- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Labeling Endogenous Proteins Using CRISPR-mediated Insertion of Exon (CRISPIE)
Published: Vol 12, Iss 5, Mar 5, 2022 DOI: 10.21769/BioProtoc.4343 Views: 8910
Reviewed by: Gal HaimovichQin TangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2021
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The CRISPR/Cas9 technology has transformed our ability to edit eukaryotic genomes. Despite this breakthrough, it remains challenging to precisely knock-in large DNA sequences, such as those encoding a fluorescent protein, for labeling or modifying a target protein in post-mitotic cells. Previous efforts focusing on sequence insertion to the protein coding sequence often suffer from insertions/deletions (INDELs) resulting from the efficient non-homologous end joining pathway (NHEJ). To overcome this limitation, we have developed CRISPR-mediated insertion of exon (CRISPIE). CRISPIE circumvents INDELs and other editing errors by inserting a designer exon flanked by adjacent intron sequences into an appropriate intronic location of the targeted gene. Because INDELs at the insertion junction can be spliced out, “CRISPIEd” genes produce precisely edited mRNA transcripts that are virtually error-free. In part due to the elimination of INDELs, high-efficiency labeling can be achieved in vivo. CRISPIE is compatible with both N- and C-terminal labels, and with all common transfection methods. Importantly, CRISPIE allows for later removal of the protein modification by including exogenous single-guide RNA (sgRNA) sites in the intronic region of the donor module. This protocol provides the detailed CRISPIE methodology, using endogenous labeling of β-actin in human U-2 OS cells with enhanced green fluorescent protein (EGFP) as an example. When combined with the appropriate gene delivery methods, the same methodology can be applied to label post-mitotic neurons in culture and in vivo. This methodology can also be readily adapted for use in other gene editing contexts.
Keywords: CRISPR/Cas9Background
Epitope tagging is widely used to label or modify a protein of interest to study its function. However, such tagging has been traditionally done via overexpression of the tagged protein from an exogenous plasmid. Protein overexpression often causes undesirable side effects, such as altered protein localization, stoichiometry, and cell physiology. To epitope-tag proteins at their endogenous levels, mouse conditional knock-in methods have been developed (e.g., Fortin et al., 2014; Melander et al., 2021). However, the cost and inflexibility of mouse genetics have limited their broad application. Knock-ins in other species also remain difficult.
To overcome these issues, recent efforts have employed the emerging CRISPR/Cas9 technology to introduce protein epitope tags. However, while successful labeling can be achieved in a fraction of cells, the previous methods have limitations. Although methods utilizing the homology-directed repair (HDR) mechanism hold the promise of precise DNA insertions (Nishiyama et al., 2017; Roberts et al., 2017), HDR is inefficient in post-mitotic cells, and is still sensitive to INDEL formation resulting from the competing non-homologous end joining (NHEJ) pathway. On the other hand, while NHEJ can achieve high repair efficiency across diverse cell types, INDEL formation at the repaired junction of double-strand breaks (DSBs) poses a significant challenge because INDELs in protein coding sequences can cause frameshift and nonsense mutations. Although this can be overcome by selecting cells with the correct editing events where growing/cloning is appropriate, its application in post-mitotic cells, such as neurons, can be problematic.
To circumvent the negative impacts of INDEL formation in exons, CRISPIE targets intronic sequences (Zhong et al., 2021). The desired epitope tag is included in a designer exon module, flanked by intronic sequences at both ends of the exon. When this donor module is inserted into the intronic targeting site, the epitope tag is included in the endogenous transcript via RNA splicing. The same splicing event also eliminates INDEL events at the junction of insertion, except for the low-probability event that the INDEL is very big and it interferes with RNA splicing. Therefore, the CRISPIE process results in epitope tagging of endogenous proteins that is nearly error-free at the mRNA level, and thus at the protein level.
Compared to previous NHEJ-based epitope insertion methods, the main differences of CRISPIE are that the donor module is a functional exon, and the targeting site is within the intron. We have also empirically found that different intronic locations give highly heterogeneous labeling efficiency. While future bioinformatic approaches could better predict labeling efficiencies, it is currently necessary to screen several editing locations within a given intron. We suggest targeting at least 100 bp away from the intron-exon junction, to avoid potential interference with the splicing of endogenous exons. When optimized, we can achieve ~40% labeling efficiency in vivo, corresponding to ~22% per allele in a diploid genome.
Here, we detail a step-by-step protocol for achieving CRISPIE labeling of human β-actin (ACTB) at its N-terminus in human U-2 OS cells. Labeling of other genes can be similarly carried out using gene-specific sgRNA targeting sequences and the appropriate donors. Although not described, labeling in post-mitotic neurons follows an essentially identical procedure barring the transfection method. This protocol should be readily adapted for other proteins, cell types, and epitope tags.
Materials and Reagents
10 cm surface treated tissue culture dishes (FisherbrandTM, catalog number: FB012924)
1.7 mL microcentrifuge tube (Thermo Scientific, catalog number: 21-402-903)
35 mm cell culture dishes (FalconTM, catalog number: 353001)
Sterile polystyrene disposable serological pipets (FisherbrandTM, catalog numbers: 13-678-11 [25 mL]; 13-678-11E [10 mL]; 13-678-11D [5 mL])
Disposable borosilicate glass Pasteur pipets (FisherbrandTM, catalog number: 13-678-20C)
Human U-2 OS cells (ATCC, catalog number: HTB-96)
pX330-U6-Chimeric_BB-CBh-hSpCas9 (Addgene, catalog number: 42230)
pKanCMV_mRuby3 (Addgene, catalog number: 172854)
Lipofectamine 2000 (Invitrogen, catalog number: 11668019)
McCoy's 5A medium (ATCC, catalog number: 30-2007)
Opti-MEM (Gibco, catalog number: 31985062)
Trypsin/EDTA (Gibco, catalog number: 25300062)
Fetal bovine serum (FBS) (Hyclone, catalog number: 26140079)
70% ethanol in deionized water
BbsI-HF (New England Biolabs, catalog number: R3539S)
Poly nucleotide kinase (PNK) (New England Biolabs, catalog number: M0201S)
Calf Intestinal Phosphatase (CIP) (New England Biolabs, catalog number: M0290S)
10× Cut Smart Buffer (New England Biolabs, catalog number: B6004S)
T4 DNA ligase buffer (New England Biolabs, catalog number: B0202S)
Quick Ligation Kit (New England Biolabs, catalog number: M2200S)
Molecular grade water (HyClone, catalog number: SH30538.03)
Carbenicillin (Sigma, catalog number: C1389-5G)
Ampicillin (Sigma, catalog number: A9518-25G)
Kanamycin (Sigma, catalog number: 11815024)
DH5α competent cells (ThermoFisher Scientific, catalog number: 18265017)
Luria-Bertani (LB) agar capsules (for making plates) (Sigma, catalog number: 113002231)
Kanamycin LB agar plates (30 μg/mL; made in-house)
Ampicillin LB agar plates (50 μg/mL; made in-house)
Tryptone (Fisher Scientific, catalog number: BP9726-500)
Sodium chloride (Thermo Scientific, catalog number: J2161836)
Yeast extract (Gibco, catalog number: 211929)
Terrific Broth (TB) powder (Sigma, catalog number: 1016290500)
Glycerol (Sigma, catalog number: G5516-500ML)
Glass beads (Fisher Scientific, catalog number: 11-312C)
Ice bucket
QiaQuick Purification Kit (Qiagen, catalog number: 28706)
Qiagen Miniprep Kit (Qiagen, catalog number: 27104)
SOC Medium (see Recipes)
TB Medium (see Recipes)
McCoy’s 5A Medium supplemented with FBS (see Recipes)
Equipment
Water baths set to 37°C and 42°C, respectively
Tissue culture incubator
Bacterial incubator
Ice machine
Vortex (Fisher)
Pipetaid (Drummond)
Biological safety cabinet
Vacuum source
Vacuum manifold (Vac-ManTM Vacuum Manifold, Promega, catalog number: A7231)
Thermocycler
Tabletop microcentrifuge
Fluorescence microscope
Software
SnapGene (SnapGene software; https://snapgene.com)
CRISPick (Broad Institute, https://portals.broadinstitute.org/gppx/crispick/public)
Procedure
Cas9 sgRNA design using CRISPick
The first CRISPIE plasmid needed is a Cas9 expression plasmid that also expresses the target-specific sgRNA. This allows the targeted genomic location to be specifically cut by Cas9. After designing the sgRNA in silico, it will be incorporated into the blank Cas9-sgRNA expression vector pX330 (Addgene #42230).
To select an sgRNA targeting sequence, we use Broad Institute CRISPick (https://portals.broadinstitute.org/gppx/crispick/public; note that other sgRNA design websites/software may also work).
On the starting page, select the desired reference genome, CRISPR mechanism, and origin species of the Cas9 enzyme (Figure 1).
Note: In our protocol, we use the ‘’Human GRCh38’ reference genome, CRISPRko, and SpyoCas9.
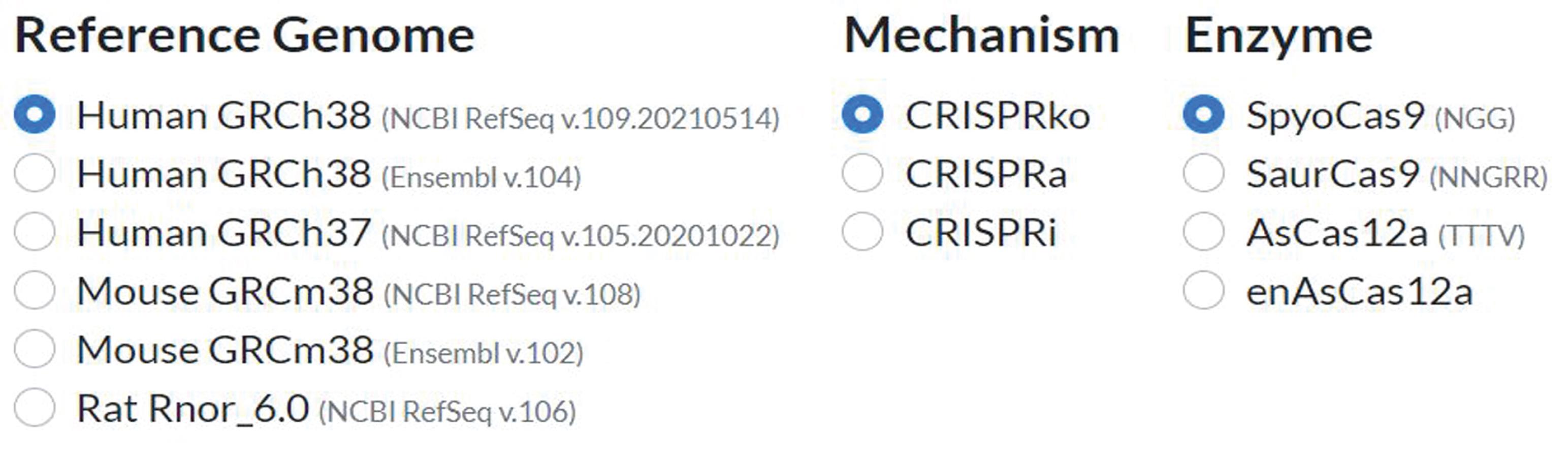
Figure 1. Specifying CRISPick parameters used in CRISPIE.Click ‘bulk mode’ and paste the target sequence into the ‘Target(s)’ box, or reference the target using the accepted target formats listed on the website.
Bulk mode allows one to paste in large/multiple target sequences (Figure 2).

Figure 2. Pasting target sequence (FASTA format) into the bulk mode input box.We recommend creating SnapGene files containing the target sequence with annotated sgRNA binding sites. This is especially useful when designing multiple sgRNAs to screen, and when analyzing the final insertion sequences.
Include an additional ~200 bp of sequence to both the 5’ and 3’ end of the target sequence, to ensure all sgRNA options are considered.
A target sequence needs to be located within the intron of a gene. The inherent advantage of CRISPIE is that INDELs at the insertion junction within the intronic region are spliced out, which is not possible when the INDELs are located within an exon. When targeting introns of a gene, it is important to choose an intron that won’t disrupt the structure of a protein after a donor module is introduced. N- and C-terminal labels are incorporated into the first or last intron of a gene, respectively. If a large stretch of coding sequence is present in the first exon of a gene, CRISPIE would have difficulty tagging its N terminus. Targeting within the gene or at the C terminus can be used as alternatives. In addition, in order to label a gene of interest at the very C-terminus, the coding sequence of the last exon of a gene needs to be included in the donor module. These are the constraints that need to be considered using CRISPIE. However, the sequence that needs to be included for CRISPIE is typically much shorter than those for HDR-based insertions.
Select the desired ‘CRISPick Quota’.
Note: If one wishes to manually select the desired sgRNAs from the output, skip this step. Otherwise, this tool uses an algorithm to recommend the top sgRNAs based on the standard criteria, such as off target effects, cut positions, etc.
Check the box ‘report unpicked sequences?’ (Figure 3).

Figure 3. Specifying the number of potential sgRNA sites by setting the CRISPick Quota. Check “Report unpicked sequences” in order to have a file of all potential sgRNAs and their characteristics (as determined by the CRISPick algorithm).After the output is displayed, download the desired files:
‘Picking Results’ displays the best sgRNAs according to the ‘CRISPick Quota’ algorithm.
‘Picking Summary’ provides a comprehensive list of possible sgRNAs in a table format with relevant sgRNA information.
‘Input Target Sequence’ contains the input file you used, for your records.
Note that if the selected sequence will be incorporated into the donor plasmid (for excising the module so it can be inserted into the targeted genomic region), it may be desirable to include the ‘context sequence’ (also listed in the Picking Results). The context sequence is the set of sequences surrounding the sgRNA-targeted sequence that may affect Cas9 binding and the endonuclease activity at the target sequence (Figure 4).
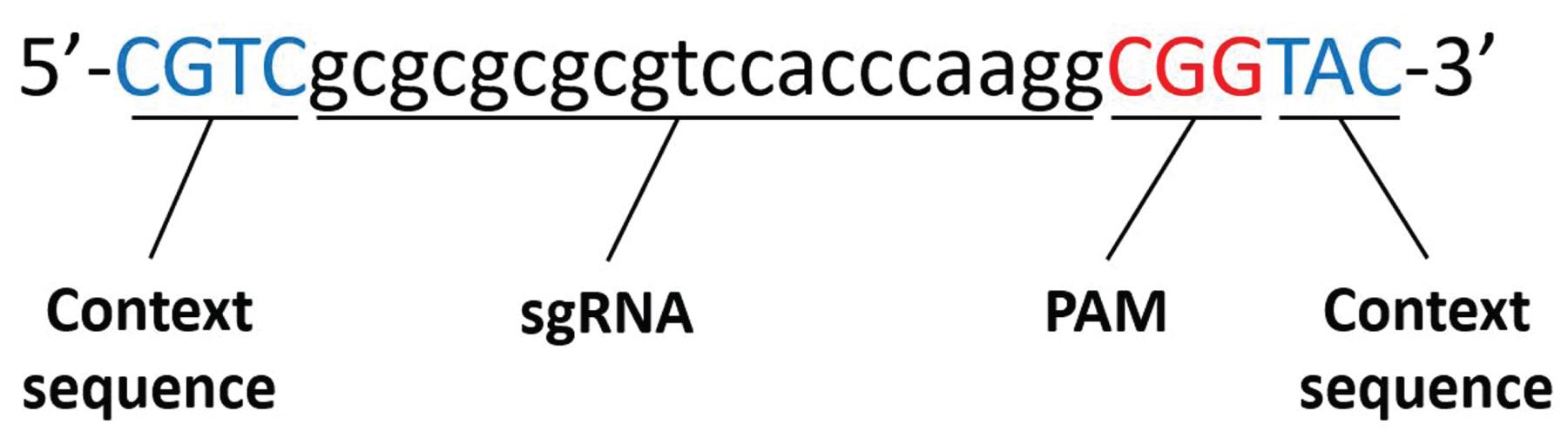
Figure 4. An example sgRNA target sequence including its context sequence. We have empirically found that the specific context sequences shown in the figure give high Azimuth scores based on CRISPick.Using a DNA design software (we use SnapGene), create a file with the desired gene of interest to be edited using CRISPIE. Annotate the target sites in the target gene.
In silico construction of Cas9-containing plasmid
Select the sgRNA sequence chosen from CRISPick (or other sgRNA design software).
To incorporate the sgRNA into the pX330 plasmid via the built-in BbsI site, add the 4 bp sequence: CACC to the 5’ end of the Forward oligo and AAAC to the 5’ end of the Reverse oligo.
This is necessary to create compatible overhangs for directional cloning of the oligos into the BbsI-digested pX330 vector.
Additionally, if the sgRNA does not start with a G, add a G at the 5’ end of the sgRNA, which is required for transcription initiation by the U6 promotor. An example is provided below for clarity (Figure 5B).
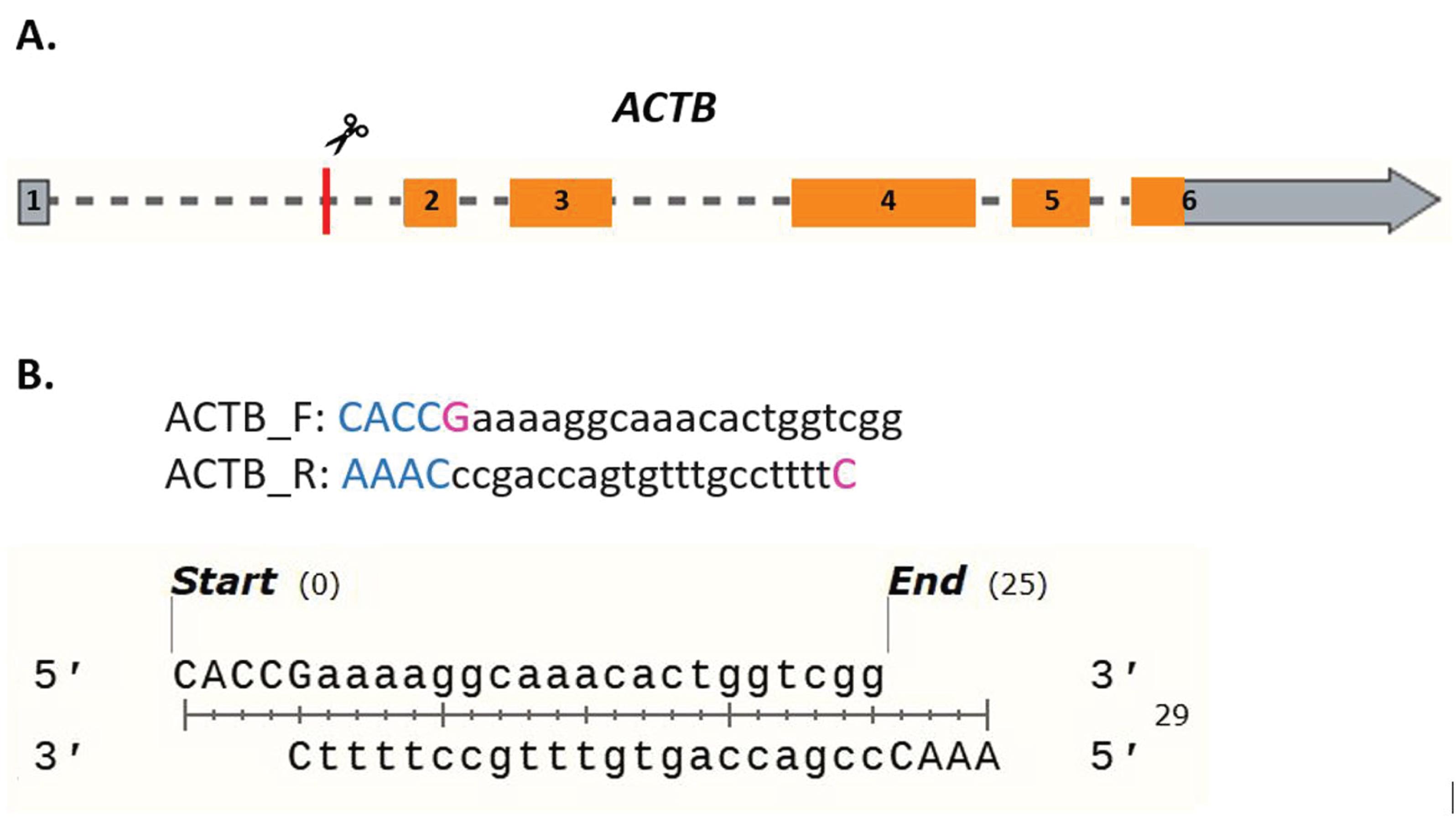
Figure 5. In silico design of sgRNAs. (A) Schematic of the human β-actin gene with exons numbered 1–6. The red bar indicates the approximate targeting site of the CRISPIE sgRNA. The coding sequence of ACTB is highlighted in orange. (B) shows both the forward and reverse primer sequences for cloning the sgRNA plasmid targeting intron 1 of human ACTB. The sgRNA sequences (black, lower-case letters), adjacent sequences for ligating into the target vector (blue capitalized letters), and the transcription initiating G (magenta) are differentially highlighted. The lower panel shows the annealed primers, which will be ligated into the pX330 vector.
Cloning the sgRNA sequence into pX330
Digestion, dephosphorylation, and purification of the vector
Digest pX330 (Addgene #42230) in a 25 μL reaction at 37°C for 3 h or overnight:
pX330 ~3 μg 10× NEB Cut Smart buffer 2.5 μL BbsI-HF 1 μL (20 units) Molecular-grade water Bring to 25 μL After digestion, add 0.6 μL of calf intestinal phosphatase (CIP) and incubate at 37°C for 30 min.
Purify the digested plasmid. We typically use the Qiaquick purification kit.
Note: We recommend the HF version of BbsI. The non-HF version may lose potency over time if stored at -20°C.
Phosphorylation and annealing of sgRNA oligos
Set up the following reaction in a PCR tube, so the polynucleotide kinase (PNK) reaction and annealing can both be performed in one step in a thermocycler:
Forward sgRNA Oligo (100 µM stock) 1 μL Reverse sgRNA Oligo (100 µM stock) 1 μL 10× T4 DNA Ligase Buffer 2 μL Molecular-grade water 15.5 μL PNK 0.6 μL Total 20 μL Carry out the following reaction protocol:
Cycle Notes 37°C for 30 min PNK reaction 95°C for 5.5 min Inactivate PNK and denature oligos -1°C every 30 s for 70 cycles Anneal oligos Hold at 4°C Dilute 2 μL of the annealing reaction in 198 μL of molecular-grade water for use in the ligation.
Ligation of annealed oligos into the digested pX330 vector
Using the NEB Quick Ligation kit, create the following reactions and incubate at room temperature for 25 min:
Vector + Ligase Control Insert + Vector NEB 2× Quick Ligase Buffer 5.5 μL 5.5 μL Digested, dephosphorylated pX330 1.2 μL 1.2 μL 1:100 diluted annealing reaction – 3.8 µL Molecular-grade water 3.8 µL – NEB Quick Ligase 0.5 μL 0.5 μL Transformation and amplification of DH5α cells with ligated plasmid using standard protocols.
We use home-made DH5α competent cells. With all tubes pre-chilled on ice, add 1–2 μL of ligation reaction (above) to 30 μL of DH5α cells. Leave on ice 20–30 min. Incubate tubes in a water bath at 42°C for exactly 1 min, then immediately place on ice for 5 min. Add 300 μL of SOC medium and incubate the tubes at 37°C shaking at 250 rpm for 1 h. Plate ~200 μL of the transformation reaction on LB agar plates (50 μg/mL Carbenicillin) and spread with glass beads. Incubate overnight at 37°C.
Select colonies to be amplified using the desired miniprep technique (e.g., Qiagen Miniprep Kit).
Sequence the miniprepped plasmid to check for the proper insertion of the sgRNA. Our lab uses Azenta Life Sciences (previously Genewiz) sanger sequencing services.
Selecting CRISPIE Donor Plasmid and Transfection Marker
The choice of which CRISPIE donor to select from the Addgene repository depends on the translational reading frame of the exons flanking the targeted intron. RNA splicing can occur both between and within individual codons of a gene. Exons have splice sites that can be located before the first, second, or third base of a codon (notated as 0, 1, and 2, respectively; Figure 6).

Figure 6. Reading frame of exons. The three possible splice junctions in terms of translational phases between two exons are shown. Each triplet codon is underlined, with the dotted underline representing a codon spanning across an intron.It is important to select a donor module that matches the reading frame of the inserted intron. Depending on the target site, one would need either a 0-0, 1-1, or 2-2 translational phase donor exon. In the case of ACTB, the start codon resides on exon 2 in the first (0) reading frame. Therefore, the 0-0 donor module is the desired plasmid for intron 1 of ACTB.
Addgene #172848 (Donor B7) contains a mEGFP with 0-0 translational phase
Addgene #172849 (Donor B8) contains a mEGFP with 1-1 translational phase
Addgene #172850 (Donor B9) contains a mEGFP with 2-2 translational phase
The donor plasmids listed above are self-excising plasmids: they express a designed sgRNA whose target site flanks the donor module within the same plasmid. However, we have empirically found that a donor module flanked by sgRNA sites identical to the genomic editing site, but positioned in an inverse orientation, gives a ~50–100% higher labeling efficiency compared to the generic donors. Presumably, this is in part because the target-specific sequence, when positioned in an inverse direction, can facilitate the insertion of donor in the forward direction, thus increasing the rate of successful editing (Figure 7).
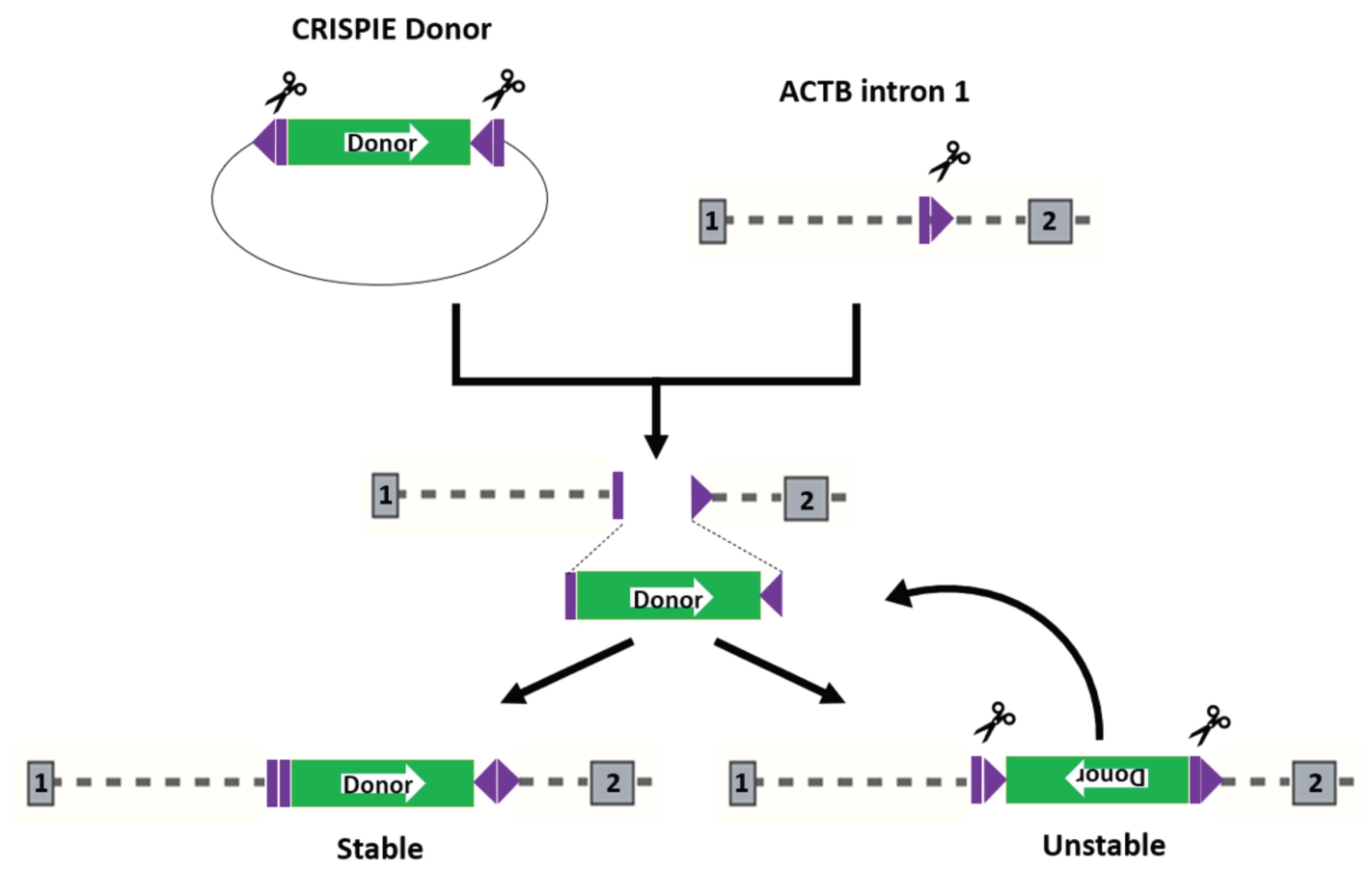
Figure 7. Opposite orientations of the sgRNA target site at the genomic locus and the donor facilitate the donor insertion in the forward orientation. After Cas9 creates a double-stranded break in genomic locus, the donor module can be inserted in either the forward or reverse orientation. Since the sgRNA sites flanking the donor module are in the opposite orientation relative to the sgRNA site at the genomic locus, forward insertion of the donor module destroys both sgRNA sites flanking the donor, resulting in a stable insertion. Inserting the donor in the reverse orientation creates functional sgRNA sites that can be subjected to further Cas9 cleavage, thus creating an unstable insertion (unless INDELs have occurred at the insertion junctions). The sgRNA binding sites are shown in purple, with the Cas9 cut site represented as the thin white line between the purple rectangle and triangle. The white arrow within the donor module represents the orientation of the donor module.Addgene #172842 is a mEGFP donor specific for the first exon of ACTB for the sgRNA shown in Figure 2.
The original donor plasmids were generated by gene synthesis (using Azenta Life Sciences, formerly Genewiz).
Other target-specific donor modules can be generated by replacing the current sgRNA targeting site of a donor plasmid with the appropriate target specific sgRNA sequence, including PAM and context sequences, using the available restriction enzyme sites. In this way, researchers can target intronic locations of other genes. Donors with different tags, such as different fluorescent proteins (FPs), can be generated by replacing the FP from existing donors.
The donor sequence include the tag-encoding exon flanked by important adjacent sequences needed for splicing and donor excision from the plasmid (Figure 8).
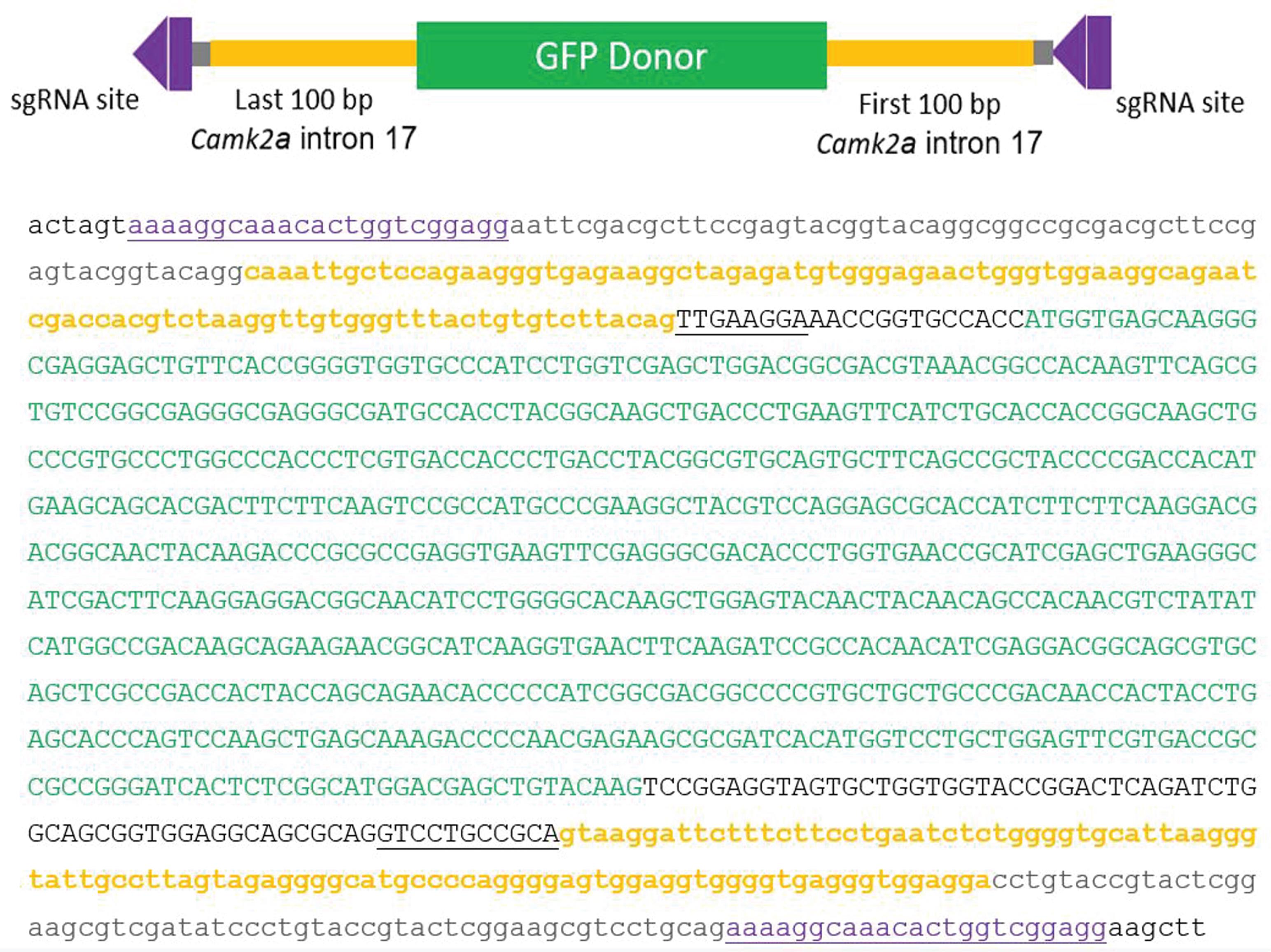
Figure 8. The 5’ to 3’ sequence of a representative CRISPIE donor module. Sequence of Donor B1 (Addgene#172842; phase 0-0) is shown. Capitalized case letters: exonic sequence; lower case letters: intronic sequence; yellow: intronic sequence from the intron 17 of mouse CaMKIIα (Camk2a); underlined: exonic sequence adjacent to Camk2a intron 17; purple: sgRNA targeting site; green: EGFP. Note, only the exonic sequence will be integrated into the final mRNA.We also use an mRuby expression vector (Addgene #172854) as the transfection marker. Other transfection markers should be characterized and optimized before incorporation into the CRISPIE procedure.
U-2 OS Lipofectamine 2000 Transfection
Culture U-2 OS cells in McCoy’s 5A medium (10% FBS). Set the incubator to 37°C and 5% CO2. The cells should be passaged every 2-3 days, when they are at ~90% confluency.
Twenty-four hours prior to transfection, seed ~1.25 × 105 cells in a 35 mm culture dish containing 2 mL of McCoy’s 5A medium (10% FBS) and let attach overnight. Swirl cells clockwise five times, anti-clockwise five times, up and down five times, and left and right five times. This ensures an even coating of cells on the bottom of the dish. We do not use antibiotics, although we have not tested whether antibiotics may affect cell growth or labeling efficiency.
Before beginning transfection, ensure that the cells are 30–50% confluent. Growth outside this range could reduce transfection efficiency.
For each transfection reaction, combine 5 µL of Lipofectamine 2000 with 145 µL of Opti-MEM medium. Mix thoroughly by vortexing. Incubate the mixture at room temperature for 5 min.
Note: Create a master mix when performing multiple transfections.
According to the manufacture’s protocol, the incubation time for this lipofectamine 2000/Opti-MEM master mix can be up to 20 min.
For each transfection reaction, combine 0.2 μg of the transfection marker (mRuby3; acquired from Addgene), 1.0 μg of the sgRNA/Cas9 plasmid (created in Procedure A–C above), and 1.0 μg of the donor module-containing plasmid (acquired from Addgene) in 150 µL of Opti-MEM into a separate tube. Negative controls can be made by excluding either the sgRNA/Cas9 plasmid or the donor module-containing plasmid from the reaction.
Note: It is important to only use trace amounts (0.2 μg) of the transfection marker to prevent masking of the fluorescence from the GFP-labeled cells by the transfection marker. Most red fluorescent proteins, including mRuby3, have bleed-through in the green channels, in part due to the bifurcating maturation of the fluorophore. At the same time, fluorescent signals of endogenously labeled proteins are often dim, and therefore can be easily obscured by the bleed-throughs from highly expressed red fluorescent proteins.
After the 5 min lipofectamine/Opti-MEM incubation is complete, combine this mix with the DNA/Opti-MEM mixture, then thoroughly pipette up and down and vortex to mix. Incubate at room temperature for 20 min. Thorough mixing is important for optimal transfection efficiency.
Allocate each transfection mixture (300 µL) to a 35 mm dish containing U-2 OS cells that were passaged 24 h prior.
Five and a half hours post-transfection, carefully aspirate off the transfection medium and add 2 mL of warmed McCoy’s 5A medium (10% FBS) to each dish. Incubate at 37°C an 5.0% CO2 for 30 min.
Note: Pay attention to suction and add medium from the side of the dish to minimize perturbation of the cells at the bottom of the dish.
Suction off media, add 2 mL of pre-warmed McCoy’s 5A medium (10% FBS) to the cells, and place them in the incubator set at 37°C and 5.0% CO2.
Note: These washing steps are essential because prolonged exposure to lipofectamine 2000 impairs U-2 OS cell health.
Protein expression can be assessed at 72 h post-transfection.
U-2 OS cell counting used to determine transfection efficiency.
Using a brightfield view at 100× magnification (a 10× air objective [0.3 NA] and 10× eyepieces), pick a region of cells that have regular, evenly spread growth.
Once the field of view (FOV) is found, zero the X and Y directions and switch to 400× magnification (40× water immersion objective [0.8 NA] and 10× eyepieces).
Switch to the red FP (RFP) filter and count any cells expressing mRuby. While still in the same FOV, switch to the GFP filter. Count the cells expressing GFP.
Note: We use an Olympus BW51XI upright microscope. However, most standard fluorescence microscopes with GFP and RFP filtersets (e.g., Chroma ET49002 and ET49005) should work. It is important to only count cells that have GFP labeling of the desired protein. In the case of actin, filaments of GFP-actin should be clearly visible (see Figure 9); avoid counting cells that have non-specific GFP fluorescence (e.g., fluorescence permeating the whole cell), which occasionally happens, possibly due to finite specificity of the Cas9 editing.
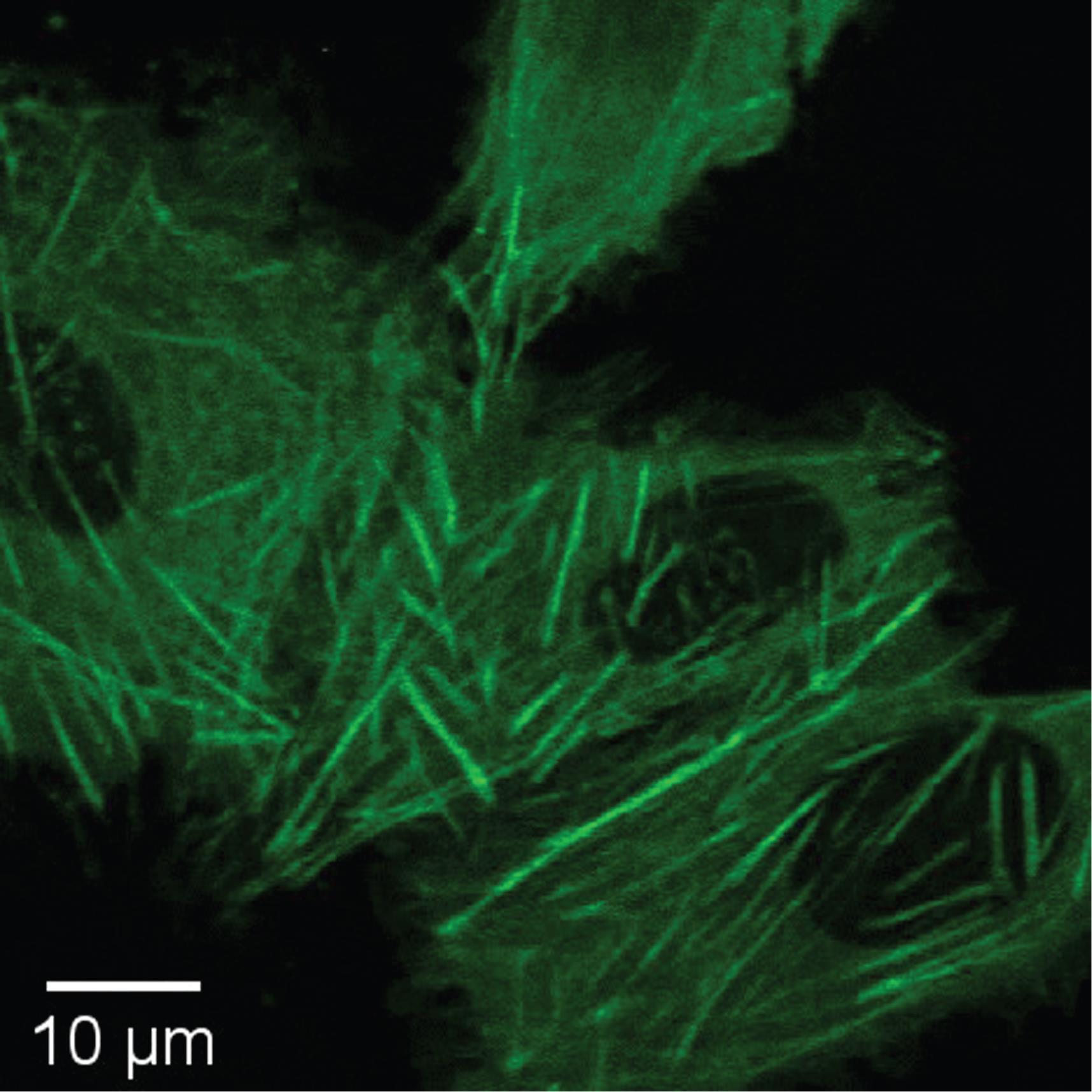
Figure 9. Example of successfully labeled U-2 OS cell. Note the GFP-labeled actin filaments and the absence of GFP in the nuclei. Only count cells that have GFP fluorescence in accordance with the characteristic morphological distribution of the labeled protein.Repeat this counting method for 12 FOV, using a pre-determined, raster pattern that is consistent across all samples. To reduce variations, we omit any FOV with <10 mRuby positive cells, and replace it with additional, pre-determined FOVs.
These cells can be imaged live or fixed, using available epi, confocal, or two-photon fluorescence microscopes.
Labeling efficiency is calculated by the number of successfully labeled cells (in the example, green) divided by the total number of transfected cells (red) in an FOV.
If determination of the labeling efficiency of each gene locus is needed, it can be derived from the labeling efficiency and the copy of target sites per cell using the equation below, assuming that the editing of each locus is an independent event.
Cell labeling efficiency = 1 – (1 – allele labeling efficiency)copy number
Assessment of the accuracy of CRISPIE editing in the genome can be achieved through multiple avenues. We typically use next generation sequencing (NGS). The genomic DNA and mRNA of GFP-positive cells are enriched by Fluorescence-Activated Cell Sorting (FACS) and subjected to RT-PCR and PCR, respectively, using the appropriate primers to detect the insertion junctions. Where needed, nested PCR can be used to increase specificity. The PCR amplicons are then sent for NGS (we uses Azenta Life Sciences, formerly known as Genewiz). As shown in the original publication (Zhong et al, 2021), the typical result is that INDELs are abundantly present in genomic DNA sequences, but are largely absent in mRNA sequences.
The primer pairs should straddle a span of ~2–300 bp at both the 5’ and 3’ insertion junctions of the donor module. This ensures that the PCR will detect relatively large insertions and deletions.
Our lab uses custom MATLAB codes to align sequences and identify editing outcomes. However, this can be done by using available software packages, or by collaborating with NGS experts.
Recipes
SOC Medium (adapted from Cold Spring Harbor, 2018)
Combine the following in 450 mL of deionized water with a stir bar (recommended):
10 g Tryptone
2.5 g Yeast Extract
0.25 g NaCl
Once the components are thoroughly mixed, add 5 mL of 250 mM KCl.
Adjust the pH of the solution to 7 with 10 N NaOH (roughly 100 μL).
After ensuring the total volume is 500 mL, autoclave for 30 min at 20 psi on a liquids cycle.
Add 2.5 mL of filter-sterilized 2 M MgCl2 solution.
Add 10 mL of filter-sterilized 1 M glucose solution.
TB Medium
Following manufacturer instructions on the bottle:
Combine 47.6 g TB pellets in 1 L of water.
Add 4 mL of glycerol.
Once dissolved, autoclave at 20 psi for 30 min.
McCoy’s 5A Medium supplemented with FBS (10% v/v)
Add 55 mL of FBS to 500 mL of McCoy’s 5A medium.
Note: Although 10% of 500 mL is 50 mL, 55 mL is added to account for the extra volume of FBS in the final volume of the medium. This is important, as altered serum concentration can have significant effects on U-2 OS cell growth.
Acknowledgments
None of this work would have been possible without the work by the authors of the original paper (Zhong et al., 2021). This work is supported by a NIH/BRAIN Initiative grant (RF1MH120119) to H.Z. and T.M., and an NIH/NINDS R01 (R01NS081071) to T.M.
Competing interests
The authors declare no competing interests.
References
- Cold Spring Harbor. (2018). SOC Medium. Cold Spring Harbor Laboratory Press.
- Fortin, D. A., Tillo, S. E., Yang, G., Rah, J. C., Melander, J. B., Bai, S., Soler-Cedeño, O., Qin, M., Zemelman, B. V. and Guo, C. (2014). Live imaging of endogenous PSD-95 using ENABLED: a conditional strategy to fluorescently label endogenous proteins. J Neurosci 34(50): 16698-712.
- Melander, J. B., Nayebi, A., Jongbloets, B. C., Fortin, D. A., Qin, M., Ganguli, S., Mao, T. and Zhong, H. (2021). Distinct in vivo Dynamics of Excitatory Synapses Onto Cortical Pyramidal Neurons and Inhibitory Interneurons. SSRN Electron J . doi:10.2139/ssrn.3837634.
- Nishiyama, J., Mikuni, T. and Yasuda, R. (2017). Virus-Mediated Genome Editing via Homology-Directed Repair in Mitotic and Postmitotic Cells in Mammalian Brain. Neuron 96(4): 755-768 e755.
- Roberts, B., Haupt, A., Tucker, A., Grancharova, T., Arakaki, J., Fuqua, M. A., Nelson, A., Hookway, C., Ludmann, S. A. and Mueller, I. A. (2017). Systematic gene tagging using CRISPR/Cas9 in human stem cells to illuminate cell organization. Mol Biol Cell 28(21): 2854-2874.
- Zhong, H., Ceballos, C. C., Massengill, C. I., Muniak, M. A., Ma, L., Qin, M., Petrie, S. K. and Mao, T. (2021). High-fidelity, efficient, and reversible labeling of endogenous proteins using CRISPR-based designer exon insertion. Elife 10: e64911.
Article Information
Copyright
![]() Wilson et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Wilson et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Wilson, E. A., Mao, T. and Zhong, H. (2022). Labeling Endogenous Proteins Using CRISPR-mediated Insertion of Exon (CRISPIE). Bio-protocol 12(5): e4343. DOI: 10.21769/BioProtoc.4343.
- Zhong, H., Ceballos, C. C., Massengill, C. I., Muniak, M. A., Ma, L., Qin, M., Petrie, S. K. and Mao, T. (2021). High-fidelity, efficient, and reversible labeling of endogenous proteins using CRISPR-based designer exon insertion. Elife 10: e64911.
Category
Cell Biology > Cell engineering > CRISPR-cas9
Cell Biology > Cell imaging > Fluorescence
Cancer Biology > General technique > Molecular biology technique
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.