- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

3D-Structured Illumination Microscopy of Centrosomes in Human Cell Lines
Published: Vol 12, Iss 6, Mar 20, 2022 DOI: 10.21769/BioProtoc.4360 Views: 4226
Reviewed by: Chiara AmbrogioSurabhi SonamAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2021
Advertisement





Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
The centrosome is the main microtubule-organizing center of animal cells, and is composed of two barrel-shaped microtubule-based centrioles embedded in protein dense pericentriolar material. Compositional and architectural re-organization of the centrosome drives its duplication, and enables its microtubule-organizing activity and capability to form the primary cilium, which extends from the mature (mother) centriole, as the cell exits the cell cycle. Centrosomes and primary cilia are essential to human health, signified by the causal role of centrosome- and cilia-aberrations in numerous congenic disorders, as well as in the etiology and progression of cancer. The list of disease-associated centrosomal proteins and their proximitomes is steadily expanding, emphasizing the need for high resolution mapping of such proteins to specific substructures of the organelle. Here, we provide a detailed 3D-structured illumination microscopy (3D-SIM) protocol for comparative localization analysis of fluorescently labeled proteins at the centrosome in fixed human cell lines, at approximately 120 nm lateral and 300 nm axial resolution. The procedure was optimized to work with primary antibodies previously known to depend on more disruptive fixation reagents, yet largely preserves centriole and centrosome architecture, as shown by transposing acquired images of landmark proteins on previously published transmission electron microscopy (TEM) images of centrosomes. Even more advantageously, it is compatible with fluorescent protein tags. Finally, we introduce an internal reference to ensure correct 3D channel alignment. This protocol hence enables flexible, swift, and information-rich localization and interdependence analyses of centrosomal proteins, as well as their disorder-associated mutations.
Keywords: Super-resolution microscopyBackground
The centrosome is an organelle composed of two barrel-shaped microtubule-based centrioles surrounded by a matrix of pericentriolar material, and functions as the main microtubule-organizing center in most animal cells. As such, the centrosome plays important roles in regulating cell shape, migration, and division. Furthermore, in quiescent cells, the mature centriole of the centrosome, the mother centriole, can transform into a basal body that gives rise to and anchors a single primary cilium (Breslow and Holland, 2019). The primary cilium is an antenna-like organelle that plays essential roles in signaling coordination during embryonic development and in tissue homeostasis (Anvarian et al., 2019). Mutations in genes that cause defects in cilia assembly or function, including a growing number of centrosomal genes, can therefore give rise to numerous human diseases and syndromes called ciliopathies, which are highly pleiotropic in nature and may affect most tissues and organs in the body (Reiter and Leroux, 2017). In addition, numerical and structural aberrations of centrosomes are a hallmark of cancer (Boveri, 1914). Centriole over-amplification is a factor promoting genomic imbalances, malignant transformation, and tumor progression as such, but may also occur in consequence of different treatment protocols. Trying to turn the bad into good, the mechanisms of how cancer cells adapt to the presence of super-numerous centrosomes during cell division are explored as a targetable cancer-specific vulnerability (Godinho and Pellman, 2014; Nigg and Holland, 2018; Mariappan et al., 2019; Sabat-Pośpiech et al., 2019; Adams et al., 2021; Goundiam and Basto, 2021). Understanding the molecular composition, structure, and function of centrosomes and cilia is therefore relevant, both from fundamental scientific and medical perspectives. Both centrioles of the centrosome contain nine triplets of microtubules termed A-, B-, and C-tubules, each harboring 13, 10, and 10 microtubule protofilaments, respectively. These are subjected to various tubulin posttranslational modifications and associate with multiple microtubule-associated proteins (MAPs) and microtubule inner proteins (MIPs), which are thought to have a stabilizing function. Other centrosomal substructures include the cartwheel, which functions as a scaffold for centriole biogenesis, and the connecting fibers or rootlet filaments that link the proximal ends of the two centrioles (Gomes Pereira et al., 2021). In addition, the most mature centriole, the mother centriole, is decorated with appendages at its distal end, called distal and subdistal appendages, which are important for cilium biogenesis and microtubule anchoring, respectively (Breslow and Holland, 2019). During ciliogenesis, the distal appendages dock to preciliary vesicles or a membrane patch on the plasma membrane, which triggers removal of the CEP97-CP110 capping complex from the distal end of the mother centriole, thereby promoting formation of the transition zone, which gates protein entrance and exit to or from the ciliary compartment; this is followed by extension of the ciliary axoneme by intraflagellar transport (IFT) (Pedersen et al., 2008; Shakya and Westlake, 2021). Concomitant with the transformation of the mother centriole to a basal body, the distal appendages are remodeled into transition fibers that anchor the basal body to the plasma membrane (Paintrand et al., 1992).
Owing to their importance for human health and disease, centrosomes and cilia have been subject to intense investigation in recent years, and significant progress has been made towards understanding the molecular composition, structural organization, and function of these organelles. For example, a landmark study by Andersen and colleagues identified and validated 23 novel components, and 41 likely candidate constituents of human centrosomes, by mass-spectrometry-based proteomics (Andersen et al., 2003); a subsequent study by the same group extended these findings to include another >150 candidate proteins, of which 22 were confirmed to be novel centrosome proteins (Jakobsen et al., 2011). Since then, technological development during the past decade and tedious efforts of many laboratories steadily extends the list of centrosomal/ciliary components and sub-organelle specific protein-interaction networks (Firat-Karalar et al., 2014; Mick et al., 2015; Gupta et al., 2015; Boldt et al., 2016; Wheway et al., 2015; Gheiratmand et al., 2019; Arslanhan et al., 2020). Concomitantly, the discovery-rate of disease -causing mutations in centrosomal or cilia protein coding genes accelerates (Best et al., 2021; Smedley et al., 2021). Standard (immuno-)fluorescence microscopy allows simultaneous localization analysis of multiple proteins, but is limited by the lack of sufficient resolution required to resolve specific association with distinct centrosomal entities. The introduction of different super-resolution fluorescence microscopy techniques rapidly advanced the field, providing up to 20 nm lateral resolution depending on the technique (Lau et al., 2012; Sonnen et al., 2012; Gartenmann et al., 2017; Sieben et al., 2018; Yang et al., 2018; Bowler et al., 2019; Chong et al., 2020; Liu et al., 2020; Zwettler et al., 2020). These can even be combined with different expansion microscopy protocols, which can isotropically disentangle the centrosome by four-, or even 10-fold (Sieben et al., 2018; Zwettler et al., 2020; Damstra et al., 2021; LeGuennec et al., 2021). Though none of these techniques may match the ultrastructural resolution of electron microscopy, they enable a more rapid assessment of relative localization of individual proteins, as well as the study of interdependencies and disease-causing mutations. A discussion of the various advantages and disadvantages of the different super-resolution-microscopy techniques is beyond the scope of this general introduction. We provide here a detailed protocol suitable (and optimized) for the analysis of centrosomal and ciliary proteins by 3D-structured illumination microscopy, which worked well for immune-based detection of most tested centrosomal/ciliary antigens in our hands, while simultaneously preserving structural organization and light-emitting capability of fluorescent protein-tags. Using this protocol, we gained important structural and functional insights into human disease-associated proteins, including CSPP1, CEP104, CEP78, and their interaction partners at the centrosome, and the primary cilium (Frikstad et al., 2019; Gonçalves et al., 2021). The detailed procedures for sample preparation and good image processing practice are generally applicable for different types of fluorescence microscopy, instruments, and software platforms. Specific procedures in image acquisition and processing are focused on the OMX V4 SIM super-resolution system, and some previous knowledge about the system may be advantageous.
Materials and Reagents
Coverslips, 10 mm in diameter, 0.17 mm thick ± 0.01 mm (Chemi-Teknik, Hecht Assistant, catalog number: 1014)
6 well cell culture plates, multidish (VWR, Nunc, catalog number: 391-8036)
hTERT RPE1 (ATCC, catalog number: CRL-4000) or other adherent cell line of interest, e.g., cell line stably expressing a specific fluorescent protein-tagged centrosomal protein (Frikstad et al., 2019; Gonçalves et al., 2021)
DMEM/F-12, GlutaMAXTM Supplement (Thermo Fisher Scientific, Gibco, catalog number: 11524436)
Fetal Bovine Serum (Thermo Fisher, catalog number: 10270106)
Penicillin/Streptavidin (Merck, catalog number: P4333)
Trypsin-EDTA solution (Merck, catalog number: T3924)
Dulbecco’s Phosphate Buffered Saline (PBS), Modified, without calcium chloride and magnesium chloride, liquid, sterile-filtered, suitable for cell culture (Merck, catalog number: D8537)
Formalin solution, neutral buffered, 10% (Merck, catalog number: HT-5011)
Methanol, ≥99.8%, HiPerSolv CHROMANORM® (VWR, catalog number: 20837.360)
Bovine Serum Albumin (Merck, catalog number: 3059)
Triton-X100 (Merck, catalog number: T9284)
Microscopy slides (Chemi-Technik, Hecht Assistant, catalog number: 43503)
ProLong Gold Antifade Mountant (Thermo Fisher Scientific, catalog number: P36930)
ProLong Diamond Antifade Mountant (Thermo Fisher Scientific, catalog number: P36965)
Antibodies:
Anti-CEP164, rabbit polyclonal (Sigma-Aldrich, catalog number: HPA037606)
AlexaFluor 488 conjugated AffiniPure Donkey-anti-Rabbit IgG (Jackson Immuno Research Labs, catalog number: 711-545-152)
DyLight 550 conjugated Donkey-anti-Rabbit IgG (Abcam, catalog number: ab96892)
AlexaFluor 647 conjugated AffiniPure Donkey-anti-Rabbit IgG (Jackson Immuno Research Labs, catalog number: 711-605-152)
Cy3 conjugated AffiniPure Donkey-anti-Mouse IgG (Jackson Immuno Research Labs, catalog number: 715-165-150)
AlexaFluor 647 conjugated AffiniPure Donkey-anti-Mouse IgG (Jackson Immuno Research Labs, catalog number: 711-605-150)
bisBenzimide H 33258 (Hoechst 33258) (Sigma-Aldrich, catalog number: 14530)
PBS-AT (see Recipes)
1.6% PFA solution (see Recipes)
Equipment
OMX V4 SIM super-resolution system and OMX analysis workstation running SoftWorx 7.1 software
Sterilized microdissectioning forceps (type Sigma-Aldrich, catalog number: F4017-1EA or similar)
Common lab equipment: steam sterilizer, cell culture incubator, safety cabinet, -20°C freezer
Software
SoftWoRx 7.1 (part of OMX V4 SIM super-resolution system)
ImageJ v1.53c in Fiji for Windows-64 package (https://fiji.sc/#) and ( https://imagej.nih.gov/ij/) run in Java1.8.0_172 (64-bit) environment
Procedure
The theoretically achievable resolution in a fluorescence microscope is a function of the wavelength utilized and the optical train, which at most imaging facilities are set configurations. Most impactful adjustable parameters for optimization in (super-resolution) imaging are the light path between lens and fluorophore (choice of: immersion oil, cover slip glass, embedding medium), and the mode of sample preparation. The choice of chemical fixative and staining conditions shall preserve the biological structure and simultaneously allow sufficient fluorescent labelling efficacy (or retain fluorescence activity, in case of fluorescent protein tags). This protocol has been optimized for the analysis of centrosomes, cilia, and the microtubule cytoskeleton on a DeltaVision OMX V4 Blaze 3D-SIM microscope, equipped with a 60×/NA1.42 oil PLAPON6 PSF lens. 3D SIM microscopes from other suppliers are expected to produce similar results. In our set-up, cells are seeded, fixated, and stained on high precision cover slips (0.17±0.01 mm), mounted using ProLong Diamond antifade mounting medium (refractive index 1.47), and imaged using an immersion oil with a refractive index of 1.516 (at 23°C; e.g., from Cargille or Leica). The localization pattern of several centrosomal reference proteins tested with our protocol are in strong agreement with recent results reported using other super-resolution imaging techniques, and at scale with previously published transmission electron microscopy data, indicative of good structural preservation and resolution. Furthermore, our fixation and mounting protocol retains the fluorescence property of e.g., mNeonGreen-fluorescent protein, enabling observation of centrosomal proteins for which compatible antibody reagents are unavailable. Finally, as reference for channel alignment and image reconstruction, we introduce multicolor labelling of the centriole distal appendage protein CEP164, which assembles a toroid of approximately 400 nm in diameter. This ensures the correct interpretation of relative localizations of centriolar proteins in multicolor labeling experiments.
Sample preparation
Cell culture and seeding
NOTE: In some experiments, longer pre-treatment times are required and cultures concomitantly expand (e.g., iterative siRNA-mediated depletion experiments). To avoid introduction of artifacts due to confluence or over-growth, cells may be cultured first without coverslips; 24 hours before the preferred end-point, cells can then be detached by trypsinization, and re-seeded on cover slips at an appropriate cell titer (see a.–c.).
Sterilize coverslips by either autoclavation or dry heat sterilization (180°C/20 min).
Prepare 6-well plate(s) with coverslips using sterilized forceps. Ensure that coverslips are well separated and do not overlap. Five round 10 mm coverslips are easily fitted.
Seed 6–8 × 104 hTERT-RPE1 cells in 2 mL of pre-warmed DMEM/F12 medium supplemented with 10% v/v Fetal Bovine Serum and 1% w/v Penicillin-Streptomycin per well.
Ensure that coverslips are completely covered and still separated.
Allow cells to attach and grow in an incubator at 37°C for 24 h.
Optionally: treat cells with serum deprivation, inhibitor(s), or transfect cells with siRNA, or plasmid DNA.
Incubate cells in a 37 °C incubator until fixation at the desired confluence (commonly 70–90% confluence end point).
Cell fixation
NOTE: All processes in living cells are temperature-sensitive. Therefore, we recommend minimizing the handling time of cells outside the incubator prior to fixation, and avoiding temperature shifts. We place culture plates on a Styrofoam-tray upon removal from the incubator to prevent rapid temperature decline. This protocol is optimized for analysis of centrosomal, ciliary, and centriolar satellite proteins; in our hands, it works for most tested antibodies. All new introduced primary antibodies require initial evaluation of staining specificity and careful titration, to reduce unspecific background binding and achieve the best possible signal-to-noise ratio.
All handling of paraformaldehyde must occur in a ventilated safety cabinet. Prepare a fresh 1.6% paraformaldehyde fixation solution by mixing 1 volume of 10% neutral buffered formalin solution (approximately 4% formaldehyde) with 1.5 volumes of PBS (ambient temperature). Typically, 2 mL of solution are required per well of a 6-well plate.
Retrieve cells from the incubator and carefully aspirate the cell culture supernatant. Avoid drying of the coverslips and rapidly add the fixation solution.
Incubate at room temperature for 10 min.
Remove the fixation solution by pipetting or aspiration. Critical step: Quickly proceed to e) to avoid drying of the specimen. Collect the solution in an appropriate container, for paraformaldehyde waste handling.
Avoid drying of coverslips and carefully wash cells once with 2 mL of PBS per well.
Aspirate PBS and post-fixate cells in 2 mL of cold methanol (-20°C).
Rapidly transfer the plates to a -20°C freezer, and incubate for 20 min.
NOTE: The duration of post-fixation was empirically determined. Longer storage of cells in methanol may be possible, but requires careful sealing of plates or dishes with parafilm to avoid evaporation. Plates should be labelled with a soft pencil, since accidental spill of methanol will wash away any marker pen labels. The combination of initial mild paraformaldehyde fixation and methanol post-fixation appears to preserve localization of microtubule-based structures and even ciliary membrane proteins, while denaturing proteins sufficiently to provide accessibility to notoriously formaldehyde-sensitive epitopes, such as the binding target of the monoclonal antibody to γ-tubulin, GTU-88 (Sigma-Aldrich, #T6557).
Immunofluorescence staining and mounting
Prepare a moist chamber, by placing a sheet of water-soaked gel blotting paper (e.g., Whatman grade GB003) in the bottom of a lightproof container with a removable lid, and cover the wet paper with a sheet of Parafilm.
For each coverslip to be stained, place a droplet of approximately 150 µL of PBS on the parafilm.
Retrieve the plate with coverslips from the freezer. Remove methanol and immediately wash coverslips (in the plate) twice with 2 mL of PBS at room temperature, avoiding complete evaporation of residual methanol and drying of the specimen.
Using pointed metal forceps, transfer one coverslip at a time to a droplet of PBS in the chamber. Place coverslip with cell-side up and gently push it down to have it covered by PBS. A scalpel may be used to aid in the collection of coverslips with the tweezers.
Incubate at room temperature for 5 min.
Carefully aspirate PBS using a flexible vacuum tube fitted with a 200 µL pipette tip, by placing the tip at the side of the coverslip. Be careful to not disturb the cell layer on the surface of the coverslip. All aspiration steps hereafter were performed in this manner.
Add 100–150 µL of blocking solution (PBS-AT) on top of each coverslip. Be careful keeping sufficient space between coverslips, to avoid overspill between adjacent coverslips.
Block (and permeabilize) the cells at room temperature for 15 min.
Prepare primary antibody dilutions in cold blocking solution (4°C) and keep on ice until use. A solution of 30 µL is optimal per 10 mm diameter coverslip. For the internal alignment reference, prepare a 1:2,000 dilution of rabbit anti-CEP164 (Sigma-Aldrich, #HPA037606).
Aspirate the blocking solution and add the primary antibody solution. The antibody solution needs to cover the complete coverslip and is kept in place by its surface tension.
Carefully close the lightproof lid to avoid evaporation and light exposure, and incubate at room temperature for 2 h.
NOTE: eventually, incubation at 4°C overnight with primary antibodies may be tested and compared to the standard protocol.
Prepare secondary antibody dilutions in PBS-AT. For the internal alignment reference, prepare one tube with donkey-anti-rabbit antibodies fluorescently labeled with either fluorophore to be used in the experiment. In our 4-channel standard set-up, we use AlexaFluor-488, DyLight550, or AlexaFluor-647-conjugated donkey anti-rabbit antibodies (all 1:1,000 in PBS-AT). In this set-up, the 405 nm laser line is used for DNA counterstaining with Hoechst 33258.
Aspirate the primary antibody solution and wash each cover slip with 1 mL of PBS using a 1 mL pipette, by iterative cycles of careful application and aspiration droplets. Leave last droplet on coverslip to prevent drying.
Aspirate the PBS droplet and apply 30 µL of appropriate secondary antibody solution to each coverslip. For the internal alignment reference, use the donkey anti-rabbit cocktail.
Incubate at room temperature for 45 min.
Wash each cover slip with 1 mL of PBS, as described in step f. Leave the last droplet on the coverslip to prevent drying.
DNA counterstain (if the 405 nm line is not used for antibody staining): Aspirate the PBS droplet and apply approximately 50 µL of DNA staining solution (0.6 µg/mL Hoechst 33258 in PBS). Incubate at room temperature for 1–2 min.
Wash each cover slip with 1 mL of PBS, as described in step f. Leave the last droplet on the coverslip to prevent drying.
Prepare an object glass with two droplets of maximal 5 µL of ProLong Diamond antifade mounting medium centrally placed above each other. Coverslips must be mounted to the central 22 × 22 mm part of a 76 × 26 mm micro slide, due to the limited travel range of the microscope stage of the OMX DV4 microscope.
Using forceps, pick one coverslip at a time and dip it ten times in a 30 mL plastic beaker filled with ddH2O to rinse off any residual salts. Dry the coverslip by carefully pushing the edge onto tissue paper. Finally, aspirate any residual water.
Drop the coverslip with the cell layer facing down onto the antifade mounting medium droplet.
Note: Be careful to avoid any bubbles. Larger bubbles may be removed by pushing them to the edges, by gently pressing with the tweezers.
Let the mounting medium cure in the dark at room temperature overnight.
Briefly control staining efficacy on standard epi-fluorescence microscope using a dry-lens (e.g., 40×/NA 0.95). IMPORTANT: Do not use immersion oil for the initial assessment! Even small residuals of immersion oils of a different make than those used for super-resolution can precipitate when mixed, and may cause damaging deposition on lenses.
Slides can be stored for several days in the dark at 4°C until imaging.
Image acquisition
This protocol is customized for a DeltaVision OMX V4 Blaze 3D-SIM microscope equipped with 6-color solid state illumination and 6 lasers (405 nm, 445 nm, 488 nm, 514 nm, 568 nm, and 642 nm), UltimateFocus Hardware Autofocus System, three high speed water-cooled PCO.edge sCMOS cameras, and a 60× NA 1.42 oil PLAPON6 PSF objective (raw pixel size=0.08 µm, reconstructed pixel size=0.04 µm). While many of the steps outlined below are specific for an OMX microscope system, it is expected that other 3D-SIM microscopes will generate comparable results. General aspects of image alignment and acquisition outlined here can serve as guidance for adaptation to other super-resolution microscopy systems. Multiple channel (color) imaging requires careful mechanical alignment of all light paths of the microscope system upon system set-up. System inert alignment differences need to be controlled and corrected prior to each imaging session, to avoid day-to-day variability. We apply a two-step alignment procedure, controlling first system-dependent parameters (X,Y,Z-alignment; chromatic aberration), using an image registration slide with a specific pattern (20×20 grid of 100 nm holes), and thereafter sample-specific aberrations, using the multicolor-labeled CEP164 internal reference control. The SIM principle requires the reconstruction of final images from multiple raw images, which were illuminated with specific illumination patterns. To achieve this, the structured illumination pattern needs to have sufficient local contrast to allow proper reconstruction. Improper embedding and refractive index mismatches can decrease local contrast, which can lead to poor reconstructions.
Inadequate image acquisition and/or processing parameters may introduce unintentionally misinterpreted artifacts, including haloing, the introduction of hammerstroke, or honeycomb effects. As a general rule, the refractive index of the immersion oil, embedding medium, and the coverslip glass need to be matched, and the signal-to-noise ratio be optimized, to avoid these artifacts (Demmerle et al., 2017; Karras et al., 2019). The protocol focuses on the selection of imaging parameters. For general user guides of the OMX V4 system, the reader is referred to the relevant facility manager, quick reference guides, and the user handbook.
X,Y-Alignment with Image alignment slide and Z-Alignment with TetraSpeck beads.
Mount the image alignment slide and locate the registration pattern.
Acquire a 3 µm Z-stack in Conventional Mode at 0.125 µm step size for all channels. Set Excitation to DIC-mode, and aim exposures at similar dynamic ranges of approximately 3,000 counts maximum.
Create OMX Image Alignment in softWoRx, using the acquired image. Apply Z-corrections if applicable, and control performance of alignment by inspection of co-localization of holes in each channel. Alignment performance is commonly best in centerfield.
Replace the Image alignment slide with a TetraSpeck-beads slide (0.1 µm) mounted with immersion oil of appropriate refractive index.
Acquire a 3 µm Z-stack in SI-mode at 0.125 µm step size using all laser lines. Aim the excitation level and exposure to achieve approximately 5000 counts in each channel.
Reconstruct the image in softWoRx.
Align the channels with OMX Align Image WITHOUT Z alignment.
Measure Chromatic correction in the reconstructed and X,Y-aligned image.
If required, shift individual channels in the Z-dimension until they line up with the same reference channel that was chosen for X,Y-alignment.
Apply Z shifts and save results.
Choose reconstructed TetraSpeck image file and Align image INCLUDING Z-alignment.
Control performance of X,Y,Z-alignment. If satisfied, Z shift values can be entered as Focus Offsets in the OMX control software and applied during acquisition.
Setting image acquisition parameters, and tuning alignment and reconstruction, using a CEP164 reference control slideThe X,Y,Z-alignment above defined corrections for the TetraSpeck beads embedded in an antifade mounting medium. The multicolor-labeled CEP164 reference control serves as an additional alignment correction tool, which detects any additional chromatic shifts that originate from the sample. In addition, it allows fine-tuning of image reconstruction parameters for avoidance of processing-related artifacts.
Carefully mount your multicolor-labeled CEP164 reference control slide with the optimal immersion oil on the stage.
Select “Sequential” imaging mode (All Z then Channel) and select SI Light Path.
Define all channels. Our standard settings for all cameras are: Med 95MHz, no binning.
Adjust acquisition parameters for each channel to achieve similar dynamic ranges, and maximum intensities in the range of 10,000 counts. Critical step: Do NOT reach saturation. The very low dark-noise in sCMOS cameras commonly results in more than sufficient signal-to-noise ratios at 3,000 counts and above. In our set-up, laser transmission is set to 100% and Gain to 1 for all channels, while signal intensity is regulated by exposure time (approximately 30 ms in our samples). We find that the best SIM reconstructions can be obtained if the signal to background ratio is at least 20:1. Importantly, correct SIM reconstruction strictly requires that NO pixel in the originally acquired image set (raw data) is saturated. Only then a correct relative scaling of all captured pixel intensity values is possible. A saturated pixel in a raw image originates from a source that has an undeterminable intensity above the saturation limit/dynamic range of the camera.
Set up an SI Experiment. Define sample thickness: for centrosomes, 4 µm is adequate. Set the step size to 0.125 µm and focus start point at middle.
Find your Region-of-interest (ROI) using Spiral Mosaic and then Go To Point tools.
Select a minimum of two examples of centrosomes with the mother centriole in parallel to the coverslip (CEP164 toroid seen from side) and two with the mother centriole perpendicular to the coverslip (toroid seen from top). For each example, use Go To Point tools, and position the CEP164 signal in the center position. Run the experiment to acquire a reference data file.
In softWoRx: open Task Builder, add reference data files from g., and enable Processing Options. Choose Processing tasks OMX SI Reconstruction with appropriate Task options parameters (Image registration, including Z-alignment parameters from 1.). These parameters are dependent on the individual 3D SIM microscope and should be determined with the help of the relevant facility managers and/or service personnel.These parameters are dependent on the individual 3D-SIM microscope and should be determined with the help of the relevant facility managers and/or service personnel.
Control the performance of the Image reconstruction (absence of processing artifacts, see above) and alignment of channels. A weak adjustment of the Wiener Filter constant (0.001 for all channels in our standard settings) can help to tune noise removal during the reconstruction. For CEP164, we expect a toroid of 400 nm diameter (distal appendages), within which under optimal conditions even individual maxima of the nine-fold symmetry may be seen. Re-adjust Z-corrections to account for chromatic aberrations.
If image reconstruction artifacts such as hatching/stripe pattern are prominent, please contact your facility manager. Too low Signal-to-Noise ratio and/or bleaching may be the cause, since they interfere with software-assisted tuning of the K0 angles. Initial angle settings are defined during instrument installation and adjusted only by trained personnel. In case of haloing, consider trying immersion oils of different refractive index. Finally, as always, imaging results depend on high signal-to-noise ratios and low unspecific staining background, all of which require empirical optimization for each new tested antibody.
Imaging of your sample
The image acquisition, alignment, and reconstruction settings optimized in the routines above will apply for the acquisition of your sample of interest, provided you use the same coverslip make and immersion oil. Center the camera on the centrosome you wish to image. As already stated, careful titration of primary antibodies and adjustment of exposure times are required to gain sufficient signal-to-noise ratio for image reconstruction. The Wiener filter may be adjusted to improve reconstruction results. mNeonGreen’s fluorescence is well conserved in this protocol, but can be boosted by staining with the mouse monoclonal antibody to mNeonGreen protein, clone 32F6 (available from Chromatek and Proteintech Group). The quantum efficiency of other commonly used fluorescent proteins (eGFP, mCherry, etc.) is too compromised by fixation and staining protocol in most cases, and demands immune-based boosting to reach sufficient signal-to-noise ratios for SIM.
Extended Image Processing
Image processing in FIJI/ImageJ
For detailed guidance in image processing, please visit manuals and discussion fora for your preferred image analysis software. We provide here some initial guidance on image analysis in ImageJ. Reconstructed and aligned images are stored at 32-bit depth by softWoRx in DeltaVision (.dv) format that comprises all relevant metadata, and can be directly imported into ImageJ and displayed as composites. Image file sizes for four channel images at 4 µm depth (4 × 33 z-sections; 1024 × 1024; 32-Bit) are about 540 MB. For qualitative and quantitative analyses, it is recommended to create a compilation (e.g., Powerpoint slide deck) of z-projections (maximum projections) of all images, to catalogue orientations and image quality. For further analysis, define a ROI comprising the centrosome and duplicate it from the original reconstructed and aligned z-stack. A 50 × 50 pixel ROI (2 × 2 × 4 µm; voxel size 0.04 × 0.04 × 0.125 µm) is about 1.3 MB for a four-channel 32-bit image, allowing data analysis on a standard computer. ImageJ routines (macros) to create and store e.g., montages, z-projections, side-views, etc., are easily recorded or coded. Document each step of your processing and analysis well, and be aware of unintended introduction of image artifacts, which may lead to misinterpretations. Standardize protocols to allow comparability.
Example analysis of centrosome region
Draw a 50 × 50 pixel box, centered on the centrosome of interest.
Duplicate the hyperstack. The nuclear counterstain is not needed in most cases, and you can restrain duplication on the centrosomal protein channels, which decreases file size. Store the hyperstack in TIFF format as a new file.
Adjust display settings for each channel to increase contrasts (minimum; maximum values for contrast range). Do not omit information. Do not over-saturate. Do not change linearity. Apply the same settings across the entire z-stack, and keep ranges for all images in one series.
Using the Orthogonal Views tool, you can concomitantly inspect individual sections projections.
Analyze multiple centrosomes of each series in both parallel or perpendicular position in respect to the coverslip, to gain maximal spatial information (lateral resolution >> axial resolution) and to familiarize yourself with your samples. Compare centrosomes of equal cell cycle phase, to control for staining artifacts.
NOTE: For visualization (not analysis!) purposes, you may want to resize the image with constraint aspect ratios by a factor of three, while including an interpolation of pixel intensities. Do not change image depth (number of sections). Bilinear and Bicubic interpolation are common tools used for digital magnification of images, without changing the original pixel size in the image. Therefore, it is important to remember that both algorithms create new pixels, to fill the distances between pixels of the original image. Simplified: the bilinear interpolation algorithm determines pixel intensities of new “fill”-pixels on the basis of the intensities of neighboring original pixels (2 × 2 matrix), calculating values on the assumption of a linear change between these. The bicubic interpolation algorithm calculates intensities of new “fill”-pixels on basis of a 4 × 4 pixel matrix, weighting pixel intensities by a non-linear polynomial interpolation. Hence, both scaling algorithms create new image information, and thus must be clearly stated in a publication. Bicubic scaling may result in smoother images, but will, due to its non-linearity, create misleading artifacts and should be avoided (see Figure 1B).
Data analysis
The power of our 3D-SIM protocol is exemplified in two published studies on human ciliopathy associated proteins (Frikstad et al., 2019; Gonçalves et al., 2021). In the first study, we generated hTERT-RPE1 cells stably expressing mNeonGreen-fusion proteins of CSPP1 and CEP104, respectively. To avoid over-expression artifacts, we used fluorescence assisted cell sorting (FACS) to select populations of cells that express the fusion protein at near-endogenous level, and eventually created monoclonal cell lines from these. Following the 3D-SIM protocol, we resolved the localization of CSPP1 and CEP104 at the distal end of the mother centriole, which are spatially separated based on their relative localization to CEP164 and γ-tubulin. Investigation of ciliated hTERT-RPE1 cells nicely resolved the axoneme tip localization of both proteins, enclosed within a discernable lumen of the ciliary membrane, and visualized by staining for the GTPase ARL13B (Proteintech, #17711-1-AP) (Frikstad et al., 2019).
In the more recent study (Gonçalves et al., 2021), we investigated the localization of CEP78 and its vision- and hearing-loss causative mutant form CEP78L150S using the same strategy. Notably, the CEP78L150S mutant lost its association with the centrosome. The interaction of CEP78 with CEP350 at the mother centriole of the centrosome is key to activation of the ubiquitin-ligase complex that facilitates dissolution of the CP110 containing capping complex, which ultimately permits axoneme extension and cilia formation. Our data analysis, exemplified in Figure 1, placed CEP78 centrally to CEP350 and sub-distally to CEP164 and CP110, which is in excellent agreement with an earlier immunogold electron microscopy study that reported CEP78 at the centriole wall in HeLa cells (Brunk et al., 2016). Thus, we superimposed our 3D-SIM data on scaled previously published TEM micrographs of centrosomes (Paintrand et al., 1992). Strikingly, overlay of orthogonal or vertical projections of CEP164 (distal appendages) and CP110 (capping complex) of our 3D-SIM images perfectly aligned on the structural features in respectively oriented TEM micrographs, without any additional digital scaling adjustments. These results indicate a very good structural preservation of centrioles in our protocol, and resolved the localization of CEP78 to the distal half of the centriole microtubule triplets, where it is partially enclosed by CEP350.
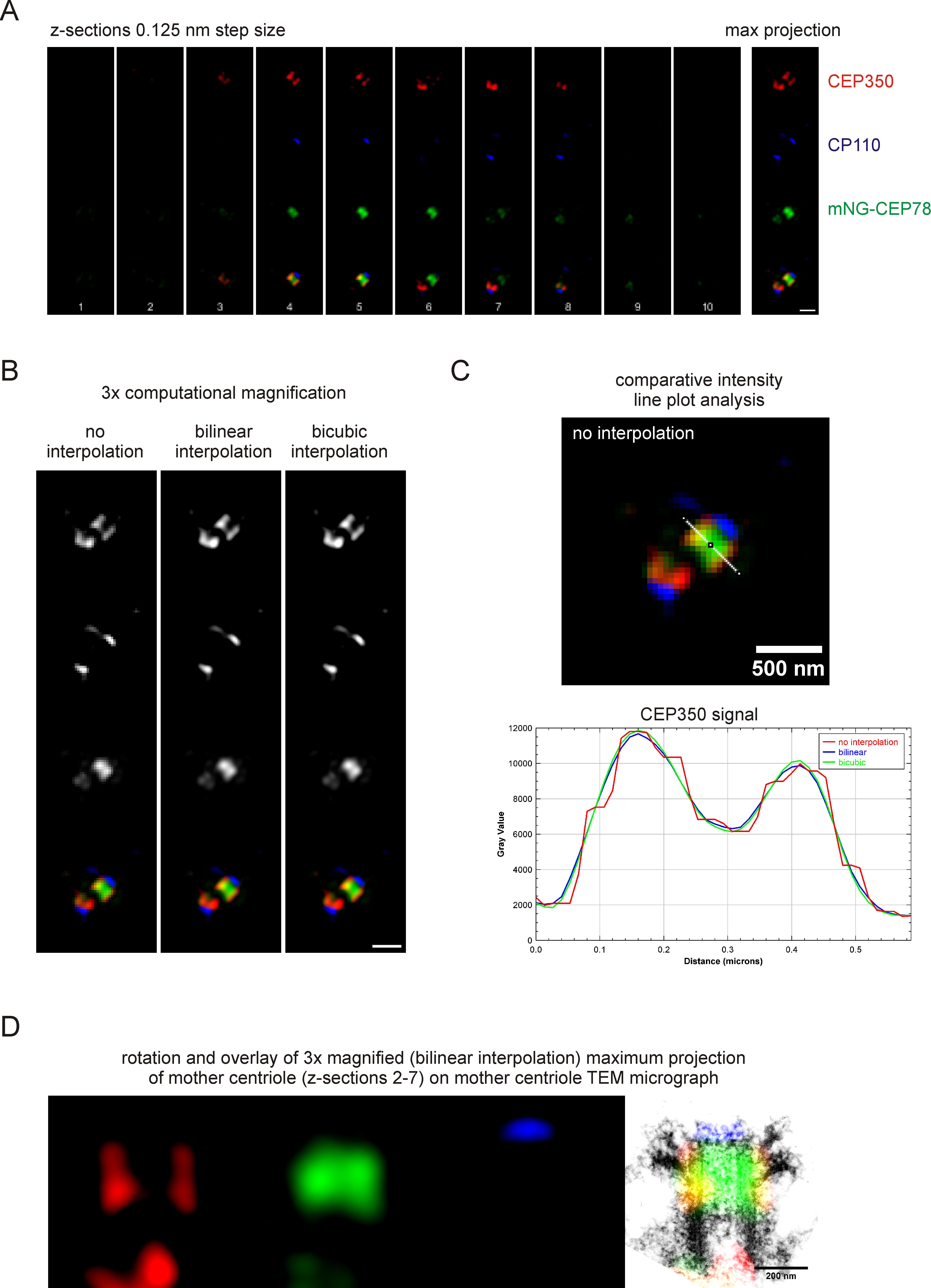
Figure 1. Example analysis of 3D-SIM images reported in Gonçalves et al. (2021). (A) Single channel 3D-SIM, and overlay images of individual z-sections through the centrosome area (50×50 pixels), as well as a maximum z-projection. (B) 3× computational magnification of 50 × 50 pixel area with no, linear, or bicubic interpolation, and maximum projection thereof. (C) Comparative line plot analysis of CEP350 signal along indicated line on micrographs of either re-sized image (example image: no interpolation). Interpolation smoothens the image but also changes signal intensities. This excludes further quantitative analysis and must be carefully evaluated to avoid unintended biological misinterpretations. (D) Projection of 3× magnified, bilinear interpolated 3D-SIM image on electron microscopy micrograph of a mother centriole (from Paintrand et al., 1992) as presented as part of Figure 4A in Gonçalves et al. (2021) .
Recipes
PBS-AT
1% Bovine Serum Albumin (BSA) w/v
0.5% Triton-X100 v/v
Dulbecco’s Phosphate Buffered Saline (PBS)
Mix together and store in 50 mL aliquots. For short term storage, keep in the fridge at 4°C. For long term storage, freeze at -20°C and thaw prior to use.
1.6% PFA solution
2 volumes of 10% Formalin solution + 3 volumes of PBS
Mix together prior to fixation.
For 12 mL:
4.8 mL of 10% Formalin solution, neutral buffered
7.2 mL of Dulbecco’s Phosphate Buffered Saline (PBS)
Acknowledgments
Sebastian Patzke acknowledges funding support by the Norwegian Cancer Society, Career Development grant (6839316). Lotte B. Pedersen acknowledges funding from Independent Research Fund Denmark (8020-00162B) and from the Carlsberg Foundation (CF18-0294). Kay O. Schink was supported by a career development grant from the Norwegian Cancer Society, a Career grant from the South-Eastern Norway Regional Health Authority (2020038), and a Research Grant from the Research Council of Norway (315103).
This protocol was applied in Gonçalves et al. (2021) and Frikstad et al. (2019).
Competing interests
The authors declare that they have no competing interests.
References
- Adams, S. D., Csere, J., D'Angelo, G., Carter, E. P., Romao, M., Arnandis, T., Dodel, M., Kocher, H. M., Grose, R., Raposo, G., et al. (2021). Centrosome amplification mediates small extracellular vesicle secretion via lysosome disruption. Curr Biol 31(7): 1403-1416.e1407.
- Andersen, J. S., Wilkinson, C. J., Mayor, T., Mortensen, P., Nigg, E. A. and Mann, M. (2003). Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426(6966): 570-574.
- Anvarian, Z., Mykytyn, K., Mukhopadhyay, S., Pedersen, L. B. and Christensen, S. T. (2019). Cellular signalling by primary cilia in development, organ function and disease. Nat Rev Nephrol 15(4): 199-219.
- Arslanhan, M. D., Gulensoy, D. and Firat-Karalar, E. N. (2020). A Proximity Mapping Journey into the Biology of the Mammalian Centrosome/Cilium Complex. Cells 9(6): 1390.
- Best, S., Lord, J., Roche, M., Watson, C. M., Poulter, J. A., Bevers, R. P. J., Stuckey, A., Szymanska, K., Ellingford, J. M., Carmichael, J., et al. (2021). Molecular diagnoses in the congenital malformations caused by ciliopathies cohort of the 100,000 Genomes Project. J Med Genet. doi: 10.1136/jmedgenet-2021-108065.
- Boldt, K., van, R. J., Lu, Q., Koutroumpas, K., Nguyen, T. M., Texier, Y., van Beersum, S. E., Horn, N., Willer, J. R., Mans, D. A. et al. (2016). An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat Commun 7: 11491. doi: 10.1038/ncomms11491.
- Boveri, T. (1914). Zur Frage der Entstehung maligner Tumoren. Fischer. Jena.
- Bowler, M., Kong, D., Sun, S., Nanjundappa, R., Evans, L., Farmer, V., Holland, A., Mahjoub, M. R., Sui, H. and Loncarek, J. (2019). High-resolution characterization of centriole distal appendage morphology and dynamics by correlative STORM and electron microscopy. Nat Commun 10(1): 993.
- Breslow, D. K. and Holland, A. J. (2019). Mechanism and Regulation of Centriole and Cilium Biogenesis. Annu Rev Biochem 88: 691-724.
- Brunk, K., Zhu, M., Bärenz, F., Kratz, A. S., Haselmann-Weiss, U., Antony, C. and Hoffmann, I. (2016). Cep78 is a new centriolar protein involved in Plk4-induced centriole overduplication. J Cell Sci 129(14): 2713-2718.
- Chong, W. M., Wang, W. J., Lo, C. H., Chiu, T. Y., Chang, T. J., Liu, Y. P., Tanos, B., Mazo, G., Tsou, M. B., Jane, W. N., et al. (2020). Super-resolution microscopy reveals coupling between mammalian centriole subdistal appendages and distal appendages. Elife 9: e53580.
- Damstra, H. G. J., Mohar, B., Eddison, M., Akhmanova, A., Kapitein, L. C. and Tillberg, P. W. (2021). Visualizing cellular and tissue ultrastructure using Ten-fold Robust Expansion Microscopy (TREx). bioRxiv: 2021.2002.2003.428837.
- Demmerle, J., Innocent, C., North, A. J., Ball, G., Müller, M., Miron, E., Matsuda, A., Dobbie, I. M., Markaki, Y. and Schermelleh, L. (2017). Strategic and practical guidelines for successful structured illumination microscopy. Nat Protoc 12(5): 988-1010.
- Firat-Karalar, E. N., Rauniyar, N., Yates, J. R., III and Stearns, T. (2014). Proximity Interactions among Centrosome Components Identify Regulators of Centriole Duplication. Curr Biol 24(6): 664-670.
- Frikstad, K. M., Molinari, E., Thoresen, M., Ramsbottom, S. A., Hughes, F., Letteboer, S. J. F., Gilani, S., Schink, K. O., Stokke, T., Geimer, S., et al. (2019). A CEP104-CSPP1 Complex Is Required for Formation of Primary Cilia Competent in Hedgehog Signaling. Cell Rep 28(7): 1907-1922.
- Gartenmann, L., Wainman, A., Qurashi, M., Kaufmann, R., Schubert, S., Raff, J. W. and Dobbie, I. M. (2017). A combined 3D-SIM/SMLM approach allows centriole proteins to be localized with a precision of ∼4-5 nm. Curr Biol 27(19): R1054-R1055.
- Gheiratmand, L., Coyaud, E., Gupta, G. D., Laurent, E. M., Hasegan, M., Prosser, S. L., Goncalves, J., Raught, B. and Pelletier, L. (2019). Spatial and proteomic profiling reveals centrosome-independent features of centriolar satellites. EMBO J 38(14): e101109.
- Godinho, S. A. and Pellman, D. (2014). Causes and consequences of centrosome abnormalities in cancer. Philos Trans R Soc Lond B Biol Sci 369(1650): 20130467.
- Gomes Pereira, S., Dias Louro, M. A. and Bettencourt-Dias, M. (2021). Biophysical and Quantitative Principles of Centrosome Biogenesis and Structure. Annu Rev Cell Dev Biol 37: 43-63.
- Gonçalves, A. B., Hasselbalch, S. K., Joensen, B. B., Patzke, S., Martens, P., Ohlsen, S. K., Quinodoz, M., Nikopoulos, K., Suleiman, R., Damsø Jeppesen, M. P., et al. (2021). CEP78 functions downstream of CEP350 to control biogenesis of primary cilia by negatively regulating CP110 levels. Elife 10: e63731.
- Goundiam, O. and Basto, R. (2021). Centrosomes in disease: how the same music can sound so different? Curr Opin Struct Biol 66: 74-82.
- Gupta, G. D., Coyaud, E., Goncalves, J., Mojarad, B. A., Liu, Y., Wu, Q., Gheiratmand, L., Comartin, D., Tkach, J. M., Cheung, S. W. et al. (2015). A Dynamic Protein Interaction Landscape of the Human Centrosome-Cilium Interface. Cell 163(6): 1484-1499.
- Jakobsen, L., Vanselow, K., Skogs, M., Toyoda, Y., Lundberg, E., Poser, I., Falkenby, L. G., Bennetzen, M., Westendorf, J., Nigg, E. A. (2011). Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO J 30(8): 1520-1535. PM:21399614
- Karras, C., Smedh, M., Förster, R., Deschout, H., Fernandez-Rodriguez, J. and Heintzmann, R. (2019). Successful optimization of reconstruction parameters in structured illumination microscopy – A practical guide. Optics Communications 436: 69-75.
- Lau, L., Lee, Y. L., Sahl, S. J., Stearns, T. and Moerner, W. E. (2012). STED microscopy with optimized labeling density reveals 9-fold arrangement of a centriole protein. Biophys J 102(12): 2926-2935.
- LeGuennec, M., Klena, N., Aeschlimann, G., Hamel, V. and Guichard, P. (2021). Overview of the centriole architecture. Curr Opin Struct Biol 66: 58-65.
- Liu, Z., Nguyen, Q. P. H., Nanjundappa, R., Delgehyr, N., Megherbi, A., Doherty, R., Thompson, J., Jackson, C., Albulescu, A., Heng, Y. M. et al. (2020). Super-Resolution Microscopy and FIB-SEM Imaging Reveal Parental Centriole-Derived, Hybrid Cilium in Mammalian Multiciliated Cells. Dev Cell 55(2): 224-236.e226.
- Mariappan, A., Soni, K., Schorpp, K., Zhao, F., Minakar, A., Zheng, X., Mandad, S., Macheleidt, I., Ramani, A., Kubelka, T. et al. (2019). Inhibition of CPAP-tubulin interaction prevents proliferation of centrosome-amplified cancer cells. EMBO j 38(2): e99876.
- Mick, D. U., Rodrigues, R. B., Leib, R. D., Adams, C. M., Chien, A. S., Gygi, S. P. and Nachury, M. V. (2015). Proteomics of Primary Cilia by Proximity Labeling. Dev Cell 35(4): 497-512.
- Nigg, E. A. and Holland, A. J. (2018). Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nat Rev Mol Cell Biol 19(5): 297-312.
- Paintrand, M., Moudjou, M., Delacroix, H. and Bornens, M. (1992). Centrosome organization and centriole architecture: their sensitivity to divalent cations. J Struct Biol 108(2): 107-128.
- Pedersen, L. B., Veland, I. R., Schroder, J. M. and Christensen, S. T. (2008). Assembly of primary cilia. Dev Dyn 237(8): 1993-2006.
- Reiter, J. F. and Leroux, M. R. (2017). Genes and molecular pathways underpinning ciliopathies.Nat Rev Mol Cell Biol 18(9): 533-547.
- Sabat-Pośpiech, D., Fabian-Kolpanowicz, K., Prior, I. A., Coulson, J. M. and Fielding, A. B. (2019). Targeting centrosome amplification, an Achilles' heel of cancer. Biochem Soc Trans 47(5): 1209-1222.
- Shakya, S. and Westlake, C. J. (2021). Recent advances in understanding assembly of the primary cilium membrane. Fac Rev 10: 16.
- Sieben, C., Douglass, K. M., Guichard, P. and Manley, S. (2018). Super-resolution microscopy to decipher multi-molecular assemblies. Curr Opin Struct Biol 49: 169-176.
- Smedley, D., Smith, K. R., Martin, A., Thomas, E. A., McDonagh, E. M., Cipriani, V., Ellingford, J. M., Arno, G., Tucci, A., Vandrovcova, J. et al. (2021). 100,000 Genomes Pilot on Rare-Disease Diagnosis in Health Care - Preliminary Report. N Engl J Med 385(20): 1868-1880.
- Sonnen, K. F., Schermelleh, L., Leonhardt, H. and Nigg, E. A. (2012). 3D-structured illumination microscopy provides novel insight into architecture of human centrosomes. Biol Open 1(10): 965-976.
- Wheway, G., Schmidts, M., Mans, D. A., Szymanska, K., Nguyen, T. M., Racher, H., Phelps, I. G., Toedt, G., Kennedy, J., Wunderlich, K. A. et al. (2015). An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. NatCell Biol 17(8): 1074-1087.
- Yang, T. T., Chong, W. M., Wang, W. J., Mazo, G., Tanos, B., Chen, Z., Tran, T. M. N., Chen, Y. D., Weng, R. R., Huang, C. E., et al. (2018). Super-resolution architecture of mammalian centriole distal appendages reveals distinct blade and matrix functional components. Nat Commun 9(1): 2023.
- Zwettler, F. U., Reinhard, S., Gambarotto, D., Bell, T. D. M., Hamel, V., Guichard, P. and Sauer, M. (2020). Molecular resolution imaging by post-labeling expansion single-molecule localization microscopy (Ex-SMLM). Nat Commun 11(1): 3388.
Article Information
Copyright
![]() Frikstad et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Frikstad et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Frikstad, K. M., Schink, K. O., Gilani, S., Pedersen, L. B. and Patzke, S. (2022). 3D-Structured Illumination Microscopy of Centrosomes in Human Cell Lines. Bio-protocol 12(6): e4360. DOI: 10.21769/BioProtoc.4360.
- Gonçalves, A. B., Hasselbalch, S. K., Joensen, B. B., Patzke, S., Martens, P., Ohlsen, S. K., Quinodoz, M., Nikopoulos, K., Suleiman, R., Damsø Jeppesen, M. P., et al. (2021). CEP78 functions downstream of CEP350 to control biogenesis of primary cilia by negatively regulating CP110 levels. Elife 10: e63731.
Category
Developmental Biology > Cell growth and fate
Cancer Biology > General technique > Molecular biology technique
Cell Biology > Cell imaging > Super resolution imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.