino, T. and Takeuchi, O. (2022). DSP-crosslinking and Immunoprecipitation to Isolate Weak Protein Complex. Bio-protocol 12(15): e4478. DOI: 10.21769/BioProtoc.4478.
- Akaki, K., Ogata, K., Yamauchi, Y., Iwai, N., Tse, K. M., Hia, F., Mochizuki, A., Ishihama, Y., Mino, T. and Takeuchi, O. (2021). IRAK1-dependent Regnase-1-14-3-3 complex formation controls Regnase-1-mediated mRNA decay. Elife 10: e71966.
Category
Molecular Biology > Protein > Protein-protein interaction
Biochemistry > Protein > Interaction > Crosslinking
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.