- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Pull-down of Biotinylated RNA and Associated Proteins
Published: Vol 12, Iss 4, Feb 20, 2022 DOI: 10.21769/BioProtoc.4331 Views: 8629
Reviewed by: Chiara AmbrogioKarem A CourtZheng Zachory WeiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2021
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Mapping networks of RNA-protein interactions in cells is essential for understanding the inner workings of many biological processes, including RNA processing, trafficking, and translation. Current in vivo methods for studying protein-RNA interactions rely mostly on purification of poly(A) transcripts, which represent only ~2–3% of total RNAs (Figure 1). Alternate robust methods for tagging RNA molecules with an RNA aptamer (e.g., MS2-, U1A- and biotin-RNA aptamer) and capturing the RNA-protein complex by the respective aptamer-specific partner are not extensively studied. Here, we describe a protocol (Figure 2) in which a biotin-RNA aptamer, referred to as the RNA mimic of biotin (RMB), was conjugated separately to two small RNA secondary structures that contribute to trafficking and translating HAC1 mRNA in the budding yeast Saccharomyces cerevisiae. The RMB-tagged RNA was expressed in yeast cells from a constitutive promoter. The biotinylated RNA bound to proteins was pulled down from the cell lysate by streptavidin agarose beads. RNA was detected by RT-PCR (Figure 3) and associated proteins by mass spectrometry (Figure 4). Our findings show that an RNA aptamer tag to RNA molecule is an effective method to explore the functional roles of RNA-protein networks in vivo.
Keywords: RNA binding protein (RBP)Background
RNA is a multifaced biomolecule with a wide range of biological functions. For example, messenger RNA (mRNA) carries the instructions for protein synthesis, ribosomal RNA (rRNA) provides the structural and enzymatic framework of the ribosome, and transfer RNA (tRNA) contains the anticodon of specific mRNA codons and delivers amino acids corresponding to the codon during translation. RNAs also play key regulatory partners for certain proteins involved in a wide variety of biological processes, including gene expression during growth, development, and cellular stress (Hudson and Ortlund, 2014). These RNA binding proteins (RBPs) contain one or more RNA-binding domains (RBD), which recognize and bind to specific sequences at their target RNA. Unraveling the specificity in RNA-protein recognition is therefore key to understanding the mechanisms by which cellular pathways are controlled and connected at the molecular level.
Several in vivo and in vitro strategies are being used to identify global RBPs, including a novel in vivo technique called RNA interactome capture (RIC) (Keene et al., 2006; Castello et al., 2013; Jazurek et al., 2016; Kastelic and Landthaler, 2017). Typically, RIC involves crosslinking proteins to mRNAs in vivo, immunoprecipitation (IP) of the mRNA-protein complex (CLIP) by using oligo(dT) magnetic beads, and identification of covalently linked proteins by mass spectrometric based methods and RNA fragments, by reverse transcriptase (RT) PCR and sequencing (Ramanathan et al., 2019) (Figure 1).

Figure 1. Schematic representation of the RNA interactome capture (RIC). Proteins are crosslinked to RNA in vivo by UV irradiation. Yellow light box: RNA binding proteins (RBPs) bound to poly(A) RNAs are shown by various colored shapes, and unbound proteins are shown by grey circles. Light green box: The RNA-protein complex is recovered from the unbound proteins by oligo-d(T) 25 beads. Proteins and RNA fragments are subjected to limited RNase treatment. Proteins are identified by quantitative mass spectrometry. RNA fragments occupied by RBP are subjected to sequencing to identify the RBP binding sites.
CLIP enables the identification of several RBPs with well-defined RNA binding domains, such as the RNA recognition motif (RRM), the zinc finger (Znf) domain, or the K-homology (KH) motif. Additionally, CLIP identifies novel RBPs that do not have a canonical RNA binding domain, but instead have intrinsically disordered regions (IDRs), such as S/R repeats, RG/RGG repeats, Q/N-rich stretches and short linear motifs (SLiMs) (Gerstberger et al., 2014, Hentze et al., 2018, Balcerak et al., 2019). These disordered regions in RBPs are often dynamic and flexible, with low-ordered secondary structures that enable them to bind to multiple RNA partners. The low-ordered structure transits to an ordered conformation upon binding to their partner RNA (Srivastava et al., 2021). Additionally, a single RBP can bind to several RNA targets (Muller-McNicoll and Neugebauer, 2013), thus necessitating the direct measurement of RNA binding by RBP to establish the functional interaction.
Many RBPs bind to the 5’- and/or 3’-unstranslated regions (UTR) of mRNAs and regulate their storage, stability, localization, and/or translational efficiency. Among RBPs that bind to the specific 3’-UTR sequence (5’-AUUUAU-3’), also known as the AU-rich element (ARE), are AUF1 (Brewer, 1991), tristetrapolin (Lai et al., 1999), and HuR (Fan and Steitz, 1998). These ARE-BPs are known to promote stabilization, degradation, or translatability of mRNAs (Otsuka et al., 2019); however, the molecular mechanisms that regulate their targets are still unknown.
Recent high-throughput sequencing technologies, including SHAPE-Seq (selective 2′-hydroxyl acylation analyzed by primer extension sequencing) and Frag-Seq (fragmentation sequencing), revealed that >90% of 5’- and 3’-UTR in mRNAs could form secondary structures, in both yeast (Kertesz et al., 2010) and human transcriptomes (Wan et al., 2014, Bevilacqua et al., 2016). Combined with CLIP and the parallel analysis of RNA structure (PARS), Groot et al. recently showed that RNA-protein interaction largely depends on their contextual structures (Sanchez de Groot et al., 2019). They also show that cellular proteins bind to single- and double-stranded RNA. To date, RBP2GO (https://rbp2go.dkfz.de) has collectively listed more than 22,000 RBPs in 13 organisms, including the budding yeast Saccharomyces cerevisiae. However, the specific RNA-protein interaction networks and physiological functions have not been fully assigned.
The CLIP method relies on oligo-dT beads, thus limiting it to poly(A) RNAs, which represent only ~2–3% of total RNAs (Djebali et al., 2012; Uszczynska-Ratajczak et al., 2018). For non-poly(A) RNAs, the potential solution is to tag them with an RNA aptamer and capture the RNA-protein complexes by the respective aptamer-specific protein. The commonly used RNA aptamers are 19-nucleotides MS2 RNA hairpin (5′-ACAUGAGGAU CACCCAUGU-3’) (Yoon et al., 2012), 15–19-nucleotides BoxB RNA hairpin (5′-NNGCCCTGAA GAAGGGCNN-3’) (Baron-Benhamou et al., 2004, Cocozaki et al., 2008), 29-nucleotides U1A RNA hairpin (5′-AGCUUAUCCA UUGCACUCCG GAUGAGCU-3′) (Katsamba et al., 2001), and 43-nucleotides biotin-RNA aptamer, referred to as the RNA mimic of biotin (RMB, 5′-ACCGACCAGA AUCAUGCAAG UGCGUAAGAU AGUCGCGGGC CGGG-3′) (Vasudevan and Steitz, 2007). The MS2 approach has been valuable in targeting and identifying the specific RNA-protein complex (Yoon et al., 2012), indicating that other tagging methods could also potentially be used. However, each of these RNA tagging methods has specific drawbacks, in that it can affect the function of the target RNA. Therefore, it is important to select a method based on the biological question being asked.
In this report, we elaborate a protocol in which an RMB was conjugated to two small RNA secondary structures that contribute to targeting and translating HAC1 mRNA (Ruegsegger et al., 2001) in the budding yeast S. cerevisiae. The first one is a 60-nucleotide RNA molecule representing the 3′-bipartite element (3′-BE) (Aragon et al., 2009), and the second one is a 40-nucleotide RNA molecule representing the 5’-UTR-intron RNA duplex (5’-RD) (Ruegsegger et al., 2001). Both 5’-RD-RMB and 3′-BE-RMB were expressed from a constitutive ADH1 promoter in yeast cells grown in the presence of 4-thiouracil (4-SU). Cells were harvested and irradiated with UV light. The 3′BE-RMB-protein and the 5’-RD-RMB-protein complexes were pulled down from the cellular lysate by streptavidin-agarose beads (Figure 2).

Figure 2. The workflow diagram of the RNA and protein pulldown and analysis. Yeast cells expressing 3’-BE-RMB or 5’-RD-RMB were grow till OD600 reached ~0.6. DTT or 4-thiouracil (4-SU) were added to the culture and the cells grown for an additional 3 hours. Cell were collected in a 50 mL Falcon tube, washed with 1× PBS, irradiated with UV light, and harvested. Cells were lysed and divided into two Eppendorf tubes. In both tubes, streptavidin agarose was added and mixed for 1 hour at 4°C. Tubes were centrifuged to separate pellet (P) and supernatant (S) fractions for subsequent RNA or protein analysis.
The RMB-tagged RNA was detected by RT-PCR (Figure 3). The covalently associated protein was visualized by Coomassie and Silver-staining methods (Figure 4) and identified by mass spectrometry (Ghosh et al., 2021). We also showed that an uncharacterized protein Pal2 specifically bound to the synthetic 3’-BE (Ghosh et al., 2021). Our findings indicate that an RNA tag to a small RNA molecule is an effective method to explore the functional roles of RNA-protein networks in vivo.
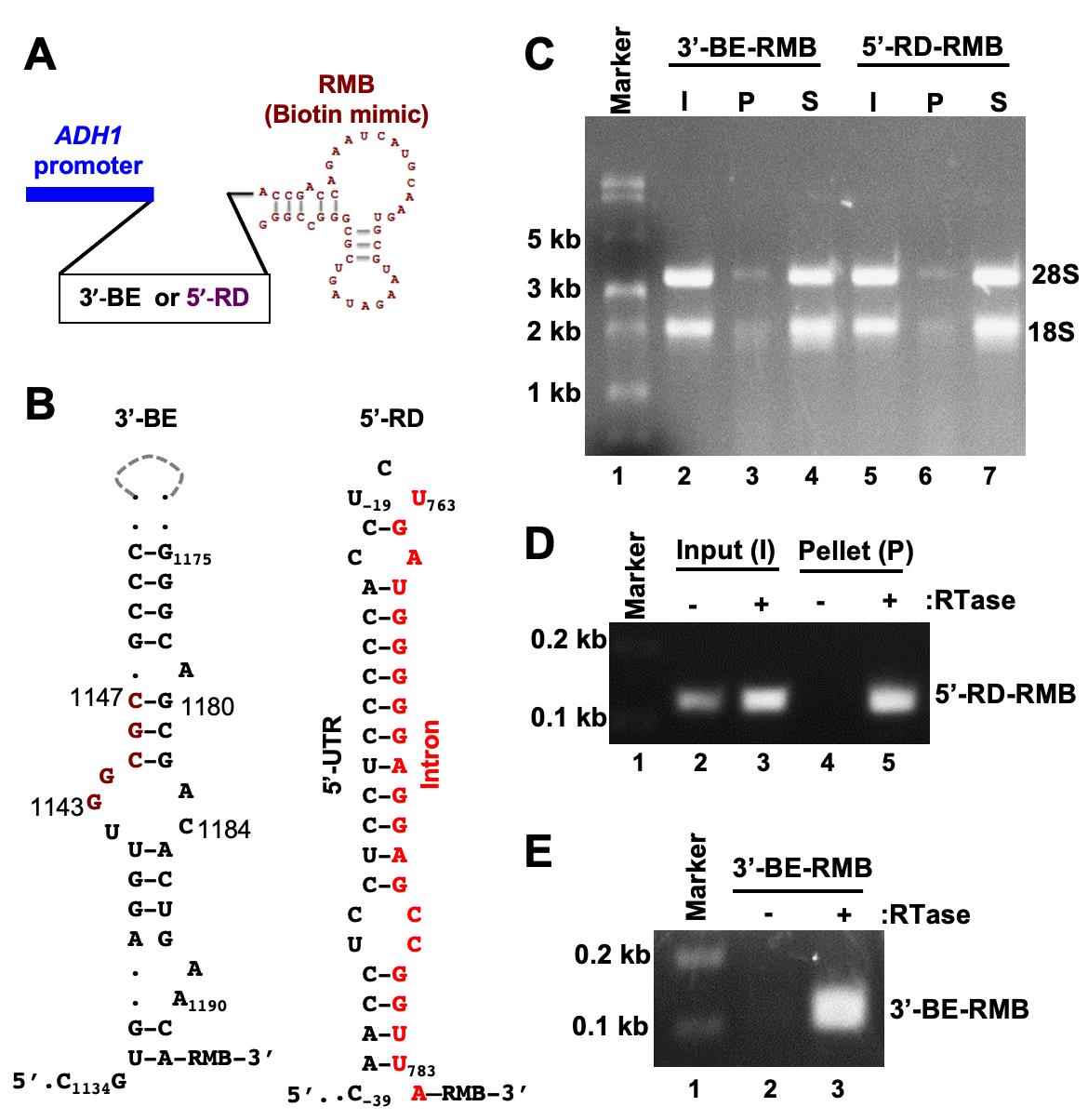
Figure 3. Pulldown of biotinylated RNA. A. The template DNA sequence of the 3’-BE- or 5’-RD RNA was placed under the control of the constitutive ADH1 promoter. The predicted structure of the RMB is shown. B. The predicted secondary structures of 3’-BE and 5’-RD RNAs are shown. The conserved RNA motif within the 3’-BE is shown in red. The 5’-RD consisting of 5’-UTR and intron of HAC1 mRNA, which are shown in black and red, respectively. The numbers indicate the nucleotide positions of HAC1 mRNA. C. Whole cell extract (WCE) from yeast cells expressing the biotinylated RNA (3’-BE-RMB or 5’-RD-RMB RNAs) was prepared and mixed with streptavidin agarose for 1 hour at 4°C. The mixture was centrifuged to separate the pellet (P) and supernatant (S) fractions. RNA was isolated using a Trizol method and equal amounts of RNA (500 ng) from the pellet and supernatant fractions were loaded onto a denaturing formaldehyde agarose gel, to monitor the integrity of RNA. As an input (I), 500 ng RNA from the WCE was also separated. D. To analyze 5’-RD-RMB mini-RNA expression, cDNA was synthesized from total RNA isolated from input (I) and pellet (P) fractions, using gene-specific primers in the presence and absence of reverse transcriptase (RTase). The cDNA was then used as a template to amplify the 5’-RD RNA. E. To analyze 5’-BE-RMB mini-RNA expression, cDNA was synthesized from total RNA isolated from the P fraction using gene-specifics primers in the presence and absence of reverse transcriptase (RTase). The cDNA was then used as a template to amplify the 5’-BE RNA.
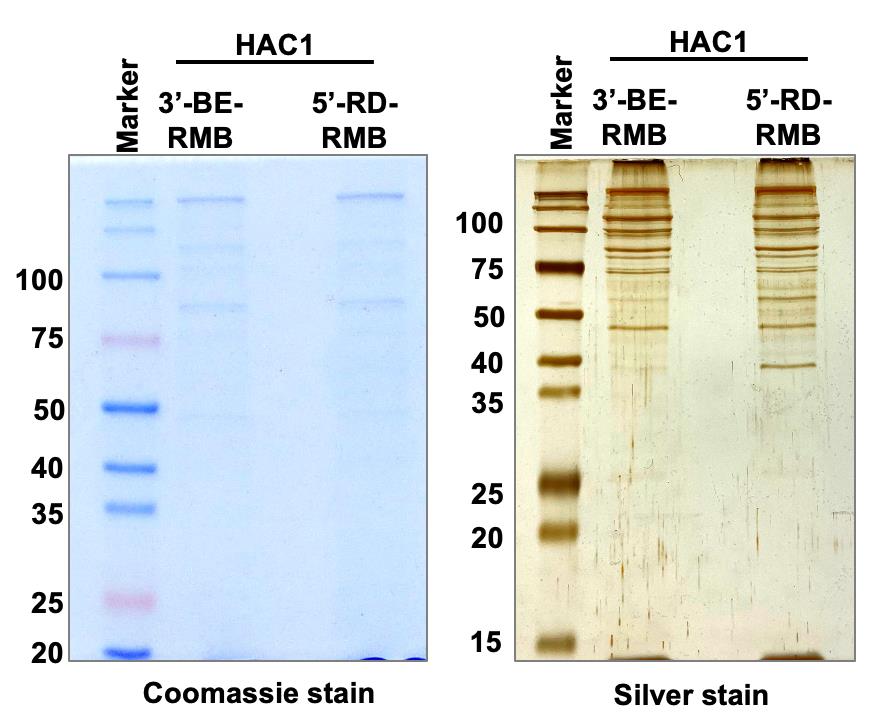
Figure 4. Pulldown of protein complex. Whole cell extract (WCE) from yeast cells expressing the biotinylated RNA (3’-BE-RMB or 5’-RD-RMB RNAs) was prepared and mixed with streptavidin agarose for 1 hour at 4°C. Proteins binding specifically to the 3’-BE- or 5’-RD-RMB mini-RNA were immuno-precipitated by streptavidin agarose and run on an SDS-polyacrylamide gel. The Coomassie blue- and Silver-stained gels are shown.
The cytosolic HAC1 mRNA in the budding yeast S. cerevisiae. contains an unusual intron. The intron is unusual because it is not spliced in the nucleus by the spliceosome, but is instead retained in the mRNA that is exported to the cytoplasm (Ruegsegger et al., 2001). This HAC1 intron interacts with its 5’-UTR to form an RNA duplex (RD), and this 5’-UTR-intron RD prevents translation initiation, thus keeping the mRNA in the translationally repressed form under normal conditions (Ruegsegger et al., 2001; Sathe et al., 2015). Under conditions of cellular stress, 3′-BE targets HAC1 mRNA to the endoplasmic reticulum (ER) stress site (Aragon et al., 2009), where the dual kinase RNase Ire1 cleaves the intron from the translationally repressed HAC1 mRNA (Cox et al., 1993; Mori et al., 1993; Gonzalez et al., 1999). However, the molecular mechanisms of 3′-BE-mediated mRNA transport and the RD-mediated translational repression are not yet defined, motivating us to identify both 3′-BE- and RD-protein complexes, and determine their functional role in targeting and translational de-repression of HAC1 mRNA.
Materials and Reagents
Materials
BasixTM 1.5 mL microcentrifuge tubes (Fisher Scientific, catalog number: 02-682-002)
FisherbrandTM 0.2 mL PCR Tubes (Fisherbrand, catalog number: 14-230-205)
50 mL centrifuge tubes (CellPro, catalog number: C5602)
50 mL centrifuge tubes (CellPro, catalog number: C5600)
250 mL Erlenmeyer glass flask (Corning, catalog number: 4980500)
150 mm × 15 mm Petri dish (Fisherbrand, catalog number: FB0875714)
4-Thiouracil (Alfa Aesar, catalog number: 591-28-6)
DL-Dithiothreitol (Sigma, catalog number: D9779)
0.5 mm zirconia beads (Fisher Scientific, catalog number: 11079105z)
Aprotinin (Sigma, catalog number: A1153)
Pepstatin A (Sigma, catalog number, P4265)
Protease inhibitor Tablet mini, EDTA-free (Thermo Fisher Scientific, catalog number: A32955)
Triton X-100 (Sigma, catalog number: T9284)
Phenylmethylsulfonyl fluoride (Sigma, catalog number: P7626)
Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad, catalog number: 5000006)
Streptavidin Sepharose® High Performance (GE Healthcare, catalog number: GE-17-5113-01)
SuperScript® III Reverse Transcriptase (Invitrogen, catalog number: 180-80-093)
RNaseOUT (Invitrogen, catalog number: P/W 100000840)
dNTPs (dATP, dGTP, dCTP, dTTP) (New England Biolabs Inc., catalog number: N0446S)
2× PCR master mixture (New England Biolabs Inc., catalog number: M0496S)
PierceTM Silver staining kit (Thermo Scientific, catalog number: 24612)
QIAzol reagent (QIAGEN, catalog number: 79306)
Chloroform (Sigma, catalog number: C2432)
Isopropanol (Sigma, catalog number: 19516)
RNase free water (Invitrogen, catalog number: 12183018)
Glycogen, RNA grade (Thermo Scientific, catalog number: R0551)
Ethanol (Pharmco Products, catalog number: 111000200)
Methanol (Fisher Chemical, catalog number: A412-4)
Acetic Acid, Glacial (Fisher Chemical, catalog number: A38-212)
Ethylenediaminetetraacetic acid (EDTA) (Sigma, catalog number: E9884)
Sodium acetate (Sigma, catalog number: S2889)
Bacteriological peptone (Thermo Scientific, catalog number: J20048-P5)
Yeast extract (Bio Basic, catalog number: 8013-01-2)
D-glucose (Sigma, catalog number: G8270)
Agar (Fisher Bioreagents, catalog number: BP9744-500)
Ultra-clear Quick dissolve Agarose (EZ Bioresearch, catalog number: S-1020-500)
Tris-Base (Fisher Bioreagents, catalog number: BP152-10)
Glycine (Sigma, catalog number: G8898)
Trichloroacetic acid (Sigma, catalog number: T6399)
Acetone (Sigma, catalog number: 179124)
Ammonium bicarbonate (AMBIC; Sigma, catalog number: A6141)
Iodoacetamide (IAA; Sigma, catalog number: I1149)
Sequencing Grade Modified Trypsin (Promega)
Lysyl-endopeptidaseR (Lys-C, FujiFilm Wako Pure Chemical Corporation)
Trifluoroacetic acid (TFA; Sigma, catalog number: T1647)
Formic acid (Sigma)
Sequencing Grade Modified Trypsin (Promega)
Sodium dodecyl sulfate (SDS) (Sigma, catalog number: 11667289001)
β-mercaptoethanol (β-ME) (Sigma, catalog number: 444203)
Diethyl pyrocarbonate (DEPC) (Sigma, catalog number: D5758)
Ethidium bromide (Sigma, catalog number: E1510)
Formaldehyde (Fisher Bioreagents: BP531-500)
Formamide (Sigma, catalog number: 47671)
DNase I (New England Biolabs Inc., catalog number: M0303S)
Yeast Nitrogen base (without amino acids) with ammonium sulphate (Sigma, catalog number: Y1251)
Amino acids (All amino acids from Sigma)
MOPS (Sigma, catalog number: M3183)
Potassium Chloride (Sigma, catalog number: P9541)
Magnesium acetate (Sigma, catalog number: M5661)
Brilliant Blue R (Sigma, catalog number: B7920)
Bromophenol blue (Sigma, catalog number: 114391)
Xylene Cyanol FF (Sigma, catalog number: X4126)
SC-uracil (complete synthetic medium without uracil)
YEPD (see Recipes)
Synthetic Complete (SC) medium with or without uracil or leucine (see Recipes)
1× PBS (see Recipes)
Buffer A (see Recipes)
Fixing solution for Coomassie staining (see Recipes)
Staining solution Coomassie staining (see Recipes)
Destaining solution Coomassie staining (see Recipes)
1× TE buffer (see Recipes)
10× MOPS buffer (see Recipes)
6× RNA loading dye (see Recipes)
Fixing solution for silver staining (see Recipes)
Sensitizer solution for silver staining (see Recipes)
Staining solution for silver staining (see Recipes)
Developer solution for silver staining (see Recipes)
D774, pRS426-ADH1-3’-BE-RMB (Ghosh et al., 2021)
D844, pRS425-ADH1-5’-RD-RMB (Ghosh et al., 2021)
X2159, MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ire1::NatMX hac1::KanMX, pRS426-ADH1-3’-BE-RMB: URA3
X2160, MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ire1::NatMX hac1::KanMX, pRS425-ADH1-5’-RD-RMB: LEU2
Equipment
Shaking incubator (Innova 42, New Brunswick Scientific)
Spectro linker- XL-1000 UV crosslinker (Spectronics Corporation)
Centrifuge (Eppendorf 5415R)
Centrifuge (Eppendorf 5810R)
UV transilluminator (UVITEC Cambridge)
Thermocycler-T100 (Bio-Rad)
NanoDrop-1000 Spectrophotometer (Thermo Scientific)
Incubator shaker (New Brunswick Innova 42)
Mini-100 Orbital shaker (Genie)
Diagger Vortex (Genie)
-80°C freezer (Sanyo)
Refrigerator (Sanyo)
Nutator shaker (Clay Adams)
High-Field Hybrid Ion Trap-Orbitrap Mass Spectrometer (Thermo Scientific Orbitrap Elite)
Procedure
Culturing yeast strains and UV-crosslinking
Day-1: Sample Growth
Inoculate a single yeast colony of the strain X2159 and X2160 in 10 mL of SC-uracil and SC-leucine medium, respectively.
Grow the yeast overnight at 30°C in a shaking incubator.
Inoculate the overnight culture to a starting OD600 value ~0.1 in 100 mL of SC-uracil or SC-leucine medium in a baffled flask.
Grow the yeast at 30°C in a shaking incubator until the OD600 value reaches ~0.6.
Add 100 µL of 0.1 M 4-thiouracil and 500 µL of 1.0 M DTT to each culture (final concentration of 4-thiouracil=100 µM and of DTT=5 mM).
Grow them for a further 3 h at 30°C in a shaking incubator.
Harvest each culture in a fresh 50 mL Falcon tube by centrifugation at 2,900 rpm (291 × g) for 10 min, using an Eppendorf 5810R.
Resuspend each cell pellet in 20 mL of 1× PBS and centrifuge at 2,900 rpm (291 × g) for 10 min, using an Eppendorf 5810R.
Discard the supernatant and resuspend the cell pellet again in 20 mL of 1× PBS.
Place each cell suspension into a petri dish (150 mm × 15 mm) and UV-crosslink for 2.5 min twice, with a 2 min interval, using a Stratalinker 2400 UV crosslinker.
Transfer the cells into a 50 mL Falcon tube and centrifuge at 2,900 rpm (291 × g) for 10 min, using an Eppendorf 5810R.
Discard supernatant and store the cell pellet at -20°C.
Preparation of whole cell extract (WCE)
Day-3: Whole cell extract preparation and fractionation
Prepare Buffer A and keep it on ice for 5–10 min.
Take out the cell pellets from -20°C and immediately add 1 mL of Buffer A.
Resuspend the cells and place them in a fresh 1.5 mL microcentrifuge.
Add 500 µL equivalent of 0.5 mm zirconia beads to each cell pellet.
Break the cells using a vortexer for 10 min at 4°C.
Centrifuge the broken cells at 10,000 rpm (9,300 × g) for 10 min at 4°C using an Eppendorf centrifuge (5415R).
Collect the supernatant (WCE) which contains RNA-protein mixture in a fresh 1.5 mL microcentrifuge tube.
Add 1 mL of Buffer A and repeat Steps B6 and B7. Collect the supernatant, and pool to the previously RNA-protein mixture.
Clarify the RNA-protein mixture by centrifugation at 13,000 rpm (15,700 × g) for 10 min at 4°C using an Eppendorf centrifuge (5415R).
Quantify protein concentration using the Bio-Rad Bradford’s reagent.
Quantify RNA using a NanoDrop spectrophotometer.
Pull down of biotinylated RNA-protein mixture from WCE
In a 1.5 mL microcentrifuge tube, mix 100 µL of streptavidin Sepharose resin with 500 µL of Buffer A and centrifuge at 2,000 rpm (400 × g) for 1 min using an Eppendorf centrifuge (5415R). Repeat this washing step at least five times.
Add 300 µg of protein from the WCE to 100 µL of washed streptavidin Sepharose resin.
Mix the reaction by inverting the tube 3–4 times and incubate at 4°C for 1 h on a nutator shaker.
Centrifuge the reaction mixture at 2,000 rpm (400 × g) for 1 min using an Eppendorf centrifuge (5415R). Discard the supernatant.
Resuspend the resin with 500 µL of Buffer A and centrifuge at 2,000 rpm (400 × g) for 1 min using an Eppendorf centrifuge (5415R). Repeat this washing step five times.
Add 50 µL of 2× SDS-loading dye to the resin.
Heat the samples for 5 min at 90°C and centrifuge at 10,000 rpm (9,300 × g) for 1 min using an Eppendorf centrifuge (5415R).
Load 15 µL of supernatant on a 10% SDS-PAGE gel.
Run the SDS-PAGE gel at a constant voltage of 120 V.
Detection of RNA binding proteins
Load the (i) protein ladder, (ii) input, and (iii) pull down samples on the SDS-PAGE gel. Run the SDS-PAGE gel at a constant voltage of 120 V.
Coomassie staining:
Fix the gel using the fixing solution for 1 h.
Remove fixing solution and stain the gel using the Coomassie staining solution for 30 min.
Remove staining solution, destain the gel in destaining solution overnight, and wash 3–4 times with double distilled water.
Silver staining:
Wash the gel in water twice for 5 min.
Fix the gel in fixing solution twice for 15 min.
Wash the gel twice in 10% ethanol for 5 min.
Wash the gel twice in water for 5 min.
Incubate the gel in sensitizer solution for 1 min.
Wash the gel twice in water for 1 min.
Incubate the gel in staining solution for 30 min.
Quickly wash the gel twice with water for 20 s.
Immediately add the developer solution and incubate until protein bands appear.
Add the stop solution, wash gently, replace with stop solution, and incubate for 10 min.
Pull down of biotinylated RNA-protein mixture and RNA isolation
Wash 200 µL of streptavidin Sepharose resin in a fresh 1.5 mL microcentrifuge tube with 1 mL of Buffer A at 2,000 rpm (400 × g) for 1 min. Repeat the washing step at least five times.
Add 300 µg of RNA from the WCE to 200 µL of washed streptavidin Sepharose resin.
Mix the reaction by inverting the tube 3–4 times and incubate at 4°C for 1 h on a nutator shaker.
After incubation, spin down at 2,000 rpm (400 × g) for 1 min. Separate pellet (P) and supernatant (S) fractions.
Wash the P fraction with 1 mL of Buffer A at 2,000 rpm (400 × g) for 1 min. Repeat the washing step five times.
Isolate RNAs from both P and S fractions using the Trizol method (see below). Also, isolate RNA from the WCE (input).
Add 1 mL of QIAzol reagent to each sample. Mix thoroughly by pipetting up and down.
Incubate on ice for 5 min.
Add 200 µL of chloroform and vortex thoroughly.
Incubate on ice for 2–3 min.
Spin down at 12,000 rpm (13,400 × g) for 15 min.
Transfer the aqueous phase containing RNA to a fresh tube.
Add 1 µL of RNase-free glycogen (20 µg) to the aqueous phase.
Add 500 µL of isopropanol to the aqueous phase, mix gently by inverting the tube, and incubate at -20°C overnight.
After incubation, spin down at 12,000 rpm (13,400 × g) for 20 min.
Discard the supernatant and resuspend the pellet in 1 ml of 75 % ethanol.
Vortex briefly and spin down at 10,000 rpm (9,300 × g) for 5 min.
Discard the supernatant and air dry the pellet for 5–10 min.
Note: DO NOT dry the pellet by vacuum centrifuge and DO NOT dry the pellet completely, to ensure the total solubilization of RNA.
Resuspend the pellet in 40 µL of RNase free water.
Incubate the RNA in 70°C for 3 min and immediately transfer to ice.
Quantify the RNA using a NanoDrop spectrophotometer.
Store the RNA at -80°C or proceed to cDNA preparation and RT-PCR.
cDNA preparation and Reverse Transcriptase (RT)-PCR
Formaldehyde gel electrophoresis: Add 0.7 g of agarose to 50 mL of 1× MOPS buffer and heat to dissolve.
Add 730 µL of formaldehyde and 1 µL of ethidium bromide (10 mg/mL) to the gel, mix gently, and pour the gel in a gel casting tray inside a chemical hood.
RNA Sample preparation and electrophoresis: Take 1 µg of RNA sample in 10 µL with 1× MOPS buffer and mix with 2 µL of 6× RNA loading dye.
Heat the sample at 70°C for 3 min in a dry bath and immediately transfer it to ice.
Load the sample onto the formaldehyde gel. Run the gel in 1× MOPS buffer at a constant voltage (80 V).
Visualize RNA the in gel using a gel documentation system.
DNase I treatment (for 10 µL reaction): Mix together 10 µg of RNA, 1 µL of 10× DNase I reaction buffer, and 1 µL of DNase I in a 10 µL reaction mixture.
Incubate at 37°C for 10 min in a thermocycler.
Add 1 µL of 0.05 M EDTA to the reaction mixture (final concentration of 5 mM).
Inactivate the reaction by heating at 75°C for 10 min.
cDNA preparation: Prepare two master mixtures (MM1 and MM2) as shown below (Table 1):
Table 1. cDNA preparation
Master mix MM1 for 5 reactions Master mix MM2 for 5 reactions) Reagent Volume (µL) Reagent Volume (µL) 10 mM dNTPs 5.0 5× RT buffer 20 *RMB-specific reverse primer (10 µM) 2.5 0.1 M DTT 5.0 DEPC water 40 RNaseOUT 1.0 RTase 1.0 * 5’-CCCGCGACTATCTTACGCACTT-3’
Add 9.5 µL of MM1 and 2 µL of RNA (1 µg/mL) in a 0.2 mL PCR tube.
Incubate the reaction mixture at 70°C for 3 min.
Immediately transfer the tube onto ice.
Add 5.4 µL of MM2 to the reaction mixture to a final volume of 16.9 µL.
Place the tube in a thermocycler with the following setting.
Temperature Time Number of cycles
cDNA synthesis 50°C 45 min 1
Inactivation 65°C 10 min 1
Store cDNA samples at -80°C or proceed to PCR amplification.
PCR amplification of 3’-BE-RMB and 5’RD-RMB
Use the following primers to amplify the 3’-BE-RMB and 5’-RD-RMB cDNA.
Primer Sequence (5’ to 3’)
Forward primer for the 3’-BE-RMB 5’-ACGGAAAATGTACCAGAGTCGACGACG-3’
Forward primer for the 5’-RD-RMB 5’-CATAACAACCTCCTCCTCCCCCACCTACG-3’
Reverse primer from the RMB sequence 5’-CCCGCGACTATCTTACGCACTT-3’
PCR reaction: To 20 µL of 2× PCR super mix, add 1 µL of forward and reverse primer and 2 µL of cDNA, and bring the volume to 40 µL with water.
PCR conditions: Run the PCR cycle program as follows: Initial denaturation at 95°C for 2 min, then 30 cycles of denaturation: 95°C for 15 s, annealing: 55°C for 30 s, extension: 72°C for 30 s.
Gel electrophoresis: Load 5 µL of PCR products on 1.5% agarose gel and run at 110 V for 30 min. Visualize the DNA bands in gel by UV light.
Biotinylated RNA-bound protein digestion and clean up for Mass Spectrometry analysis
Take the remaining 45 µL of protein-RNA mixture from Section C, step 6 (in 2× SDS-loading dye) and dilute this to 900 µL with MilliQ water in a 1.5 mL microcentrifuge tube.
Immediately add 100 µL of 100% TCA solution, mix, and incubate on ice for 30 min.
Add 400 µL of cold acetone, mix, and incubate on ice for an additional 30 min.
Pellet the precipitated proteins by centrifugation at 16,000 × g for 10 min.
Carefully discard 1,200 µL of the supernatant with a P1000 pipette. Then, use a P200 pipette with a gel loading tip to remove the remaining 200 µL of supernatant, without disturbing the tiny pellet formed on the side of the tube facing away from the center of the centrifuge rotor. Of note, if <2–5 µg of protein is recovered, no pellet will be visible.
Add 800 µL of cold acetone, vortex, and centrifuge at 16,000 × g for 8 min.
Remove supernatant in two stages as in Step 5, but only 700 µL with the P1000 and 100 µL with the P200 pipette.
Add 800 µL of cold acetone, vortex, and centrifuge at 16,000 × g for 8 min.
Remove supernatant as in Step 7, and leave the tube open to air-dye for 5-30 min (lay the tube sideways to prevent any air-drop contamination).
Solubilize the pellet in 15 µL of 8 M urea in 50 mM AMBIC, pH 8.5, whenever the sample contains 10-80 µg of total protein. For samples with <10 µg protein, use half the volume for all the steps. For samples with >100 µg of protein, solubilize those with 8 M urea in 50 mM AMBIC, pH 8.5, to 4 µg/µL final concentration, and take 15 µL (60 µg) of that solution for the next steps.
If the pellet is not solubilized completely within 30 minutes at room temperature (RT), proceed to the next step to facilitate solubilization by reducing the cysteine disulfide bridges.
Add 2.5 µL of 25 mM DTT in 25 mM AMBIC, pH 8.5, and 42.5 µL of 25 mM AMBIC, pH 8.5.
Incubate the tube at 56°C for 15 min.
Cool to RT and add 3.0 µL of 55 mM iodoacetamide (IAA) in 25 mM AMBIC for 15 min (alkylation).
Terminate the alkylation reaction by adding 8 µL of 25 mM DTT. Considering a sample with 10–80 µg of protein, the total volume would be 71 µL [15 µL of 8 M urea (Step 10), 2.5 µL of 25 mM DTT and 42.5 µL of 25 mM AMBIC (Step 12), 3.0 µL of 55 mM IAA (Step 14), and 8 µL of 25 mM DTT (Step 15)].
Prepare the Trypsin/Lys-C protease mixture in 25 mM AMBIC, pH 8.5, with equal amounts (100 ng/µL) of Trypsin (Sequencing-Grade-Modified Trypsin) and Lys-C (Lysyl Endopeptidase). Add the Trypsin-Lys-C protease mixture to the protein substrate in a ratio of 1:30 (for example, 30 µg protein substrate will have 1 µg protease mixture).
Use 25 mM AMBIC, pH 8.5, to bring the total volume to 100 µL (50 µL for samples with <10 µg of protein substrate).
Incubate at 37°C overnight.
Terminate the digestion with 2.5% TFA to 0.3% final concentration (acidification).
Perform solid phase peptide extraction (according to the manufacturer’s protocol) prior to loading on the instrument. Bond Elute OMIX C18 (10–100 µL; 80 µg capacity) pipette tips from Agilent are preferred, due to binding capacity, reproducibility with small substrate amounts, and ease of use.
Dry the clean peptide samples and reconstitute in 0.1% formic acid to ~0.5–1 µg/µL final peptide concentration.
Inject 0.5 to 2 µL of sample for the nano LC-MS/MS analysis, depending on the sample complexity and instrument used.
Analyze the sample on a hybrid linear ion trap-orbitrap mass spectrometer, where speed of MS/MS identifications in the ion trap is combined with high mass accuracy for the precursor peptides in the orbitrap. A nanoflow system for the chromatographic reverse-phase separation is also advised for greater sensitivity.
Set the Ion trap-Orbitrap as follows: MS1 resolution: 120–240k, mass range: 350–1,600 m/z, CID- or HCD-type MS/MS of 20–30 most intense ions detected in MS1 scan, limited redundancy with dynamic exclusion, and MIPS filter mode-ON.
Convert raw data files to mgf. file format using the Trans Proteomic Pipeline (Seattle Proteome Center, Seattle, WA) and search with the Mascot search engine (Matrix Science), or use the Proteome Discoverer suite (Thermo Fisher Scientific) without file conversion.
Use Mascot search engine with static cysteine carbamidomethylation and dynamic methionine oxidation, plus asparagine and glutamine deamidation as possible modifications.
Set the peptide tolerance at 10ppm and the fragment mass tolerance at 0.6Da (Ion Trap instruments).
Representative data: Experimental details used to validate the system are provided in the original published research article (Ghosh et al., 2021). In brief, we identified a previously uncharacterized protein called Pal2, which was bound to the 3’-BE-RMB. This was further confirmed by an electrophoretic mobility shift assay. The results can be accessed through the following link: https://pubmed.ncbi.nlm.nih.gov/34035143/.
Recipes
YEPD
Reagent Quantity
Bacteriological peptone 20 g
Yeast extract 10 g
H2O 960 mL
Sterilize by autoclaving
Add 50 mL of 40% dextrose
Synthetic Complete (SC) medium with or without uracil or leucine
Reagent Quantity
Yeast Nitrogen base (without amino acids) with ammonium sulphate 6.7 g
Amino acid mix without uracil or leucine (Homemade) 2 g
H2O 960 mL
Sterilize by autoclaving
Add 50 mL of 40% dextrose
1× PBS pH 7.4
138 mM NaCl
2.7 mM KCl
10 mM Na2HPO4
1.8 mM KH2PO4
Adjust pH to 7.4 with HCl
Buffer A
20 mM Tris-HCl, pH 7.4
200 mM KCl (potassium chloride)
5 mM MgOAc (magnesium acetate)
1 mM DTT (dithioerythritol)
1 mM PMSF (phenylmethylsulfonyl fluoride)
1 µg/mL Aprotinin
1 µg/mL Pepstatin A
Protease inhibitor Tablet mini (1 tablet per 10 mL)
0.1% Triton-X-100
Fixing solution for Coomassie staining
50% Methanol
10% glacial acetic acid
Staining solution Coomassie staining
0.1% Coomassie brilliant blue-R250
50% Methanol
10% Glacial acetic acid
Destaining solution Coomassie staining
40% Methanol
10% Glacial acetic acid
1× TE buffer
10 mM tris-HCl pH 7.5
1.0 mM EDTA pH 8.0
10× MOPS buffer
0.2 M MOPS, pH 7.0
20 mM sodium acetate
10 mM EDTA pH 8.0
Dissolve 41.8 g MOPS (Sigma) in 700 mL of sterile water and adjust the pH to 7.0 with 2 N NaOH.
Add 20 mL of 1 M sodium acetate, 20 mL of 0.5 M EDTA pH 8.0, and adjust the volume to 1,000 mL.
Store the buffer at room temperature
6× RNA loading dye
95% formamide
0.025% bromophenol blue
0.025% xylene cyanol FF
5 mM EDTA pH 8.0
Store at room temperature.
Fixing solution for silver staining
30% ethanol
10% acetic acid solution
Sensitizer solution for silver staining
50 μL of Sensitizer (PierceTM Silver staining kit) in 25 mL of water
Staining solution for silver staining
0.5 mL of Enhancer (PierceTM Silver staining kit) in 25 mL of Stain
Developer solution for silver staining
0.5 mL of Enhancer (PierceTM Silver staining kit) in 25 mL of Developer
Acknowledgments
This work was supported by grants from the National Institutes of Health (NIGMS) (1R01GM124183) and the UWM Graduate School (Discovery and Innovation Grant). This protocol is derived from the original research article published by Ghosh et al. (2021) in Science Signaling (https://doi.org/10.1126/scisignal.aaz4401).
Competing interests
Authors declare that they have no conflict of interests.
References
- Aragon, T., van Anken, E., Pincus, D., Serafimova, I. M., Korennykh, A. V., Rubio, C. A. and Walter, P. (2009). Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature 457(7230): 736-740.
- Balcerak, A., Trebinska-Stryjewska, A., Konopinski, R., Wakula, M. and Grzybowska, E. A. (2019). RNA-protein interactions: disorder, moonlighting and junk contribute to eukaryotic complexity. Open Biol 9(6): 190096.
- Baron-Benhamou, J., Gehring, N. H., Kulozik, A. E. and Hentze, M. W. (2004). Using the lambdaN peptide to tether proteins to RNAs. Methods Mol Biol 257: 135-154.
- Bevilacqua, P. C., Ritchey, L. E., Su, Z. and Assmann, S. M. (2016). Genome-Wide Analysis of RNA Secondary Structure. Annu Rev Genet 50: 235-266.
- Brewer, G. (1991). An A + U-rich element RNA-binding factor regulates c-myc mRNA stability in vitro. Mol Cell Biol 11(5): 2460-2466.
- Castello, A., Horos, R., Strein, C., Fischer, B., Eichelbaum, K., Steinmetz, L. M., Krijgsveld, J. and Hentze, M. W. (2013). System-wide identification of RNA-binding proteins by interactome capture. Nat Protoc 8(3): 491-500.
- Cocozaki, A. I., Ghattas, I. R. and Smith, C. A. (2008). Bacteriophage P22 antitermination boxB sequence requirements are complex and overlap with those of lambda. J Bacteriol 190(12): 4263-4271.
- Cox, J. S., Shamu, C. E. and Walter, P. (1993). Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73(6): 1197-1206.
- Djebali, S., Davis, C. A., Merkel, A., Dobin, A., Lassmann, T., Mortazavi, A., Tanzer, A., Lagarde, J., Lin, W., Schlesinger, F. (2012). Landscape of transcription in human cells. Nature 489(7414): 101-108.
- Fan, X. C. and Steitz, J. A. (1998). Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J 17(12): 3448-3460.
- Gerstberger, S., Hafner, M. and Tuschl, T. (2014). A census of human RNA-binding proteins. Nat Rev Genet 15(12): 829-845.
- Ghosh, C., Uppala, J. K., Sathe, L., Hammond, C. I., Anshu, A., Pokkuluri, P. R., Turk, B. E. and Dey, M. (2021). Phosphorylation of Pal2 by the protein kinases Kin1 and Kin2 modulates HAC1 mRNA splicing in the unfolded protein response in yeast. Sci Signal 14(684).
- Gonzalez, T. N., Sidrauski, C., Dorfler, S. and Walter, P. (1999). Mechanism of non-spliceosomal mRNA splicing in the unfolded protein response pathway. EMBO J 18(11): 3119-3132.
- Hentze, M. W., Castello, A., Schwarzl, T. and Preiss, T. (2018). A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol 19(5): 327-341.
- Hudson, W. H. and Ortlund, E. A. (2014). The structure, function and evolution of proteins that bind DNA and RNA. Nat Rev Mol Cell Biol 15(11): 749-760.
- Jazurek, M., Ciesiolka, A., Starega-Roslan, J., Bilinska, K. and Krzyzosiak, W. J. (2016). Identifying proteins that bind to specific RNAs - focus on simple repeat expansion diseases. Nucleic Acids Res 44(19): 9050-9070.
- Kastelic, N. and Landthaler, M. (2017). mRNA interactome capture in mammalian cells. Methods 126: 38-43.
- Katsamba, P. S., Myszka, D. G. and Laird-Offringa, I. A. (2001). Two functionally distinct steps mediate high affinity binding of U1A protein to U1 hairpin II RNA. J Biol Chem 276(24): 21476-21481.
- Keene, J. D., Komisarow, J. M. and Friedersdorf, M. B. (2006). RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat Protoc 1(1): 302-307.
- Kertesz, M., Wan, Y., Mazor, E., Rinn, J. L., Nutter, R. C., Chang, H. Y. and Segal, E. (2010). Genome-wide measurement of RNA secondary structure in yeast. Nature 467(7311): 103-107.
- Lai, W. S., Carballo, E., Strum, J. R., Kennington, E. A., Phillips, R. S. and Blackshear, P. J. (1999). Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol Cell Biol 19(6): 4311-4323.
- Mori, K., Ma, W., Gething, M. J. and Sambrook, J. (1993). A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 74(4): 743-756.
- Muller-McNicoll, M. and Neugebauer, K. M. (2013). How cells get the message: dynamic assembly and function of mRNA-protein complexes. Nat Rev Genet 14(4): 275-287.
- Otsuka, H., Fukao, A., Funakami, Y., Duncan, K. E. and Fujiwara, T. (2019). Emerging Evidence of Translational Control by AU-Rich Element-Binding Proteins. Front Genet 10: 332.
- Ramanathan, M., Porter, D. F. and Khavari, P. A. (2019). Methods to study RNA-protein interactions. Nat Methods 16(3): 225-234.
- Ruegsegger, U., Leber, J. H. and Walter, P. (2001). Block of HAC1 mRNA translation by long-range base pairing is released by cytoplasmic splicing upon induction of the unfolded protein response. Cell 107(1): 103-114.
- Sanchez de Groot, N., Armaos, A., Grana-Montes, R., Alriquet, M., Calloni, G., Vabulas, R. M. and Tartaglia, G. G. (2019). RNA structure drives interaction with proteins. Nat Commun 10(1): 3246.
- Sathe, L., Bolinger, C., Mannan, M. A., Dever, T. E. and Dey, M. (2015). Evidence That Base-pairing Interaction between Intron and mRNA Leader Sequences Inhibits Initiation of HAC1 mRNA Translation in Yeast. J Biol Chem 290(36): 21821-21832.
- Srivastava, A., Yesudhas, D., Ahmad, S. and Gromiha, M. M. (2021). Understanding disorder-to-order transitions in protein-RNA complexes using molecular dynamics simulations. J Biomol Struct Dyn: 1-11.
- Uszczynska-Ratajczak, B., Lagarde, J., Frankish, A., Guigo, R. and Johnson, R. (2018). Towards a complete map of the human long non-coding RNA transcriptome. Nat Rev Genet 19(9): 535-548.
- Vasudevan, S. and Steitz, J. A. (2007). AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell 128(6): 1105-1118.
- Wan, Y., Qu, K., Zhang, Q. C., Flynn, R. A., Manor, O., Ouyang, Z., Zhang, J., Spitale, R. C., Snyder, M. P., Segal, E., et al. (2014). Landscape and variation of RNA secondary structure across the human transcriptome. Nature 505(7485): 706-709.
- Yoon, J. H., Srikantan, S. and Gorospe, M. (2012). MS2-TRAP (MS2-tagged RNA affinity purification): tagging RNA to identify associated miRNAs. Methods 58(2): 81-87.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Uppala, J. K., Ghosh, C., Sabat, G. and Dey, M. (2022). Pull-down of Biotinylated RNA and Associated Proteins. Bio-protocol 12(4): e4331. DOI: 10.21769/BioProtoc.4331.
- Ghosh, C., Uppala, J. K., Sathe, L., Hammond, C. I., Anshu, A., Pokkuluri, P. R., Turk, B. E. and Dey, M. (2021). Phosphorylation of Pal2 by the protein kinases Kin1 and Kin2 modulates HAC1 mRNA splicing in the unfolded protein response in yeast. Sci Signal 14(684).
Category
Molecular Biology > RNA > RNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.