- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Activation of Mitochondrial Ca2+ Oscillation and Mitophagy Induction by Femtosecond Laser Photostimulation
Published: Vol 12, Iss 7, Apr 5, 2022 DOI: 10.21769/BioProtoc.4369 Views: 2410
Reviewed by: Giusy TornilloAnonymous reviewer(s)
Original research article
The authors used this protocol in:
Jun 2021
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Ultra-precise stimulation solely to individual mitochondria, without any influence to the whole cell, is quite difficult by traditional biochemical reagents. In mitophagy research, the mitochondria and even the whole cell usually suffer irreversible and great damage caused by treatment with potent chemicals. In this protocol, we present the technical procedures of our developed noninvasive ultra-precise laser stimulation (UPLaS) technology, which introduces precise stimulation to individual mitochondria, to excite mitochondrial Ca2+ (mitoCa2+) oscillations, with little perturbation to mitochondrial membrane potential (MMP), or mitochondrial reactive oxygen species (mitoROS). The mitoCa2+ oscillation by UPLaS was able to initiate the PINK1/Parkin pathway for mitophagy. This protocol has good potential to benefit researches on mitophagy and mitochondrial diseases.
Graphic abstract:
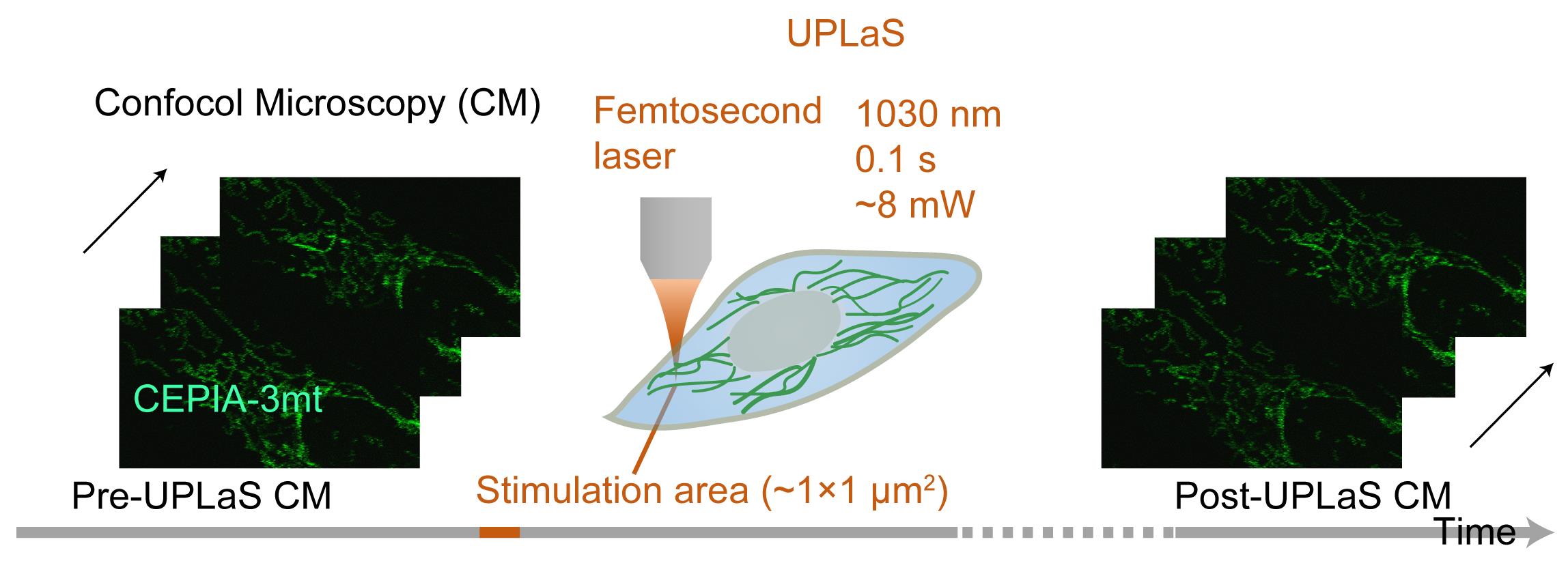
Figure 1. Flowchart of the UPLaS technology. The femtosecond laser (1030 nm, 1 MHz, 220 fs) can stimulate individual mitochondria (1 μm2) for a short period (0.1 s), whereas confocal microscopy (CM) provides continuous cell imaging to monitor molecular dynamics in real time, before and after UPLaS.
Background
Precise stimulation of individual mitochondria, without perturbation to the whole cell or damage to mitochondria, is of great significance to mitochondrial research (Russell et al., 2020). Moreover, the isolated modulation of mitochondrial membrane potential (MMP), mitochondrial reactive oxygen species (mitoROS), or mitochondrial Ca2+ (mitoCa2+), without perturbation to the other two, might provide more insights into mitochondria function. Mitophagy is abnormal in Parkinson's disease (PD) (Liu, J. et al., 2019; Ge et al., 2020). The mechanism of mitophagy and its dysregulation is essential to the better understanding of PD. In previous works, where PINK1/Parkin mediated mitophagy has been studied (Jin and Youle, 2013; Lazarou et al., 2015; Pickrell and Youle, 2015; Chung et al., 2020), carbonyl cyanide m-chlorophenyl hydrazine (CCCP) was extensively used to depolarize mitochondria and trigger mitophagy. However, the completely depolarization of MMP and significant high-level mitoROS are irreversible, and prevent further investigation of the mechanism of action of mitochondria.
Femtosecond laser has advanced biological researches by providing efficient multiphoton excitation, while maintaining good biological safety (Smith et al., 2005, 2008; Iwanaga, et al., 2006; Liu, X. et al., 2009; Zhao et al., 2009; Wang et al., 2014). Specifically, a transient tightly-focused femtosecond laser irradiation to a localized subcellular structure has been demonstrated to be a highly efficient optical method to directly induce molecular signaling events in single cells (He et al., 2012; Wang et al., 2014, 2018; Cheng et al., 2021). In this protocol, we present a noninvasive ultra-precise laser stimulation (UPLaS) technology to individual mitochondria that excites mitoCa2+ oscillations, with little perturbation to MMP or mitoROS. This UPLaS technology is achieved by coupling a femtosecond laser to a confocal microscope, to provide single target cells with a short flash photostimulation. The mitoCa2+ oscillation induced by UPLaS initiates the PINK1/Parkin pathway for mitophagy.
The UPLaS technology provides a noninvasive method for simultaneous photostimulation and confocal microscopy of subcellular organelles at a resolution of 628.3 nm. This method is highly flexible, as it can be demonstrated on any two-photon microscope system, and any subcellular organelle can be stimulated by this system.
Materials and Reagents
Hela cell culture
P100 mm Petri dish (Corning, catalog number: 3262)
35 mm Glass Bottom Cell Culture Dish (Nest, catalog number: 801002)
µ-Dish 35 mm, high Grid-500 Glass Bottom (IBidi, catalog number: 81168)
Hela cells (National Collection of Authenticated Cell Cultures, Manufacturer, Brand, catalog number: TCHu187)
DMEM (Hyclone, catalog number: SH30243.01)
Fetal Bovine Serum (FBS; Gibco, catalog number: 10099141)
Penicillin/Streptomycin (Gibco, catalog number: 10378016)
Phosphate-buffered saline (PBS; Hyclone, catalog number: SH30256.01)
0.25% Trypsin-EDTA (Gibco, catalog number: 25200056)
Hela culture medium (see Recipes)
SH-SY5Y cell culture
SH-SY5Y cells (COBIOER BIOSCIENCES, catalog number: CBP60913)
MEM, no glutamine (Gibco, catalog number: 11090081)
Ham's F-12 Nutrient Mix (Gibco, catalog number: 11765054)
GlutaMAXTM (Gibco, catalog number: 35050061)
Sodium Pyruvate (Gibco, catalog number: 11360070)
MEM NEAA (Gibco, catalog number: 11140050)
SH-SY5Y culture medium (see Recipes)
Cell Transfection and loading
CEPIA-3mt (Addgene, Plasmid #58219)
Parkin-mCherry (Addgene, Plasmid #23956)
LC3-GFP (Addgene, Plasmid #11546)
jetPRIME (Polyplus, catalog number: 101000046)
MitoTrackerTM Green (Invitrogen, catalog number: M7514)
MitoTrackerTM Red CMXRos (Invitrogen, catalog number: M7512)
Tetramethylrhodamine, Methyl Ester, Perchlorate (TMRM) (Invitrogen, catalog number: T668)
MitoSOXTM Red Mitochondrial Superoxide Indicator (Invitrogen, catalog number: M36008)
Dimethyl sulfoxide (DMSO,Sigma-Aldrich, catalog number: D9170 )
Immunofluorescence
4% Paraformaldehyde Fix Solution (Beyotime, catalog number: P0099)
Triton X-100 (Sigma-Aldrich, catalog number: 9036-19-5)
NON-Fat Powdered Milk (Sangon Biotech, catalog number: A600669-0250)
Tween-20 (Sigma-Aldrich, catalog number: P1379)
anti-PINK1 antibody (Abcam, catalog number: ab216144)
antibody anti-Rabbit IgG H&L (Sigma, catalog number: AP307P)
Antifade Mounting Medium (Beyotime, catalog number: P0126)
Equipment
Femtosecond-laser (MenloSystems, Type: BlueCut OEM Seed, Serial No: 111; Type: BlueCut Amp, Serial No: 116; 1030 nm, 1 MHz, 220 fs)
Confocal microscope (Olympus, model: FV1200)
Laser power meter (COHERENT, Item #: 1098297, Model: FIELDMATE, Serial #: 0763K14R)
Software
FV1200 (Olympus)
ImageJ
GraphPad Prism 7
Adobe Illustrator CC 2017
Procedure
Cell culture and transfection
Hela cell passage
Hela cells were cultured in DMEM supplemented with 10% FBS and 1% Penicillin/Streptomycin (hitherto referred to as DMEM10).
Remove the DMEM10 culture medium from the 100 mm Petri cell culture dish when cell density reaches approximately 80%.
Wash the Petri dish three times with 1.5 mL of PBS. Remove the PBS.
Slowly add 1 mL of trypsin, and place the dish back into the incubator at 37°C for 1 min.
Remove the trypsin. Add 2–3 mL of DMEM10 culture medium. Pipette the medium several times to help the cells detach. Transfer the medium containing cells to a 15 mL tube.
Seed ~25,000 cells into a 35 mm glass bottom Petri dish, and add DMEM10 culture medium up to 1.5 mL.
Place the dish containing cells back in the incubator. Incubate the cells at 37°C for 24 h before transfection, staining, or UPLaS experiments.
Cell transfection
Dilute 1 μg of DNA (CEPIA-3mt, Parkin-mCherry, or LC3-GFP) in 200 μL of jetPRIME buffer.
Vortex for 10 s and spin down.
Add 2 μL of jetPRIME reagent (DNA/jetPRIME ratio 1:2).
Vortex for 1 s, spin down, and incubate at room temperature (RT) for 10 min.
Add the transfection mix to the cells cultured in 2 mL of DMEM10 culture medium in a 35 mm Petri dish.
Replace DMEM10 culture medium with 2 mL of fresh DMEM10 culture medium 4 h after transfection.
Check transfection efficiency 24–48 h after transfection. Normally, select several fields of view (FOV) by fluorescence microscopy, the transfection is ideal when 60–80% of cells have been transfected successfully.
Note: According to the actual transfection efficiency and cell density, 1–3 μg of DNA can be added, while maintaining the DNA/jetPRIME ratio previously used. The culture and transfection procedures for SH-SY5Y cells are the same as that of Hela cells. The SH-SY5Y full serum culture medium (MEM/F12-10 culture medium) contains 43.5 mL of MEM, 43.5 mL of Ham’s F-12 Nutrient Mix, 10 mL of FBS, 1 mL of GlutaMAX, 1 mL of Sodium Pyruvate, 1 mL of MEM NEAA, and 1 mL of (1×) Penicillin/Streptomycin. The culture medium for SH-SY5Y cells in all culture and transfection steps is MEM/F12-10.
Setting up the UPLaS system
This UPLaS system consists of a commercial confocal microscope and a femtosecond laser. In Figure 2, a fiber femtosecond laser (1,030 nm, 1 MHz, 220 fs, 1 W) is incorporated into the excitation path of a confocal system.
Turn on the confocal microscope.
Fix the Galvo to the center of the FOV.
Turn on the femtosecond laser.
Note: Please set the femtosecond laser power at a low level (~50 mW, tested between Shutter and RM3 by a laser power meter) for the process of adjusting the optical path.
Use the reflective mirrors (RM1 and RM2, shown in Figure 2B) to direct the femtosecond laser beam through a mechanical shutter. Open the shutter.
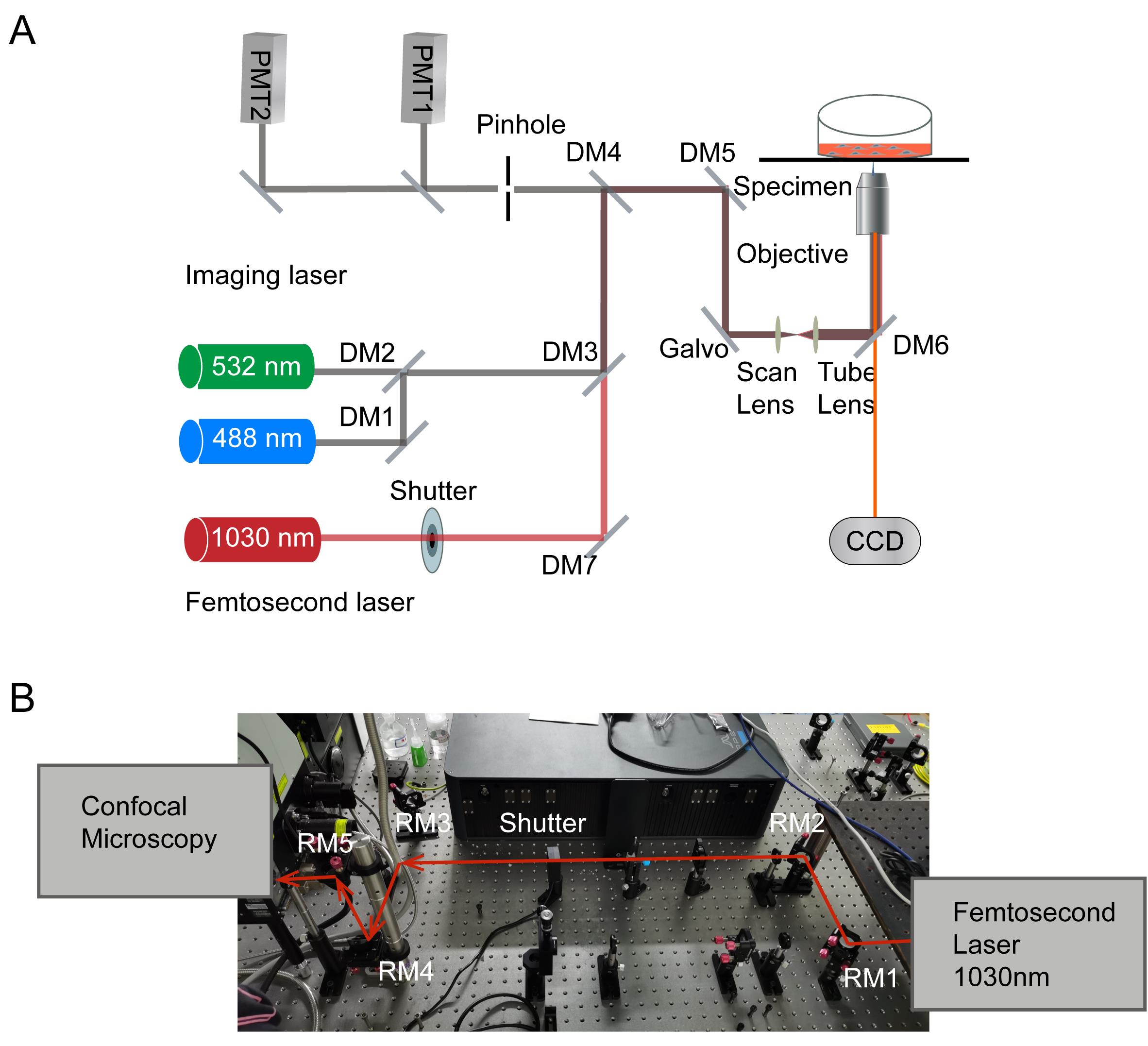
Figure 2. The photostimulation scheme established on a femtosecond laser coupling into a confocal microscope. (A) Optical paths of (B) the UPLaS system. The femtosecond laser is aligned into the microscope scanning system through RM4 and RM5, to form a UPLaS system. A CCD camera is used to provide a bright-felid image to align the optical path. DM = dichroic mirror, RM = reflective mirror. The wavelength of confocal scanning laser is 488 nm/532 nm, and the typical collection wavelength interval of fluorescence is <560 nm/560–625 nm/> 625 nm. The fiber femtosecond laser (1,030 nm, 120 fs, 50 MHz, 1 W) can be replaced by a Ti = Sapphire laser (810 nm, 80 MHz, 65 fs, 1 W), or other commercial femtosecond laser oscillators.Steer RM4 and RM5 to make the femtosecond laser beam coincide with the scanning laser beam.
Measure the transmission efficiency of the objective of the femtosecond laser.
Note: It will use the power at specimen to illustrate the related experiment procedures below.
Turn off the shutter, femtosecond laser, and confocal microscope until experiments start.
Notes:
The shutter is synchronized with the confocal scanning process. It opens at the pre-designed time which is set through the confocal imaging controlling software. The stimulation area can be pre-assigned manually in the confocal imaging controlling software to any form like line, polygon, or circle. Thus, the sample is only stimulated by the femtosecond laser when the confocal scanning process enters a given imaging frame. Along with femtosecond laser stimulation, confocal microscopy provides continuous cell imaging to monitor molecular dynamics in real time.
It can be replaced by a Ti: Sapphire laser or other commercial femtosecond oscillators in the NIR range. The laser power and some other photostimulation parameters need to be tuned because the optical parameters (pulse width, wavelength, and repetition rate) vary a lot in different femtosecond lasers, which thus induce different multiphoton excitation efficiencies. Especially if the wavelength is changed, the power needs to be adjusted carefully. Our experience is that, if the wavelength reaches 800 nm, the power needs to be reduced a lot. If the wavelength is as short as 700 nm, the cells will be extremely sensitive, and this is not recommended.
Activation of mitoCa2+ oscillation by UPLaS
Transfecting cells with fluorescent Ca2+ indicated protein
Prepare Hela cells transfected with CEPIA-3mt, as described in step A.
Note: For a 35 mm glass Petri dish, 1 μg of plasmid CEPIA-3mt is enough. The amount of plasmid can be adjusted according to the actual transfection efficacy and cell density, as mentioned in step A.2.g.
Replace the medium 4 h after transfection, and incubate for 24 h before UPLaS experiments.
Activation of Ca2+ oscillation by UPLaS.
Turn on the femtosecond laser 30 min before the start of the experiment, to stabilize the laser energy output and ensure the shutter is closed.
Turn on the laser-scanning confocal microscope and open the microscope software. Set the excitation laser at 473 nm. Set the power level of the 473 nm laser at 0.1 mW. Set the image size to 512 × 512 pixels. Set the interval time of each pixel as 2 μs. Set the total imaging frames at approximately 100, to provide ~3 min of continuous microscopy for any individual experiment.
Take out the Petri dish containing cells transfected with CEPIA-3mt from the incubator, and place the cells into the 37°C incubator mounted over the microscope stage.
Select the target cell, and acquire fluorescent images of CEPIA-3mt with the 473 nm laser, as shown in Figure 1.
Select the target mitochondria and define the parameters of stimulation.
Set a stimulation point (1 × 1 μm2) in a submicron region, in a single mitochondrial tubular structure
Set the total stimulation time at 0.1 s. Synchronize the shutter of femtosecond laser with the confocal scanning according to the predefined photostimulation area, which is only open when the laser scanning drops in the stimulation frame, and closes immediately when it goes out.
Set the power of the femtosecond laser to 8 mW (1,030 nm) at the sample.
Set the scanning and stimulation time sequence.
Scan the targeted cells during Pre-UPLaS CM and Post-UPLaS CM with a 473 nm laser. Stimulate the cells during UPLaS with the femtosecond laser.
Click on the XY-T button to start the continuous microscopy imaging progress.
Wait until the imaging process is finished and save the imaging data.
Turn off the femtosecond laser and confocal microscope after the experiment.
Detecting the change in MMP and mitoROS
Detecting MMP change in cells stimulated by UPLaS
Prepare Hela cells following step A.1.
Dissolve 5 mg of TMRM into 200 μL of DMSO, to a stock concentration of 50 mM.
Dissolve 1 μL of TMRM into 1 mL of DMSO to 50 μM.
Prepare the TMRM staining solution: dilute 1 μL of TMRM (50 μM) in 1mL of DMEM10 culture medium, to a final concentration of 50 nM.
Incubate the cells in the TMRM staining solution at 37°C for 20 min.
Replace the TMRM staining solution with 2 mL of DMEM10 culture medium.
Use the UPLaS system to stimulate a single mitochondria and detect the change in MMP, following step C.2.
Note: In step C.2, excite the fluorescence of TMRM with the 543 nm laser.
Detecting mitoROS change in cells stimulated by UPLaS
Prepare Hela cells following step A.1.
Dissolve 50 μg of MitoSOX in 6.6 μL of DMSO, to a stock concentration of 10 mM.
Prepare the MitoSOX staining solution: dilute 1 μL of MitoSOX (10 mM) into 1 mL of DMEM10 culture medium, to a final concentration of 10 μM.
Incubate the cells in the MitoSOX staining solution at 37°C for 20 min.
Replace the MitoSOX staining solution with 2 mL of DMEM10 culture medium.
Use the UPLaS system to stimulate a single mitochondria, and detect the change in mitoROS following step C.2.
Note: In step C.2, excite the fluorescence of MitoSOX with the 543 nm laser.
Detecting the recruitment of Parkin
Transfecting cells with fluorescent Parkin indicated protein
Seed ~25,000 cells into a 35 mm glass bottom Petri dish with grids, and transfect cells with Parkin-mCherry, as described in step A.
Note: For a 35 mm Petri dish, 1 μg of plasmid DNA Parkin-mCherry is enough. The amount of plasmid can be adjusted according to the actual transfection efficacy and cell density as mentioned in step A.2.g.
Replace the DMEM10 culture medium with 2 mL of fresh DMEM10 culture medium 4 h after transfection, and incubate for 24 h before UPLaS experiments.
Loading cells with MitoTracker
Prepare Hela cells transfected with Parkin-mCherry, following step E.1.
Dissolve 50 μg of MitoTracker Green in 74.4 μL of DMSO, to a stock concentration of 1 mM.
Dissolve 1 μL of MitoTracker Green (1 mM) in 10 μL of DMSO, to 100 μM.
Prepare the MitoTracker Green staining solution: dilute 1 μL of MitoTracker Green (100 μM) into 1 mL of DMEM10 culture medium, to a final concentration of 100 nM.
Incubate the cells in the MitoTracker Green staining solution at 37°C for 20 min.
Replace the MitoTracker Green staining solution with 2 mL of DMEM10 culture medium.
Detecting the recruitment of Parkin to the mitochondria induced by UPLaS
Use the UPLaS system to stimulate a single mitochondria tubular structure, and acquire fluorescent images of Parkin-mCherry and MitoTracker Green, following step C.2.
Note: In step C.2, excite the fluorescence of Parkin-mcherry with the 543 nm laser and MitoTracker Green with the 473 nm laser. Set the total imaging frames at 2, to acquire an image of Parkin distribution before UPLaS.
Place the cells back into the incubator.
After 30 min, put the dish with grids on the microscope stage. Locate the selected grid and the cell that was stimulated by the femtosecond laser.
Start single frame confocal scanning. Locate the mitochondrion which is stimulated by the femtosecond laser. Save the Parkin distribution fluorescent picture of photostimulation for further data analysis.
Place the cells back into the incubator again, and repeat steps E.3.c and d, after 1 h, and 2 h of stimulation.
Turn off the femtosecond laser and confocal microscope after the experiment.
Detecting the change in PINK
Cell Preparation and Stimulation
Prepare Hela cells seeded into a 35 mm glass bottom Petri dish with grids, as describe in step A.1.
Dissolve 50 μg of MitoTracker Red in 94 μL of DMSO, to a stock concentration of 1 mM.
Dissolve 1 μL of MitoTracker Red (1 mM) in 10 μL of DMSO, to 100 μM.
Prepare the MitoTracker Red staining solution: dilute 1 μL MitoTracker Red (100 μM) in 1 mL of DMEM10 culture medium, to a final concentration of 100 nM.
Incubate the cells in the MitoTracker Red staining solution at 37°C for 20 min.
Replace the MitoTracker Red staining solution with 2 mL of DMEM10 culture medium.
Use the UPLaS system to stimulate an individual mitochondrion, as described in step E.3.
Note: In step E.3, excite the fluorescence of MitoTracker Red with the 543 nm laser.
Place the dish back in the incubator. Incubate the cells for 5 min, 30 min, and 1 h before immunofluorescence (IF) microscopy.
Immunofluorescence (IF) microscopy of PINK1 in cells with UPLaS
Wash: Take the dish containing cells with photostimulation treatment in step F.1 out of the incubator. Remove DMEM10 culture medium. Wash the cells with PBS three times. Remove PBS.
Fix: Add 1 mL of 4% paraformaldehyde (PFA) at 4°C into the dish. Fix the cells with 4% PFA for 10 min. Remove the PFA. Wash the cells with PBS twice for 5 min each time. Remove PBS.
Permeabilise: Incubate the cells in 1 mL of 0.1% Triton X-100 in PBS at RT for 15 min. Remove the Triton X-100 buffer. Wash the cells with PBS twice for 5 min each time. Remove PBS.
Block: Incubate the cells in 1 mL of 1% non-fat powdered milk in PBS at RT for 30 min. Remove the blocking buffer.
Primary antibody incubation: Dilute anti-PINK antibody in PBS with 0.1% Tween-20 (1:500). Incubate the cells in primary antibody buffer at 4°C for 12 h. Remove the primary antibody incubation buffer.
Secondary antibody incubation: Dilute the secondary antibody, anti-Rabbit IgG H&L, in PBS with 1% Tween-20 (1:10,000). Incubate the cells in the secondary antibody buffer at RT for 2 h. Remove the secondary antibody incubation buffer.
Wash the cells with PBS. Remove PBS.
Sealing: add the antifade mounting medium into the Petri dish to maintain the fluorescence.
Place the treated dish on the microscope stage. Locate the selected grid and the cell which was treated with the femtosecond laser.
Start single frame confocal scanning. Excite the fluorescence of Anti-PINK with the 473 nm laser, and excite the fluorescence of MitoTracker Red with the 543 nm laser.
Save the fluorescent images of stimulated cells for further data analysis.
Detecting the change in mitophagy
Transfecting cells with fluorescent autophagosomes indicated protein
Prepare Hela cells seeded into a 35 mm glass bottom Petri dish with grids and transfect cells with LC3-GFP as described in step A.
Note: For a 35 mm Petri dish, 1 μg of DNA plasmid LC3-GFP is enough. The amount of plasmid can be adjusted according to the actual transfection efficacy and cell density as mentioned in step A.2.g.
Replace the DMEM10 culture medium with 2 mL of fresh DMEM10 culture medium 4 h after transfection, and incubate for 24 h before UPLaS experiments.
Loading cells with MitoTracker Red
Prepare Hela cells transfected with LC3-GFP, following step E.1.
Dissolve 50 μg of MitoTracker Red in 94 μL of DMSO, to a stock concentration of 1 mM.
Dissolve 1 μL of MitoTracker Red (1 mM) in 10 μL of DMSO, to 100 μM.
Prepare the MitoTracker Red staining solution: dilute 1 μL of MitoTracker Red (100 μM) in 1 mL of DMEM10 culture medium, to a final concentration of 100 nM.
Incubate the cells in the MitoTracker Red staining solution at 37°C for 20 min.
Replace the MitoTracker Red staining solution with 2 mL of DMEM10 culture medium.
Detecting the formation of autophagosomes induced by UPLaS
Carry out UPLaS experiment on cells transfected with LC3-GFP and loaded with MitoTracker Red, as described in step C.2.
Note: In step C.2, excite the fluorescence of MitoTracker Red with the 543 nm laser and LC3-GFP with the 473 nm laser.
Place the cells back into the incubator.
After 30 min, place the dish on the microscope stage. Locate the selected grid and the cell which was stimulated by the femtosecond laser.
Start single frame confocal scanning. Locate the mitochondrion which was stimulated by the femtosecond laser. Save the LC3-GFP distribution fluorescent images of photostimulation cell for further data analysis.
Data analysis
The steps of processing fluorescence images
Acquire fluorescence images according to confocal microscope software FV1200 (Olympus).
Choose File > Open.
Select an experiment file and click on Open.
Choose File > Export > File type: TIFF.
Choose a destination folder and click on Save, as shown in Figure 3.
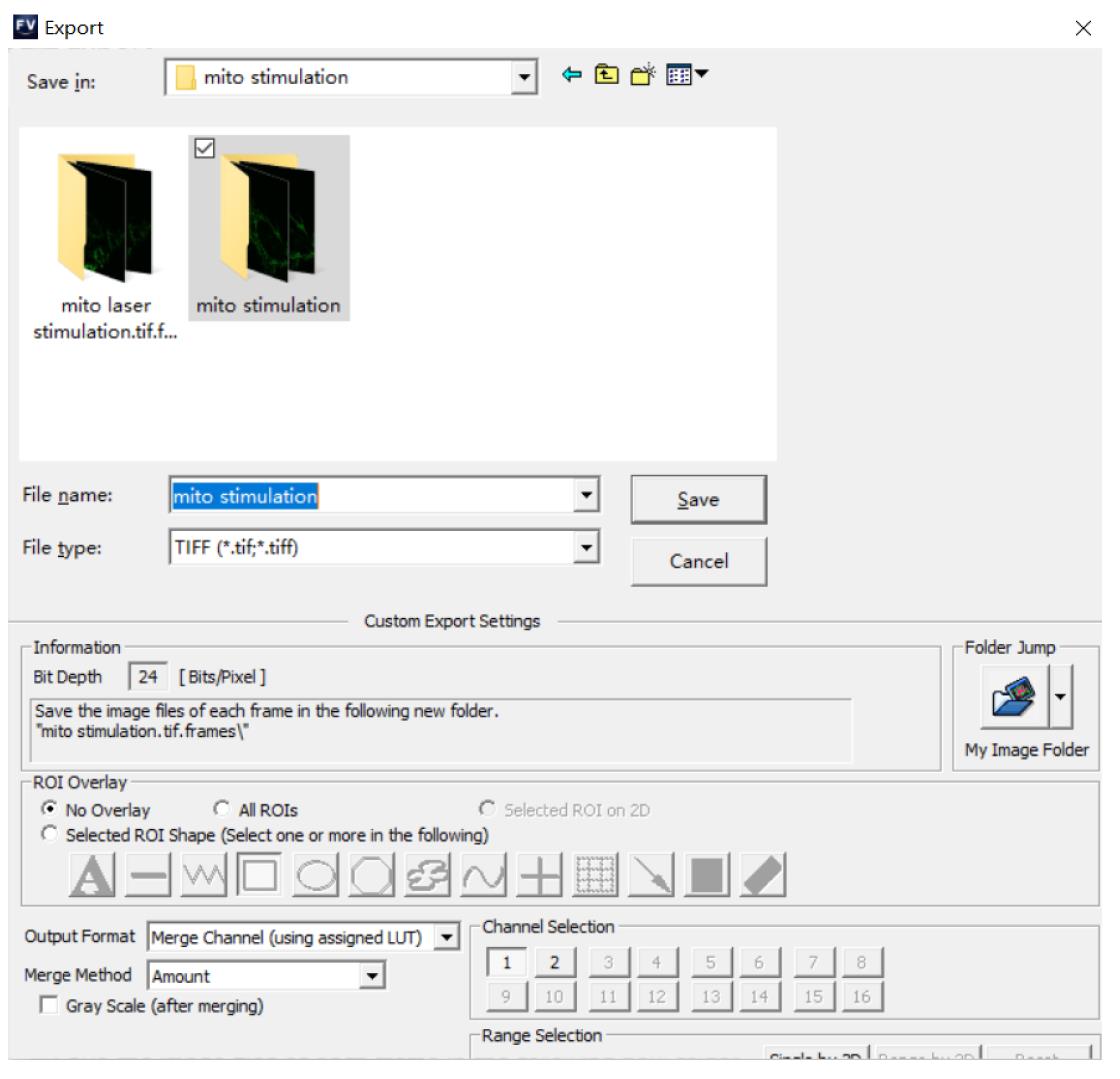
Figure 3. Exporting an experiment file as TIFF image series.Acquire fluorescence intensity by ImageJ software.
Drag your file folder into ImageJ. Select Convert to RGB and Use Virtual Stack. Click on Yes, as shown in Figure 4A. All images in this folder are shown as a stack (Figure 4B).
Click on the ‘polygon’ button (Figure 4C). To create a polygon selection, click repeatedly with the mouse to create line segments. When finished, click in the small box at the starting point (or double click), and ImageJ will automatically draw the last segment (Figure 4D). The points that define a polygon selection can be moved, and modifier keys can be used to delete or add new vertices to the polygon.
Choose Image > Stacks > Measure Stack (Figure 4E), to display the mean value of all images in this stack (Figure 4F).
Copy the mean values of all images from ImageJ and the time sequence recorded by FV1200 to GraphpPad Prism 7. Plot the charts using GraphPad Prism 7.
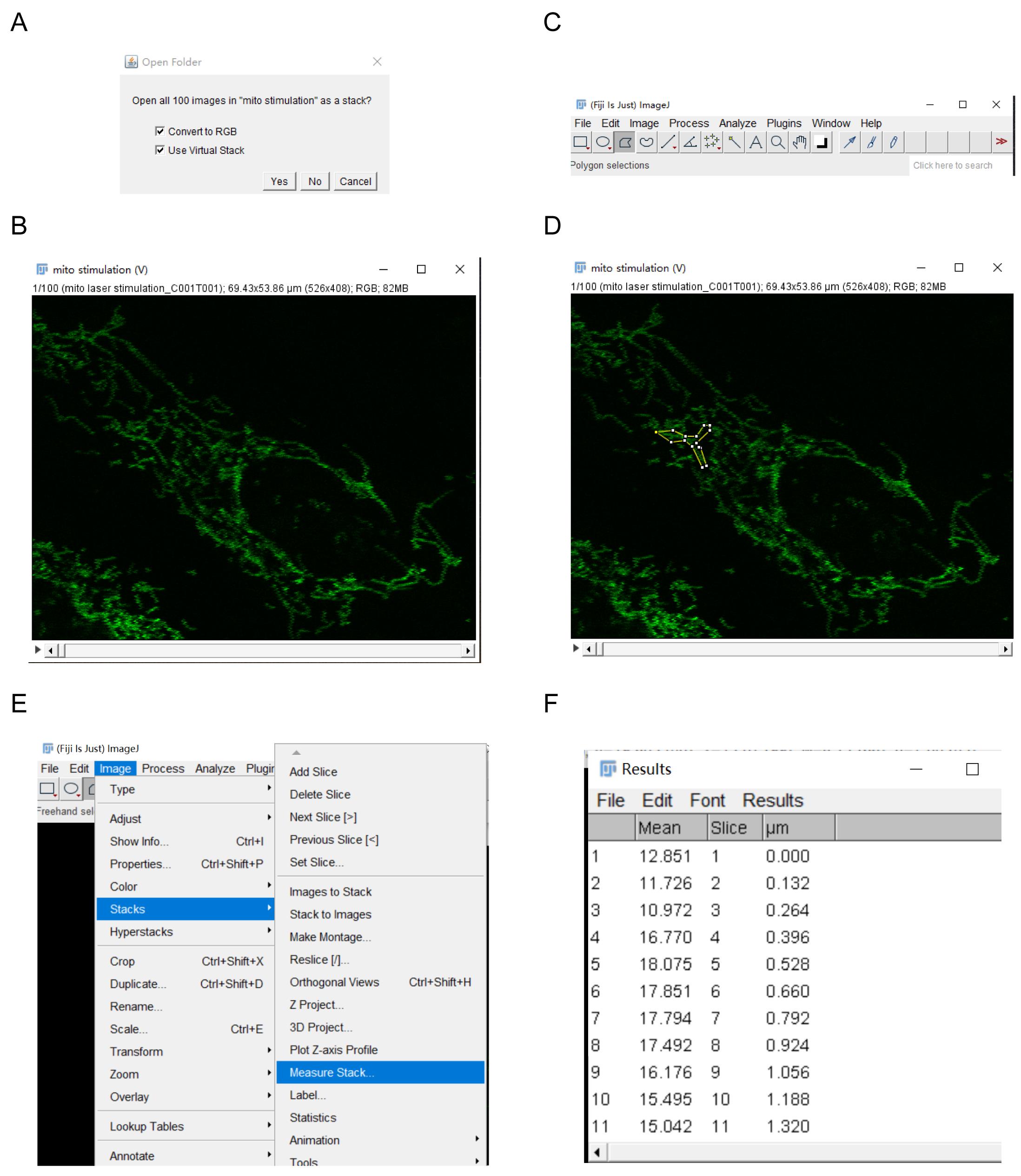
Figure 4. The workflow of acquiring fluorescence intensity using ImageJ software.Representative data
As shown in Figures 1 and 2, we established a noninvasive ultra-precise laser stimulation (UPLaS) technology by aligning a femtosecond laser into the optical path of a confocal microscope. This UPLaS method enables spatiotemporal-specific stimulations to target individual mitochondria. As shown in Figure 5, the UPLaS evoked localized mitoCa2+ oscillations in a single mitochondrial tubular structure, with transient, moderate, and fast-recovered MMP depolarization, while the mitoROS increased there, without any perturbation to its neighbors.
The time-lapse dynamics of Parkin-mCherry indicate the most significant Parkin recruitment to the stimulated mitochondria was presented 1 h after UPLaS. Then, we measured the colocalization of PINK1 [visualized by immunofluorescence (IF) microscopy] and mitochondria (indicated by MitoTracker Red). The IF microscopy of PINK1 was performed at 5, 30, and 60 min after UPLaS. The level of PINK1 on the stimulated mitochondria was higher 1 h after UPLaS. Ultimately, the mitochondria treated with UPLaS all presented consistent coincidence with autophagosomes. The detailed analysis can be found at https://doi.org/10.1038/s41419-021-03913-3.
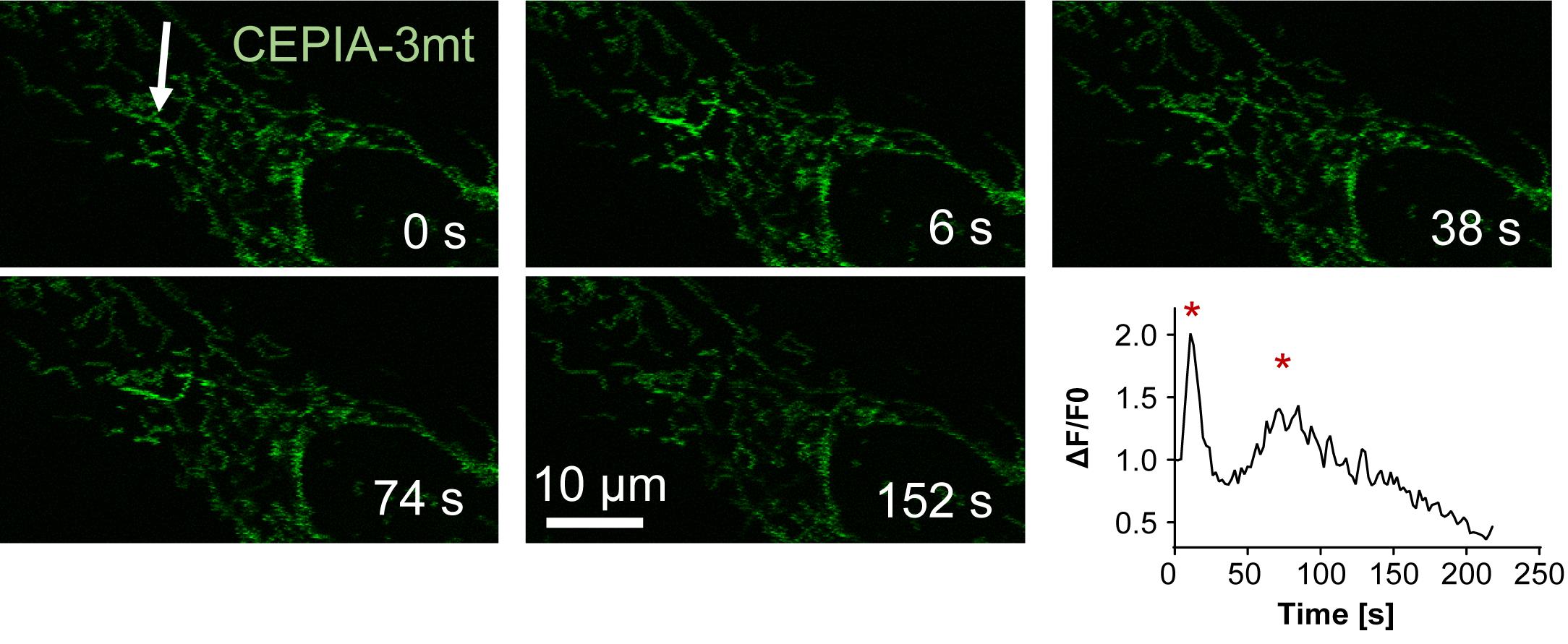
Figure 5. UPLaS excites mitoCa2+ oscillations in the target mitochondria. The individual mitochondrion stimulated by UPLaS. The responses of mitoCa2+ to UPLaS (at t = 5 s) are indicated by CEPIA-3mt. The white arrow points at the single mitochondria tubular structure stimulated by the femtosecond laser.Each independent experiment should be executed with at least three experimental repeats.
Notes
In our experiment, we align the femtosecond laser into the confocal microscope. The function of two-photon microscope can realize the UPLaS technology completely.
We conducted the same UPLaS experiments on Hela cells and SH-SY5Y cells. The culture and transfection processes were all the same as that of Hela cells.
Turn off the femtosecond laser and confocal microscope after the experiment.
Keep the power of visible lasers (473 nm and 543 nm) as low as possible when using TMRM.
Recipes
Hela culture medium (DMEM10 culture medium)
500 mL of DMEM
10 mL of (1×) Penicillin/Streptomycin
50 mL of Fetal Bovine Serum
SH-SY5Y culture medium (MEM/F12-10 culture medium)
43.5 mL of MEM, no glutamine
43.5 mL of Ham’s F-12 Nutrient Mix
10 mL of FBS
1 mL of GlutaMAX
1 mL of Sodium Pyruvate
1 mL of MEM NEAA
1 mL of (1×) Penicillin/Streptomycin
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC 92054105, 62022056 and 61975118), Science & Technology Commission Shanghai Municipality 18QA1402300, and Innovation Research Plan supported by Shanghai Municipal Education Commission ZXWF082101 to H.H.
Original research paper Yu et al. (2021): Mitochondrial Ca2+ oscillation induces mitophagy initiation through the PINK1-Parkin pathway. https://doi.org/10.1038/s41419-021-03913-3.
Competing interests
The authors declare no competing financial interests.
Ethics
This study was approved by the Ethic Committee of the School of Biomedical Engineering at Shanghai Jiao Tong University.
References
- Cheng, P., Tian, X., Tang, W., Cheng, J., Bao, J., Wang, H., Zheng, S., Wang, Y., Wei, X., Chen, T., et al. (2021). Direct control of store-operated calcium channels by ultrafast laser. Cell Res 31(7): 758-772.
- Chung, E., Choi, Y., Park, J., Nah, W., Park, J., Jung, Y., Lee, J., Lee, H., Park, S. and Hwang, S. (2020). Intracellular delivery of Parkin rescues neurons from accumulation of damaged mitochondria and pathological α-synuclein. Sci Adv 6(18): eaba1193.
- Ge, P., Dawson, V. L. and Dawson, T. M. (2020). PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson's disease. Mol Neurodegener 15(1): 20.
- He, H., Li, S., Wang, S., Hu, M., Cao, Y. and Wang, C. (2012). Manipulation of cellular light from green fluorescent protein by a femtosecond laser. Nat Photonics 6(10): 651-656.
- Iwanaga, S., Smith, N., Fujita, K. and Kawata, S. (2006). Slow Ca2+ wave stimulation using low repetition rate femtosecond pulsed irradiation. Opt Express 14(2): 717-725.
- Jin, S. M. and Youle, R. J. (2013). The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 9(11): 1750-1757.
- Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., Sideris, D. P., Fogel, A. I. and Youle, R. J. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524(7565): 309-314.
- Liu, J., Liu, W., Li, R. and Yang, H. (2019). Mitophagy in Parkinson’s disease: from pathogenesis to treatment. Cells 8(7): 712.
- Liu, X., Lv, X., Zeng, S., Zhou, W. and Luo, Q. (2009). Noncontact and nondestructive identification of neural circuits with a femtosecond laser. Appl Phys Lett 94(6): 061113.
- Pickrell, A. M. and Youle, R. J. (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85(2): 257-273.
- Russell, O. M., Gorman, G. S., Lightowlers, R. N. and Turnbull, D. M. (2020). Mitochondrial diseases: hope for the future. Cell 181(1): 168-188.
- Smith, N. I., Iwanaga, S., Beppu, T., Fujita, K., Nakamura, O. and Kawata, S. (2005). Femtosecond laser-induced calcium release in neural-type cells. In: Nanobiophotonics and Biomedical Applications II, International Society for Optics and Photonics. Proc SPIE 5705, https://doi.org/10.1117/12.596923.
- Smith, N. I., Kumamoto, Y., Iwanaga, S., Ando, J., Fujita, K. and Kawata, S. (2008). A femtosecond laser pacemaker for heart muscle cells. Opt Express 16(12): 8604-8616.
- Wang, S., Liu, Y., Zhang, D., Chen, S. c., Kong, S. K., Hu, M., Cao, Y. and He, H. (2018). Photoactivation of Extracellular-Signal-Regulated Kinase Signaling in Target Cells by Femtosecond Laser. Laser Photonics Rev 12(7): 1700137.
- Wang, Y., He, H., Li, S., Liu, D., Lan, B., Hu, M., Cao, Y. and Wang, C. (2014). All-optical regulation of gene expression in targeted cells. Sci Rep 4: 5346.
- Yu, Z., Wang, H., Tang, W., Wang, S., Tian, X., Zhu, Y. and He, H. (2021). Mitochondrial Ca2+ oscillation induces mitophagy initiation through the PINK1-Parkin pathway. Cell Death Dis 12(7): 632.
- Zhao, Y., Zhang, Y., Liu, X., Lv, X., Zhou, W., Luo, Q. and Zeng, S. (2009). Photostimulation of astrocytes with femtosecond laser pulses. Opt Express 17(3): 1291-1298.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Tian, X. and He, H. (2022). Activation of Mitochondrial Ca2+ Oscillation and Mitophagy Induction by Femtosecond Laser Photostimulation. Bio-protocol 12(7): e4369. DOI: 10.21769/BioProtoc.4369.
Category
Biological Engineering > Biomedical engineering
Biophysics > Biophotonics
Cell Biology > Cell signaling > Autophagy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.