- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CRISPR/Cas9 Gene Editing of HeLa Cells to Tag Proteins with mNeonGreen
Published: Vol 12, Iss 10, May 20, 2022 DOI: 10.21769/BioProtoc.4415 Views: 4270
Reviewed by: Antoine de MorreeOlli MatilainenAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2021

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Subcellular localization dynamics of proteins involved in signal transduction processes is crucial in determining the signaling outcome. However, there is very limited information about the localization of endogenous signaling proteins in living cells. For example, biochemical mechanisms underlying the signaling pathway from epidermal growth factor (EGF) receptor (EGFR) to RAS-RAF and ERK1/2/MAPK are well understood, whereas the operational domains of this pathway in the cell remain poorly characterized. Tagging of endogenous components of signaling pathways with fluorescent proteins allows more accurate characterization of their intracellular dynamics at their native expression levels controlled by endogenous regulatory mechanisms, thus avoiding possible tainting effects of overexpression and mistargeting. In this study, we describe methodological approaches to label components of the EGFR-RAS-MAPK pathway, such as Grb2, KRAS, and NRAS, with the fluorescent protein mNeonGreen (mNG) using CRISPR/Cas9 gene-editing, as well as generation of homozygous single-cell clones of the edited target protein.
Keywords: mNeonGreenBackground
The epidermal growth factor (EGF) receptor (EGFR) is activated by EGF or other ligands at the cell surface, which triggers the RAS-RAF-MAPK/ERK1/2 signaling pathway involved in cell proliferation, differentiation, survival, and motility (Karnoub and Weinberg, 2008). Activated EGFR is endocytosed and accumulated in early endosomes, where it is either recycled back to the plasma membrane or targeted for lysosomal degradation. Whether active EGFR continues to signal through the RAS-ERK1/2 axis from endosomes to sustain ERK1/2 activation remains controversial and is under debate in the literature (reviewed in von Zastrow and Sorkin, 2021). Evidence for the endosomal signaling to ERK1/2 is based on the detection of components of the ERK1/2 pathway in endosomes in cells stimulated with EGF (Pol et al., 1998; Howe et al., 2001; Jiang and Sorkin, 2002; Lu et al., 2009; Schmick et al., 2014). However, these observations were made either with overexpressed recombinant proteins or with chemically fixed cells or by subcellular fractionation, approaches that may not correctly report subcellular localization of endogenous proteins. We initially studied the spatio-temporal dynamics of the endogenous tagged EGFR and components of the ERK1/2 pathway by fluorescently tagging these proteins using gene editing techniques utilizing zinc finger nucleases (ZFN) and transcription activator-like effector nucleases (TALEN) (Pinilla-Macua et al., 2017, 2016). Subsequently, we switched to using the CRISPR-Cas9 system (Surve et al., 2019; Surve et al., 2021) for gene-editing. We selected for these studies a subvariant of HeLa cells that we consistently used in the laboratory because they express EGFR at levels similar to those in many mammalian cells in vivo, and because we have rigorously characterized the EGFR endocytic trafficking system in these cells.
CRISPR-Cas9 has proven to be versatile and efficient method of gene editing compared to ZFNs and TALEN, and is widely used for gene knockouts, gene activation, gene inhibition, and knock-ins. The fascinating discovery and evolution of CRISPR technology is beyond scope of this article, and excellent reviews on this topic can be found elsewhere. We used this technology to tag KRAS, NRAS, and Grb2. Generally, there are two main components of CRISPR-Cas9 technology: i) Cas9—a RNA guided endonuclease, ii) a short non-coding guide RNA consisting of a target complementary CRISPR RNA (crRNA) and a trans-activating crRNA (tracrRNA) that shuttles Cas9 to a specific site. These components are delivered inside the cells via plasmids or via direct delivery of an in vitro generated complex of purified Cas9 and purified sgRNA (Ran et al., 2013). Following the delivery, gRNA through Watson-Crick base pairing binds to its target DNA sequence, and Cas9 makes a double stranded break (DSB) near the PAM site of gRNA. The DSB is either repaired by the error prone non-homologous end joining (NHEJ) mechanism or by precise homology dependent repair (HDR) mechanism. In case of gene tagging, an additional component is provided in the form of a repair template, i.e., a donor DNA containing sequences of a fluorescent protein or small tags such as His, HA, or MYC, which through HDR allows the insertion of the tag of interest. HDR mechanism of repair is inherently inefficient, resulting in lower yields of clones of cells with the in-frame incorporated tag compared to generation of knockout clones, which generally involves NHEJ. The protocol here describes the variations (Table 1) in the approach by which gene tagging (knock-in) can be achieved in transformed cell lines like HeLa.
Materials and Reagents
CellTrics 50 μm sterile Disposable Filters (Sysmex, catalog number: 04-004-2327)
96-well plate (Ibidi, 15 μ-Plate 96 Well Black)
12-well cell culture plate (Corning, catalog number: 3513)
25 or 75 cm2 cell culture flasks (Corning, catalog number: 430639 or 430641U)
15 mL tube conical base (Sarstedt, catalog number: 62.554.205)
DNA oligonucleotide primers from IDT
GeneArt Precision gRNA Synthesis kit (Invitrogen, catalog number: A29377)
Tracr Fragment + T7 Primer Mix (contains the universal PCR amplification primers and the 80-nt constant region of the crRNA/tracrRNA) (Invitrogen, catalog number: A29377)
PhusionTM High-Fidelity PCR Master Mix (2×) (Invitrogen, catalog number: A29377)
Nuclease free water
dNTP mix (10 mM each of dATP, dGTP, dCTP, dTTP) (NEB, catalog number: N0447)
5× TranscriptAidTM Reaction Buffer (Invitrogen, catalog number: A29377)
TranscriptAidTM Enzyme Mix (Invitrogen, catalog number: A29377)
DNase I (Invitrogen, catalog number: A29377)
Single stranded DNA (ssDNA) oligos from IDT
NEBuilder® HiFi DNA Assembly Master Mix (NEB, catalog number: E2621)
pSpCas9(BB)-2A-Puro (PX459) plasmid (Addgene plasmid # 62988)
BbsI (NEB, catalog number: R0539)
NEB Buffer 2 (NEB, catalog number: B7002S)
NEB® 5-α Competent E. coli (High Efficiency) (NEB, catalog number: C2987)
Ampicillin Agar plates and Kanamycin Agar plates
NEB buffer 2.1 (NEB, catalog number: B7202)
QIAquick PCR Purification Kit (Qiagen, catalog number: 28104)
Synthetic double stranded donor DNAs from IDT
Phusion High-Fidelity DNA Polymerase (NEB, catalog number: M0530)
Phusion GC buffer 5× (NEB, catalog number: B0519)
Zero BluntTM TOPOTM PCR Cloning Kit (Invitrogen, catalog number: 45-1245)
EcoRI-HF (NEB, catalog number: R3101)
CutSmart Buffer 10× (NEB, catalog number: B6004)
QIAquick Gel Extraction Kit (Qiagen, catalog number: 28704)
XbaI (NEB, catalog number: R0145)
Easy-Fusion Halo plasmid (Addgene plasmid # 112850)
EcoRI-HF (NEB, catalog number: R3101)
AvaI (NEB, catalog number: R0152)
HincII (NEB, catalog number: R0103)
DMEM (Gibco, catalog number: 11965-092) with 10% FBS (Invitrogen, catalog number: 16140071)
DPBS (without Ca2+ or Mg2+) (Gibco, catalog number: 14190-144)
Trypsin (Gibco, catalog number: 25200-056)
Neon Transfection System (Invitrogen, catalog number: MPK5000)
Neon Transfection System 10 μL Kit (Invitrogen, catalog number: MPK1025)
Buffer R
Puromycin (Sigma-Aldrich, catalog number: P8833)
Platinum Cas 9 (Invitrogen, catalog number: A36498)
BD FACSAria III sorter fitted with 488 100 mW Trigon laser and 100 μm nozzle (BD Biosciences)
Nocodazole (Sigma-Aldrich, catalog number: M1404)
Lipofectamine 3000 (Invitrogen, catalog number: L3000001)
OptiMEM (Invitrogen, catalog number: 31985062)
Recombinant KRAS rabbit monoclonal antibody (Thermo Fisher Scientific, catalog number: 703345)
Cell Sorting buffer (see Recipes)
TGH buffer (see Recipes)
Software
https://www.benchling.com/. Benchling is a cloud-based informatics platform that is free to academic researchers. It provides design tools for DNA, oligonucleotides, and amino acids.
https://portals.broadinstitute.org/gppx/crispick/. The website is hosted by the Genetic Perturbation Platform of the Broad institute, MA, USA.
http://chopchop.cbu.uib.no/. CHOPCHOP is a web-based tool, designed by the Valen group at the University of Bergen, Norway. The website is for non-profit and academic use only.
https://nebuilder.neb.com. The web tool can be used to design primers for HiFi DNA assembly or Gibson assembly.
Procedure
Experimental Design
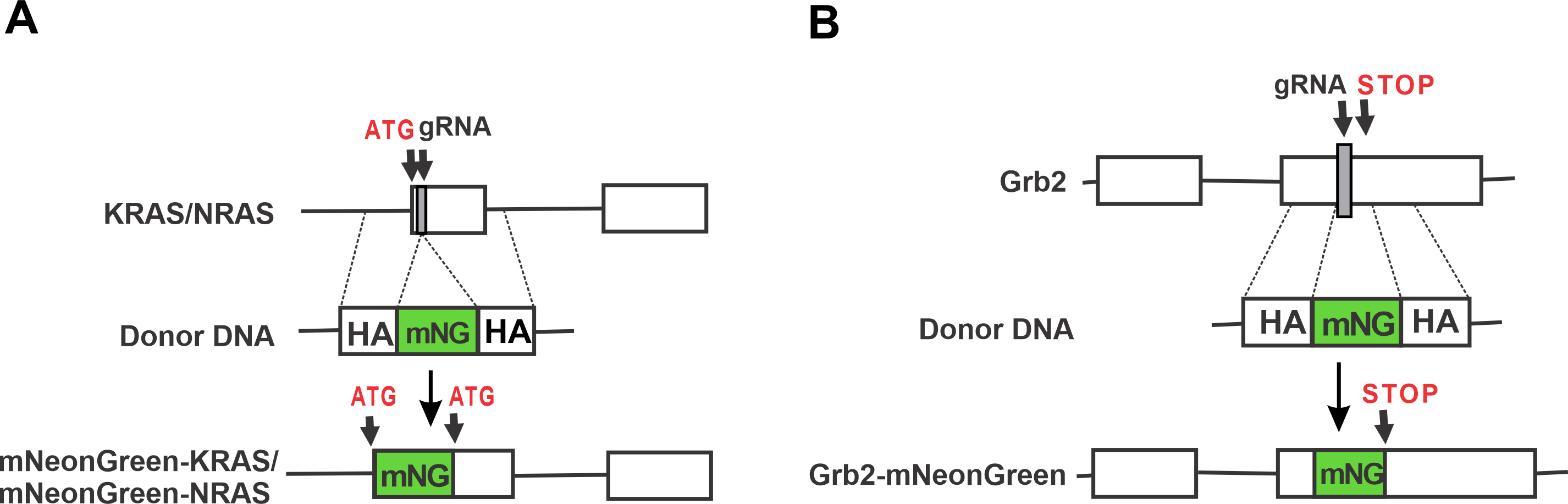
Figure 1. Schematics of insertion of mNG in KRAS, NRAS, and Grb2. mNG sequence was inserted at amino terminus of KRAS and NRAS genes (A) and at carboxy terminus of Grb2 gene (B). Donor DNAs were generated for all three genes with homology arms ranging from 500 to 1,000 bp. ATG, Start codon; STOP, Stop codon; HA, Homology Arm; mNG, mNeongreen; gRNA-guide RNA; empty boxes represent exons of KRAS, NRAS and Grb2.
Table 1. Sources of the components used to tag the genes
| KRAS | NRAS | Grb2 | |
|---|---|---|---|
| Cas9 | Commercial purified enzyme | Commercial purified enzyme | Plasmid -PX459 |
| gRNA | In vitro transcribed | In vitro transcribed | Cloned in Plasmid- PX459 |
| Donor | Synthetic gene fragment | Synthetic gene fragment | Homology arms and mNG fragments cloned in a plasmid |
| Delivery method | Electroporation | Electroporation | Transfection via Lipofectamine |
Generation of gRNAs for KRAS, NRAS, and Grb2
In-vitro Synthesis of gRNAs for tagging KRAS and NRAS
Identification of the target gRNA
We used amino termini of both KRAS and NRAS genes to tag with mNeonGreen fluorescent protein sequence because all RAS proteins are processed at their carboxy termini (Figure 1). Several guide RNA design tools are available online. We routinely use Benchling, Broad Institute GPP (now CRISPick), and CHOPCHOP, among others, to design target gRNAs. Using these tools, two targets for both KRAS and NRAS were identified, which were very close to the Start codons of both the genes. Order two complementary primers for each target (Table 2).
Table 2. Primers for in vitro translation of gRNAs
Primer Sequence 5’--- 3’ Kras_1F TAATACGACTCACTATAGGAATATAAACTTGTGGTAGT Target 1 Kras_1R TTCTAGCTCTAAAACACTACCACAAGTTTATATTC Target 1 Kras_2F TAATACGACTCACTATAGAATGACTGAATATAAACTTG Target 2 Kras_2R TTCTAGCTCTAAAACCAAGTTTATATTCAGTCATT Target 2 Nras_1F TAATACGACTCACTATAGGACTGAGTACAAACTGGTGG Target 1 Nras_1R TTCTAGCTCTAAAACCCACCAGTTTGTACTCAGTC Target 1 Nras_2F TAATACGACTCACTATAGAATGACTGAGTACAAACTGG Target 2 Nras_2R TTCTAGCTCTAAAACCCAGTTTGTACTCAGTCATT Target 2 Dilute target primers to a stock solution 100 μM in 1× TE buffer.
Prepare a mixture of target primers of 10 μM stock solution by adding 10 μL each of the 100 μM forward and reverse target primer to 80 μL of nuclease-free water.
Prepare the 0.3 μM target primer mix working solution by diluting 3 μL of the 10 μM target primer mix stock solution in 97 μL of nuclease-free water.
Assembly of the gRNA DNA templates
This step generates the DNA template required for the in vitro transcription of the gRNAs. Thaw the reagents from GeneArt Precision gRNA synthesis kit on ice. Mix and centrifuge contents of all the vials. Set up the following PCR assembly reaction in a 25-μL volume, adding the reaction components in the order given (Table 3).
Table 3. Components to make gRNA DNA template
PhusionTM High-Fidelity PCR Master Mix (2×) 12.5 μL Tracr Fragment + T7 Primer Mix 1 μL 0.3 μM Target F/R oligonucleotide mix 1 μL Nuclease free water 10.5 μL Since the PCR product is expected to be very short (120 bp), perform a two-step assembly PCR using following parameters (Table 4).
Table 4. PCR parameters to generate gRNA DNA template
Cycle Step Temperature Time Cycles Initial denaturation 98°C 10 s 1× Denaturation 98°C 5 s 32× Annealing 55°C 15 s 32× Final extension 72°C 1 min 1× Hold 4°C Hold 1× In vitro transcription (IVT) of gRNAs
Use the gRNA DNA template generated in step A.1.b for in vitro transcription of gRNAs. Set up the reaction as follows in the order given (Table 5).
Table 5. Components of IVT reaction
NTP mix (100 mM each of ATP, GTP, CTP, UTP) 8 μL gRNA DNA template (from PCR assembly) 6 μL 5× TranscriptAidTM Reaction Buffer 4 μL TranscriptAidTM Enzyme Mix 2 μL Mix the contents thoroughly and centrifuge. Carry out the IVT reaction for 3 h at 37°C.
Removal of the DNA template by DNase I digestion
It is helpful to remove DNA from IVT reaction so that it does not interfere with the subsequent applications of the RNA transcript.
Incubate the IVT reaction mix with 1 μL of DNase I (1 U/μL) immediately after the IVT reaction and incubate at 37°C for 15 min.
Purification of gRNAs using the columns and the buffers supplied in the kit
Dilute the IVT reaction to 200 μL with nuclease-free water and add 100 μL of Binding Buffer. Mix by pipetting.
Add 300 μL of ethanol (>96%) and mix by pipetting.
Transfer the mixture to the GeneJETTM RNA Purification Micro Column (preassembled with a collection tube) and centrifuge for 30–60 s at 14,000 × g. Discard the flow-through
Wash the bound RNA with 700 μL Wash Buffer 1 and Wash Buffer 2.
Centrifuge the empty purification column for an additional 60 s at 14,000 × g to completely remove any residual Wash Buffer.
Transfer the purification column to a clean 1.5-mL Collection Tube.
Add 10 μL of nuclease-free water to the center of the purification column filter, and centrifuge for 60 s at 14,000 × g to elute the RNA.
Store eluted gRNA at –20 °C until use. For prolonged storage, store the gRNA at -80°C.
Note: Follow all the necessary precautions while working with RNA to avoid its degradation or contamination and refer to the manufacturer’s instructions for detailed guidelines for generating gRNAs.
Cloning of gRNAs for tagging Grb2
Identification of the target gRNA
Grb2 was tagged with mNG at its carboxy terminal. Cloning of Grb2 gRNAs was performed using NEBuilder HiFi DNA assembly mix which allows assembly of a linear plasmid and a single stranded DNA fragment, just as multiple DNA fragments assembly. Similar to RAS proteins, two target gRNAs for Grb2 were identified using Benchling and Broad Institute GPP (now CRISPick). The following target gRNA sequence oligos were ordered as ssDNA from IDT (Table 6). Underlined sequences represent the actual target gRNA sequence flanked by overlapping sequences around BbsI restriction enzyme cut site from PX459.
Table 6. ssDNA oligos for Grb2 gRNA
grb2-RNA_T1 ATCTTGTGGAAAGGACGAAACACCGGGTTAGACGTTCCGGTTCACGGGTTTTAGAGCT AGAAATAGCAAGTT grb2- gRNA_T2 ATCTTGTGGAAAGGACG AAACACCGGGCTTAG ACGTTCCGGTTCACGGTTTTAGAGCTAGAAATAGCAAGTT Digestion of PX459 with BbsI
Linearize PX459 plasmid by digestion with BbsI at 37°C for 4 h (Table 7). Purify the digested plasmid using QIAquick PCR purification kit.
Table 7. PX459 digestion set up
PX459 Plasmid (500 ng/μL) 10 μL 10× Buffer 2.1 2 μL BbsI (10 U/μL) 1 μL ddH2O 7 μL Ligation of ssDNA into digested PX459
Centrifuge ssDNA oligo pellet and resuspend it in 1× TE to a final concentration of 100 μM, which is further diluted to a working stock of 0.4 μM in NEB Buffer 2. Set up the ligation reaction of linearized PX459 and ssDNA oligos in the order given below (Table 8).
Table 8. HiFi DNA assembly set up
Linear PX459 plasmid (50 ng) 1 μL 0.4 μM ssDNA stock 5 μL ddH2O 4 μL NEBuilder® HiFi DNA Assembly Master Mix 10 μL Incubate assembly reaction at 50°C for 1 h and transform 2 μL of the reaction mix into NEB 5-α Competent E. coli cells. Select the ampicillin resistant colonies by plating transformed E. coli cells onto Ampicillin agar plates.
Screening and confirmation of the positive clones
Select a few colonies from the ampicillin plates for plasmid isolation. Confirm the positive clones containing gRNA sequences by Sanger sequencing.
Generation of donor DNAs for tagging KRAS, NRAS, and Grb2
Cloning of donor DNA for tagging KRAS and NRAS
Donor DNAs with mNG sequence flanked by 5’ and 3’ homology arms of approximately 500 b each were obtained from IDT as gene fragments. The fragments were amplified using primers and cloned into pCRBluntII-TOPO plasmid using TOPO PCR cloning kit.
Amplification of the donor DNAs
Centrifuge vials containing gene fragments to collect the precipitate in 50 μL 1× TE. Amplify the synthetic DNA using Phusion polymerase (Tables 10 and 11) and primers mentioned below (Table 9).
Table 9. Primers for amplification of KRAS and NRAS synthetic donor DNA
Primers 5’------------3’ KRas_5HAF AGGTGGGGGTCCACTAGGAA KRas_3HAR CATATGATGTCACAATACCAAGAAAC NRas_5HAF CAGAGGCAGTGGAGCTTG NRas_3HAR ACAATCAGACAGTCTCGCTACTAT Table 10. PCR set up for amplification of KRAS and NRAS synthetic donor DNA
KRAS donor DNA NRAS Donor DNA Donor gene fragment 5 μL 5 μL Phusion GC Buffer 5× 10 μL 10 μL 10mM dNTP 1 μL 1 μL 5HAF 2.5 μL 2.5 μL 3HAR 2.5 μL 2.5 μL Phusion Polymerase 1 μL 1 μL Nuclease free water 28 μL 28 μL Table 11. PCR parameters for amplification of KRAS and NRAS synthetic donor DNA
Cycle Step Temperature Time Cycles KRAS donor NRAS donor Initial denaturation 98°C 98°C 1 min 1× Denaturation 98°C 98°C 15 s 30× Annealing 61.6°C 64.7°C 15 s 30× Extension 72°C 72°C 1 min 30× Final extension 72°C 72°C 10 min 1× Hold 4°C 4°C Hold 1× Perform gel purification of 1.68 kb of PCR products from KRAS and NRAS amplified donor DNAs.
Ligation of amplified donor DNAs and pCRBluntII-TOPO plasmid
Set up ligation reaction of gel eluted KRAS and NRAS donor DNAs and pCRII-Blunt-TOPO plasmid in a 6 μL volume as follows (Table 12).
Table 12. Donor DNA and pCRBluntII-TOPO ligation set up
KRAS Donor NRAS Donor Gel eluted fragment 2 μL 2 μL Salt Solution 1 μL 1 μL Nuclease free water 2 μL 2 μL pCR-II-Blunt-TOPO 1 μL 1 μL Incubate the ligation reaction at room temperature for 5 min. Transform 1–2 μL of the ligation mixture into competent E. coli and plate on kanamycin containing plates for selection.
Screening and confirmation of donor DNAs
Select few colonies from kanamycin plates and grow them to isolate plasmid. Confirm the positive clones by EcoRI digestion and by Sanger sequencing to make sure there are no changes in the donor sequence.
Digestion and gel elution of donor DNA sequences from positive clones
The fragments are inserted in pCRII-Blunt-TOPO in such a way that EcoRI sites lie at the ends of the insert, allowing recovery of the insert by EcoRI digestion. Digest the positive clones containing donor sequences with EcoRI as follows (Table 13).
Table 13. Digestion of pCR-II-Blunt-TOPO-Donor plasmid
KRAS NRAS pCR-II-Blunt-TOPO-Donor (1 μg/μL) 20 μL 20 μL CutSmart buffer 10× 5 μL 5 μL EcoRI-HF (10 U/μL) 3 μL 3 μL Nuclease free water 22 μL 22 μL Carry out the digestion for 3 h at 72°C. Gel elute the 1.68 kb bands from both the samples. Store the eluted donor fragments at -20°C.
Note: The donor PCR fragments were cloned to facilitate the confirmation of correct donor sequence using Sanger sequencing and to avoid repeated amplification. Purified PCR fragments can be used directly as donor DNAs, thus reducing the time of cloning.
Cloning of donor DNA for tagging Grb2
To clone donor DNA for Grb2 gene editing, 5’ and 3’ homology arms (HA) were amplified from HeLa genomic DNA using primers designed by the NEbuilder algorithm (NEB), whereas KRAS donor DNA was used as a template to amplify mNG ORF. The primers add overlapping sequences to the resulting PCR fragments and these fragments are then assembled with Easy-Fusion Halo plasmid using NEBuilder® HiFi DNA Assembly Master Mix (Table 14).
Table 14. Component Fragments for HiFi DNA assembly
Name Length (bp) Produced by 5HA fragment 793 PCR 3HA fragment 743 PCR mNG fragment 755 PCR EasyFusion halo plasmid as backbone vector 2,927 Restriction Digestion Amplification of homology arms (HA) and mNG ORF
Design primers with overlapping sequences using NEBuilder algorithm (Table 15). Amplify all three fragments required for the construction of the donor DNA as follows (Tables 16, 17, and 18).
Table 15. Primers for amplification of Grb2 homology arms and mNG ORF
Primer 5’……………..3’ Grb2-5HA_f TATCGATAAGCTTGATATCGAGAGGCAGTGTGTAGCCAG Grb2-5HA_r CTCCTCCTAAGACGTTCCGGTTCACGGG Grb2-Neon_f CCGGAACGTCTTAGGAGGAGGAGGATCAG Grb2-Neon_r TTGACTCTTACTTGTACAGCTCGTCCATG Grb2-3HA_f GCTGTACAAGTAAGAGTCAAGAAGCAATTATTTAAAG Grb2-3HA_r CCACCGCGGTGGCGGCCGCTTCTCGAACTCCTGACCTTG Table 16. PCR set up for amplification of Grb2 HAs
5HA Fragment 3HA Fragment HeLa genomic DNA (100 ng/μL) 3 μL 3 μL Phusion GC Buffer 5× 10 μL 10 μL 10 mM dNTP 1 μL 1 μL Grb2-5/3HA_f 2.5 μL 2.5 μL Grb2-5/3HA_r 2.5 μL 2.5 μL Phusion Polymerase 1 μL 1 μL Nuclease free water 30 μL 30 μL Table 17. PCR set up for amplification of mNG ORF
mNG Fragment mNG-KRAS donor (100 ng/μL) 3 μL Phusion GC Buffer 5× 10 μL 10mM dNTP 1 μL Grb2-Neon_f 2.5 μL Grb2-Neon_r 2.5 μL Phusion Polymerase 1 μL Nuclease free water 30 μL
Table 18. PCR parameters for amplification of Grb2 HAs and mNG ORFCycle Step Temperature Time Cycles 5HA 3HA mNG Initial denaturation 98°C 98°C 98°C 1 min 1× Denaturation 98°C 98°C 98°C 15 s 30× Annealing 64.4°C 60°C 59.6°C 15 s 30× Extension 72°C 72°C 72°C 25 s 30× Final extension 72°C 72°C 72°C 10 min 1× Hold 4°C 4°C 4°C Hold 1× Gel elute the PCR products using QIAquick Gel Extraction Kit and estimate the DNA concentration of the eluted fragments.
Restriction enzyme digestion of EasyFusion-Halo plasmid.
Digest EasyFusion-Halo plasmid with EcoRI and XbaI to remove the Halo insert at 37°C for 4 h (Table 19).
Table 19. Digestion of EasyFusion-Halo plasmid to remove Halo insert
EasyFusion-Halo (5 μg) 10 μL CutSmart buffer 10× 2 μL EcoRI 2 μL XbaI 2 μL Nuclease free water 2 μL Gel elute the 2.9 kb fragment of the digested plasmid using QIAquick Gel Extraction Kit and estimate the DNA concentration of the eluted fragment.
HiFi DNA assembly ligation reaction
Mix all four fragments required for the generation of Grb2 donor DNA to a final volume of 10 μL. Thaw HiFi DNA assembly mix on ice and vortex thoroughly. Add equal volume of HiFi DNA assembly master mix to the fragments (Table 20).
Table 20. Ligation set up for the generation of Grb2 donor DNA
0.1 pmol of 5HA 50 ng 0.8 μL 0.1 pmol of mNG 50 ng 0.9 μL 0.1 pmol of 3HA 50ng 1.1 μL 0.1 pmol of digested vector 185 ng 2 μL HIFi DNA assembly master mix 10 μL Nuclease free water 5.2 μL Carry out the ligation reaction at 50°C for 1 h. Transform 2 μL of the reaction mix into competent E. coli. Screen several colonies for positive clones containing all three fragments (5HA-mNG-3HA) using restriction enzyme digestion and Sanger sequencing.
Delivery of gRNAs and Donor DNAs in HeLa cells
Electroporation of RNP complex to tag KRAS and NRAS
Grow HeLa cells in DMEM with 10% FBS to 90% confluency in a 75 cm2 flask. Treat the cells with 100 ng/mL Nocodazole for 12 h.
On the day of the electroporation, thaw and store buffer R, KRAS and NRAS specific in vitro transcribed gRNA, and donor DNAs on ice. Dilute gRNAs and donor DNA to appropriate concentrations in nuclease free water.
Wash away Nocodazole with 1× DPBS. Split the cells with Trypsin and inactivate it by the addition of FBS containing DMEM. Centrifuge the single cell suspension at 300 × g for 5 min. Resuspend the pellet in 10 mL of DMEM with 10% FBS. Perform cell counting using hemocytometer.
Prepare RNP mixture as follows (Table 21), one for each gene-editing reaction in a 1.5 mL microcentrifuge tube.
Table 21. Components of RNP mixture for electroporationBuffer R 1 μL Platinum Cas9 (1 μg) 0.5 μL (1 μg) gRNA (500 ng) 2 μL Incubate RNP complex at room temperature for 20 min.
While the RNP is incubating, prepare electroporation set up and keep cells ready for the electroporation by centrifuging them at 300 × g for 5 min. Aspirate DPBS and resuspend the cell pellet to a final cell density of 8 × 107 cells/ml in buffer R.
Add 3 mL of Electrolytic buffer (Buffer E) into the Neon Pipette station.
Add 5 μL of cells (final 4 × 105 cells) and 1 μg of respective donor DNA fragments to the RNP mixture. Make sure the volume of the final mixture is 10 μL. Aspirate the final mix in Neon Tip using Neon Pipette without incorporating air bubbles and electroporate at pulse voltage of 1,005 V, pulse width of 35 ms for 2 pulses.
Transfer the electroporated cells in a 12-well plate containing prewarmed DMEM with 10% FBS without antibiotics and continue incubating the plate at 37°C in a CO2 incubator.
Transfection of gRNA and donor to tag Grb2
Grow HeLa cells in DMEM with 10% FBS to 90% confluency in 12-well plates. Treat the cells with 100 ng/mL Nocodazole for 12 h.
On the day of the transfections, dilute PX459-Grb2-gRNAs and Grb2-mNG donor DNA plasmids to appropriate concentrations in nuclease free water.
Wash away Nocodazole with 1× DPBS. Add 1 mL of fresh FBS containing DMEM.
Prepare DNA-lipid mixture as follows (Table 22), in a 1.5 mL microcentrifuge tube in the order given.
Table 22. Components of transfection mixture to tag Grb2
Tube 1 OptiMEM 50 μL PX459-Grb2-gRNA (250 ng) 0.5 μL Grb2-mNG donor (750 ng) 1 μL P3000 reagent 2 μL Tube 2 OptiMEM 50 μL Lipofectamine 3000 Reagent 1.5 μL Add contents from Tube 1 to Tube 2. Incubate the mixture at RT for 10 min.
Add plasmid-lipid mixture to the cells dropwise.
Next day after the transfection, treat transfected cells with 2 μg/mL of puromycin for 48 h to enrich transfected cells by killing un-transfected cells.
Remove puromycin containing DMEM, wash the cells with DPBS twice and allow the cells to recover. Split the surviving cells and expand them for flow sorting analysis.
Flow Sorting of mNG positive cells
Expand tagged HeLa cells in DMEM with 10% FBS in 25 or 75 cm2 flasks.
On the day of sorting, split the cells using Trypsin and inactivate it with FBS containing DMEM. Centrifuge the cells and wash once with PBS. Resuspend the final pellet in Cell sorting buffer.
Pass the cell suspension through disposable filters to get rid of cell aggregates just before the sorting. Use positive (parental HeLa cells transiently or stably expressing mNG) and negative (parental untagged HeLa cells) controls to calibrate the sorting instrument.
Collect single sorted cells in 96-well plate or in a 15 mL conical tube as a pooled population with prewarmed DMEM with 10% FBS and antibiotics to prevent contamination.
Confirmation of positive clones
Freeze at least one vial of single cell clones as a backup before using them for confirmation.
Clones can be confirmed by Western blot analysis using protein-specific antibody or mNG antibody. The correct fusion protein should have an apparent molecular mass ~25 kD larger than the parental protein. It is also important to sequence the region around the site of mNG sequence insertion to make sure there are no unwanted insertions or deletions during homology repair.
Note: Head-to-head comparison of efficiency of different delivery methods was not performed. In all the cases, the yield of mNG positive cells as determined during flow sorting was 0.1% to 0.5%. Since we were interested in single cell colonies for screening, one 96 well plate of single sorted cells was sufficient to screen them. It should be noted that flow sorting could inadvertently result in clones expressing higher yields of fusion protein.
Data analysis
Characterization of the tagged cell line clones
Select several colonies arising from flow sorted population. Process the colonies for DNA isolation for Sanger sequencing and for making lysates for Western blot analysis. To generate PCR products for Sanger sequencing, design primers that can amplify the region beyond the homology arms. The sequencing should reveal the correctly inserted mNG sequence as well as linkers and start/stop codons. Western blot should reveal the correct molecular weight of the fusion protein. For example, a screen of HeLa/mNG-KRAS clones using Western blot is shown in Figure 2. The colonies were lysed in TGH buffer, and the lysates were electrophoresed and transferred onto nitrocellulose membrane. We detected two homozygous clones in 24 colonies screened. Load lysates from parental cell line as control for the WT untagged protein. It is essential to make sure that off target changes occurring during Cas9 mediated gene editing do not affect the cells in such a way that they are unfit for the study. Since our focus is to study EGFR-RAS-MAPK pathway, we chose the clones that exhibited similar ERK1/2 phosphorylation kinetics in HeLa parental and tagged KRAS and NRAS cells in presence of EGF (Surve et al., 2021).
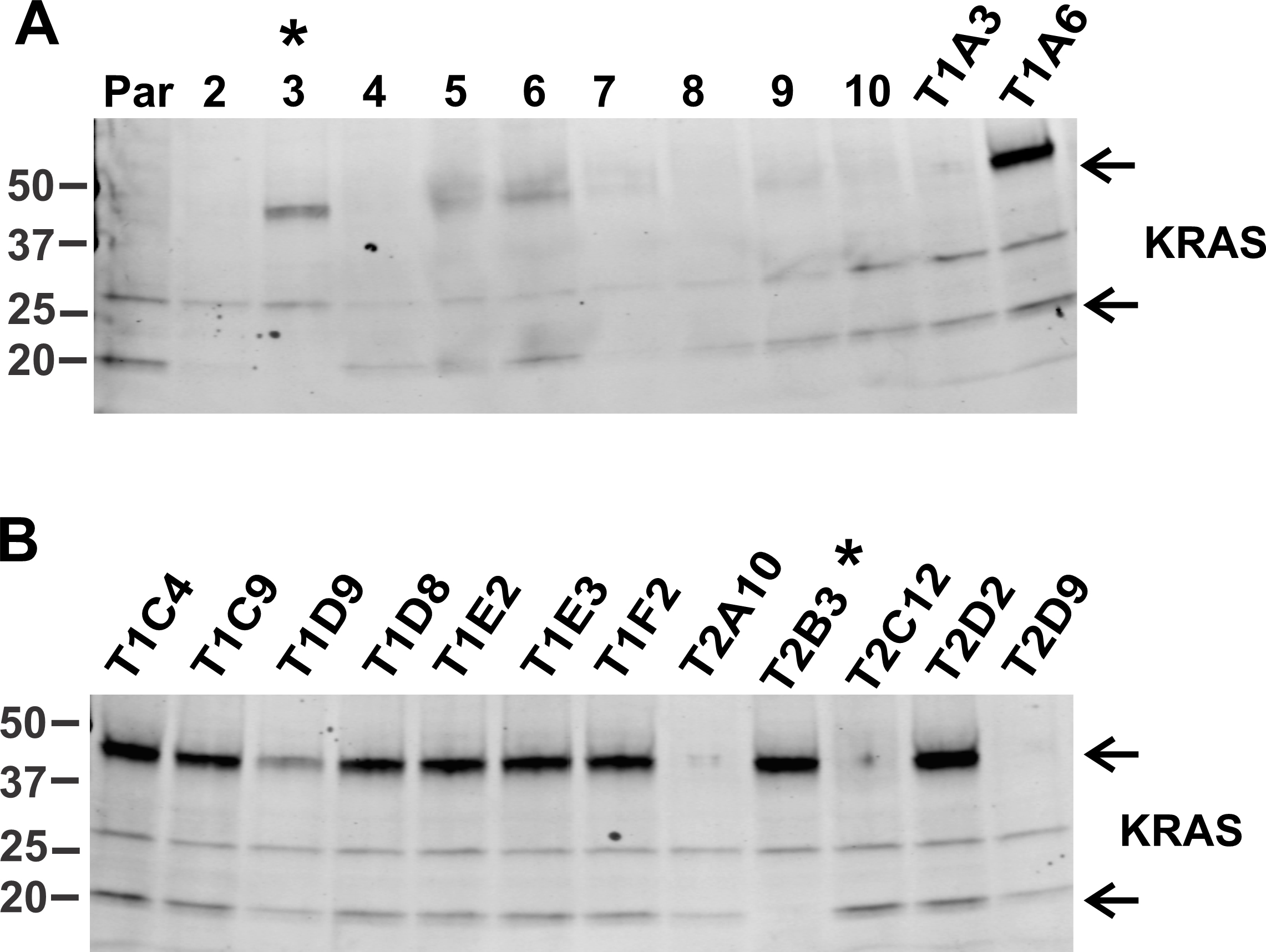
Figure 2. Screening of HeLa/mNG-KRAS single cell clones. The single cell colonies were obtained either from a 10-cm plate where transfected cells were diluted or from a flow cytometer sorted population in a 96 well plate. First screening was done by checking cell fluorescence using a confocal microscope. Second screening was performed by probing lysates of parental (Par) HeLa and single-cell clones of HeLa/mNG-KRAS cells using Western blotting (WB) with KRAS antibodies to determine the zygosity of the clones expressing mNG-KRAS. Clones from 2 to 10 (in A) were obtained from 10 cm plate, whereas remaining clones were obtained via flow sorting. Clone names starting with T1 are from Target 1 gRNA, whereas those with T2 are the results of Target 2 gRNA transfection. Top arrow in A and B indicate mNG-KRAS fusion protein, whereas lower arrow indicates untagged KRAS protein. Clones with asterisk are homozygous clones as they are missing a band corresponding to the untagged native KRAS.Examples of localization of endogenous tagged KRAS, NRAS and Grb2
After the screening and confirmation of the tagged cell lines, we imaged living tagged cell lines with spinning disc confocal microscope (Figure 3).
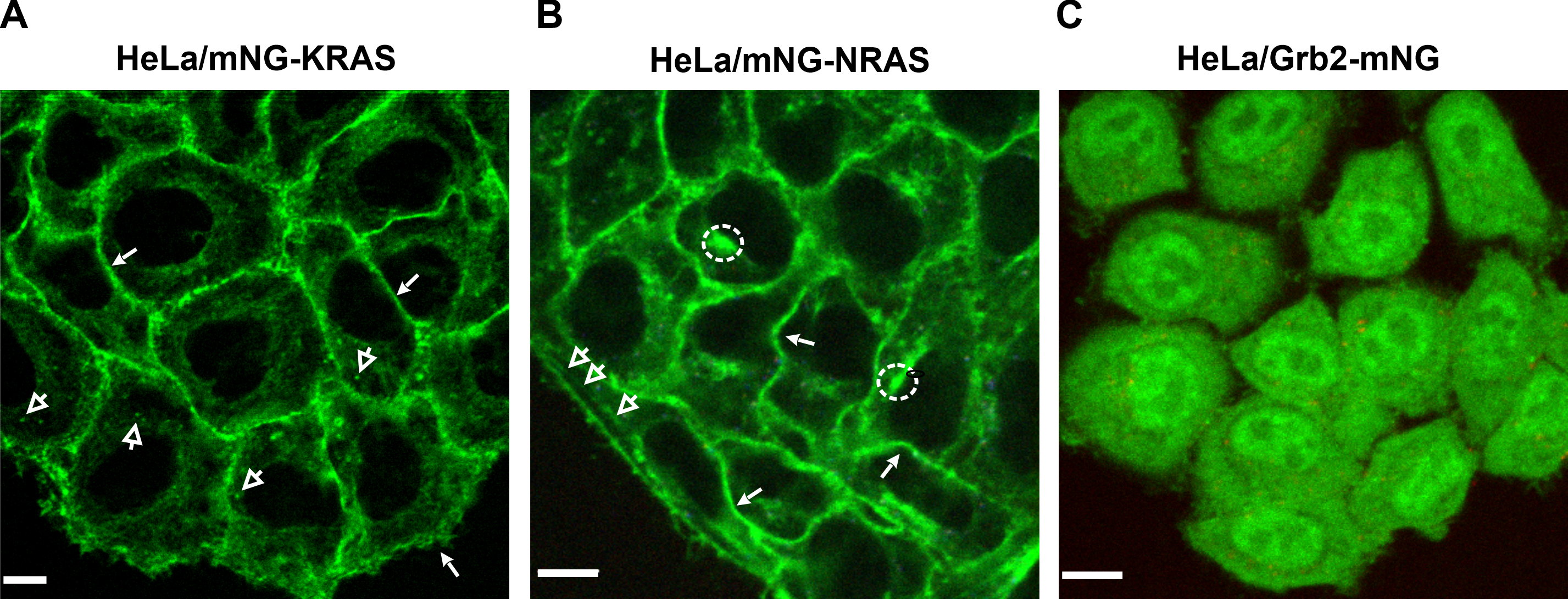
Figure 3. Localization of endogenous tagged KRAS, NRAS, and Grb2 with mNG in HeLa cells. Live cell imaging was performed using a spinning disc confocal microscope through 488 nm (green; mNG) channel. Single confocal sections are shown. A. mNG-KRAS localized mostly at the plasma membrane (closed arrows), and a small proportion also localized in vesicles (open arrows). B. Majority of mNG-NRAS localized at the plasma membrane. mNG-NRAS was also in the juxta-nuclear region (dotted circle) and tubular shaped structures (open arrows). C. Grb2-mNG localized to cytoplasm and nucleus.
Recipes
Cell Sorting buffer
1× PBS (Ca/Mg++ free)
2% heat-inactivated FBS
1 mM EDTA
TGH buffer
1% Triton X-100
10% glycerol
50 mM HEPES
2 mM EGTA
Phosphatase and protease inhibitors
Acknowledgments
Authors thank Drs Feng Zhang, Broad Institute, Cambridge, MA, and Janet Rossant, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, Toronto, ON, Canada, for plasmids. This study was supported by NIH grants CA089151 and GM124186. S. Surve was also supported by the fellowship from National Cancer Center. This protocol was adapted from Surve et al. (2021).
Competing interests
The authors declare no competing financial interests.
References
- Howe, C. L., Valletta, J. S., Rusnak, A. S. and Mobley, W. C. (2001). NGF signaling from clathrin-coated vesicles: evidence that signaling endosomes serve as a platform for the Ras-MAPK pathway. Neuron 32(5): 801-814.
- Jiang, X. and Sorkin, A. (2002). Coordinated traffic of Grb2 and Ras during epidermal growth factor receptor endocytosis visualized in living cells. Mol Biol Cell 13(5): 1522-1535.
- Karnoub, A. E. and Weinberg, R. A. (2008). Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 9(7): 517-531.
- Lu, A., Tebar, F., Alvarez-Moya, B., Lopez-Alcala, C., Calvo, M., Enrich, C., Agell, N., Nakamura, T., Matsuda, M. and Bachs, O. (2009). A clathrin-dependent pathway leads to KRas signaling on late endosomes en route to lysosomes. J Cell Biol 184(6): 863-879.
- Pinilla-Macua, I., Grassart, A., Duvvuri, U., Watkins, S. C. and Sorkin, A. (2017). EGF receptor signaling, phosphorylation, ubiquitylation and endocytosis in tumors in vivo. Elife 6: e31993.
- Pinilla-Macua, I., Watkins, S. C. and Sorkin, A. (2016). Endocytosis separates EGF receptors from endogenous fluorescently labeled HRas and diminishes receptor signaling to MAP kinases in endosomes. Proc Natl Acad Sci U S A 113(8): 2122-2127.
- Pol, A., Calvo, M. and Enrich, C. (1998). Isolated endosomes from quiescent rat liver contain the signal transduction machinery. Differential distribution of activated Raf-1 and Mek in the endocytic compartment. FEBS Lett 441(1): 34-38.
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A. and Zhang, F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8(11): 2281-2308.
- Schmick, M., Vartak, N., Papke, B., Kovacevic, M., Truxius, D. C., Rossmannek, L. and Bastiaens, P. I. H. (2014). KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell 157(2): 459-471.
- Surve, S., Watkins, S. C. and Sorkin, A. (2021). EGFR-RAS-MAPK signaling is confined to the plasma membrane and associated endorecycling protrusions. J Cell Biol 220(11).
- Surve, S. V., Myers, P. J., Clayton, S. A., Watkins, S. C., Lazzara, M. J. and Sorkin, A. (2019). Localization dynamics of endogenous fluorescently labeled RAF1 in EGF-stimulated cells. Mol Biol Cell 30(4): 506-523.
- von Zastrow, M. and Sorkin, A. (2021). Mechanisms for Regulating and Organizing Receptor Signaling by Endocytosis. Annu Rev Biochem 90: 709-737.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Surve, S. and Sorkin, A. (2022). CRISPR/Cas9 Gene Editing of HeLa Cells to Tag Proteins with mNeonGreen. Bio-protocol 12(10): e4415. DOI: 10.21769/BioProtoc.4415.
- Surve, S., Watkins, S. C. and Sorkin, A. (2021). EGFR-RAS-MAPK signaling is confined to the plasma membrane and associated endorecycling protrusions. J Cell Biol 220(11).
Category
Cell Biology > Cell engineering > CRISPR-cas9
Cell Biology > Cell signaling > Intracellular Signaling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.