- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Restimulation-Induced Cell Death (RICD): Methods for Modeling, Investigating, and Quantifying RICD Sensitivity in Primary Human T Cells via Flow Cytometric Analysis
Published: Vol 12, Iss 4, Feb 20, 2022 DOI: 10.21769/BioProtoc.4326 Views: 4914
Reviewed by: Giusy TornilloAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2021
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
When the body mounts an immune response against a foreign pathogen, the adaptive arm of the immune system relies upon clonal expansion of antigen-specific T cell populations to exercise acquired effector and cytotoxic functions to clear it. However, T cell expansion must be modulated to effectively combat the perceived threat without inducing excessive collateral damage to host tissues. Restimulation-induced cell death (RICD) is an apoptotic program triggered in activated T cells when an abundance of antigen and IL-2 are present, imposing a negative feedback mechanism that constrains the growing T cell population. This autoregulatory process can be detected via increases in caspase activation, Annexin V binding, and loss of mitochondrial membrane potential. However, simple changes in T cell viability through flow cytometric analysis can reliably measure RICD sensitivity in response to T-cell receptor (TCR) restimulation. This protocol describes the in vitro polyclonal activation, expansion, and restimulation of human primary T cells isolated from donor peripheral blood mononuclear cells (PBMC). This simple procedure allows for accurate quantification of RICD via flow cytometry. We also describe strategies for interrogating the role of specific proteins and pathways that may alter RICD sensitivity. This straightforward protocol provides a quick and dependable tool to track RICD sensitivity in culture over time while probing critical factors that control the magnitude and longevity of an adaptive immune response.
Graphic abstract:
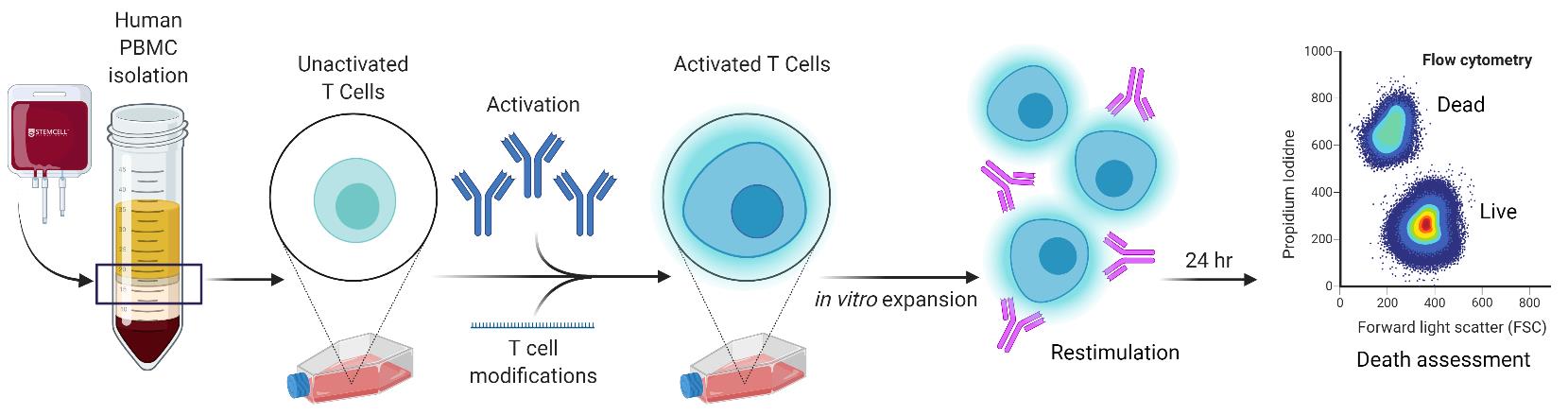
In-vitro simulation of restimulation-induced cell death in activated human T cells.
Background
Adaptive immunity requires the selective engagement of highly specific T cell clones to respond to “foreign invaders”, including microbial pathogens and tumor cells. An effective T cell response demands a fine balance of T cell receptor (TCR) and costimulatory/coinhibitory signals in order to regulate both expansion and contraction. The ability to raise powerful “armies” of CD4 and/or CD8 effector T cells from naïve or memory compartments ultimately determines whether a foreign invader will be neutralized or eliminated. Concurrently, the immune system must regulate these deadly forces to guard against deleterious immunopathologies associated with lymphoproliferation, hyperinflammation, and autoimmunity. Several checkpoints and signaling molecules help determine the size and duration of a T cell effector response after activation, including propriocidal apoptotic programs. One such self-regulatory pathway used to constrain expanding T cell populations is restimulation-induced cell death (RICD) (Nagy et al., 2010; Snow et al., 2010). RICD relies on the presence of antigen and interleukin-2 (IL-2) to induce programmed cell death in a subset of activated T cells following TCR re-engagement (Figure 1). This is distinct from cytokine withdrawal-induced death, an intrinsic apoptotic program triggered by IL-2 deprivation after pathogen clearance, and responsible for most effector cell contraction (Snow et al., 2010). Understanding the mechanisms that govern RICD sensitivity can help to elucidate if and how abnormal T cell dynamics might contribute to lymphoproliferative disorders, lymphomas, autoimmune diseases, infections, and other states of immune dysregulation.
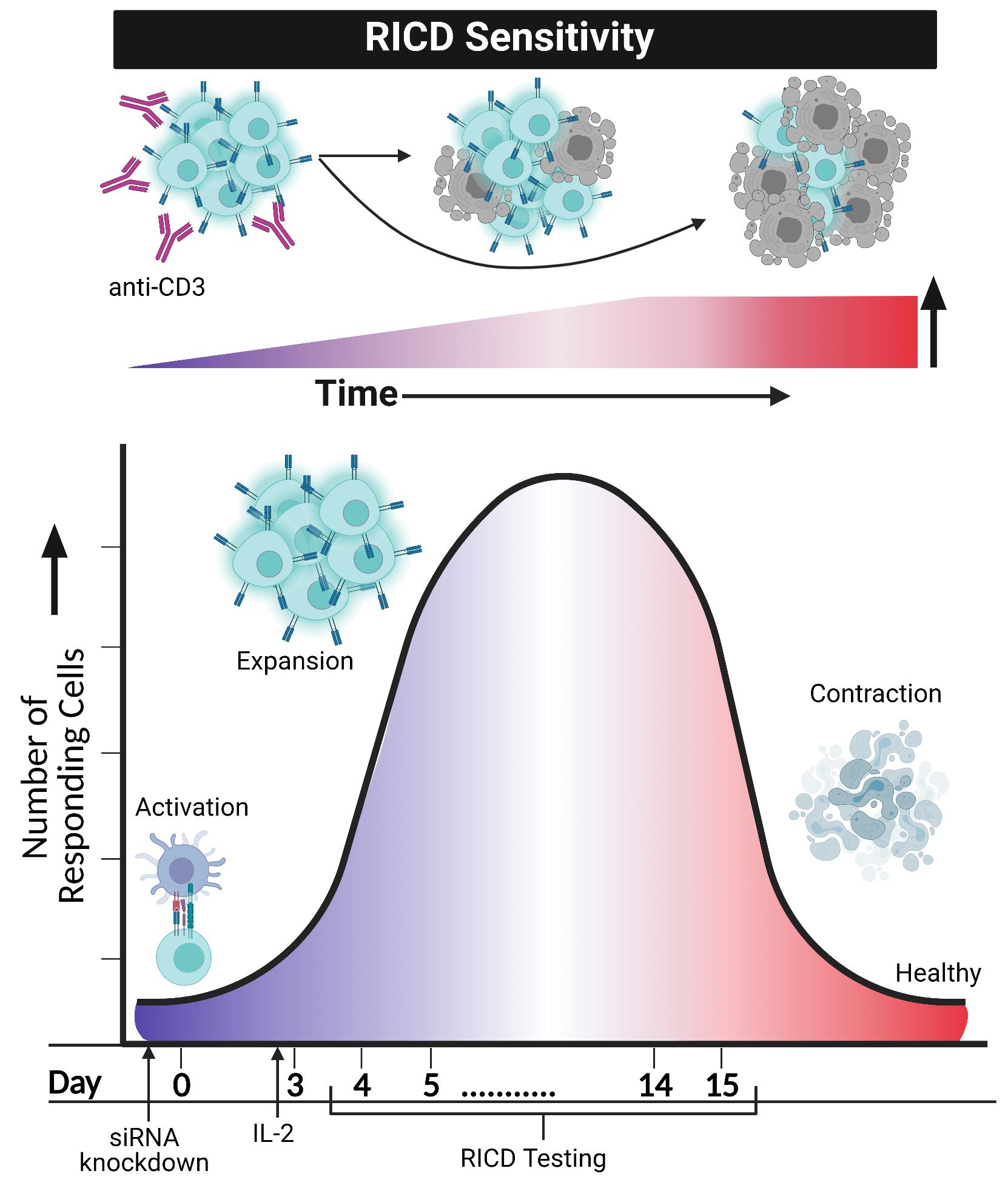
Figure 1. RICD sensitivity in activated T cells increases over time and may be imitated and investigated in an in vitro format. Restimulating activated T cells with anti-CD3 in the presence of IL-2 will cause a certain percentage of T cells to induce apoptosis. Anti-CD28 does not seem to be a contributor to this phenomenon, despite its requirement for T cell activation. Simulation of RICD through in vitro assays may be performed, testing cells anywhere from day 4 post-activation to day 15. Modulation of T cells, such as siRNA knockdown of various proteins, can occur before day 0 activation with CD3/CD28. After IL-2 addition to in vitro cultures of T cells on day 3, RICD may be tested in “early stage” effector cells (day 4) or “late stage” effector cells (day 14). Comparing control and siRNA knockdown cells helps elucidate mechanistic and molecular requirements for RICD sensitivity. Figure created using Biorender.com.
Apoptosis is utilized through T cell development and differentiation to dispose of nonfunctional, autoreactive, or excess cells (Zhang et al., 2005). Therefore, delineating mechanisms of T cell homeostasis and RICD is critical for understating T cell-driven immunopathology. In humans, autoimmune lymphoproliferative syndrome (ALPS) and X-linked lymphoproliferative disease 1 (XLP-1) are associated with apoptosis defects linked to loss-of-function or null mutations in Fas/FasL and SAP (SLAM-associated protein), respectively (Snow et al., 2009; Nagy et al., 2010; Nourse et al., 2010). ALPS patients exhibit enlarged lymph nodes and spleens, with a predilection for autoimmunity and lymphoma development. XLP-1 patients experience an overreactive and life-threatening accumulation of CD8 effector T cells in response to infection by Epstein Barr virus (EBV), which we specifically linked to RICD resistance in the absence of SAP (Snow et al., 2009). Aside genetic pre-dispositions, the tumor microenvironment also forces antigen-specific TILs to more readily succumb to apoptosis (including RICD), limiting tumor control and highlighting the significance of this pathway in multiple environments.
Early descriptions of RICD were termed “activation-induced cell death (AICD)”, initially observed as a decrease in growth of murine T cell hybridomas after antigen stimulation in a dose-dependent manner (Ashwell et al., 1987a and 1987b). However, further studies introduced “RICD” as a more relevant descriptor for this phenomenon, which only occurs in previously activated and proliferating effector T cells that experience TCR re-ligation in the presence of IL-2 (Snow et al., 2010). Early investigations indicated that RICD was specifically restricted to cycling T cells and attributed to TCR-induced FASL induction, which triggered autocrine/paracrine FAS-dependent apoptosis particularly in CD4 T cells (Boehme and Lenardo, 1993; Brunner et al., 1995; Dhein et al., 1995; Lenardo et al., 1999). Additional pro-apoptotic molecules, such as BIM and perforin/granzymes, were later implicated in RICD of CD8 T cells (Mateo et al., 2007; Snow et al., 2008). Our subsequent work defined how SAP (SH2D1A) potentiates TCR signal strength by displacing SHP-1 (SH2 domain-containing protein tyrosine phosphatase) and recruiting more LCK (lymphocyte-specific protein tyrosine kinase) to the SLAM family receptor NK, B, and T cell antigen (NTB-A), as well as inhibiting diacylglycerol kinase alpha (DGKa), to promote the expression of several pro-apoptotic molecules (FASL, BIM, NOR1, NUR77) involved in executing RICD (Katz et al., 2014; Ruffo et al., 2016). Moreover, natural suppression of SAP and FASL expression in regulatory T cells renders them resistant to RICD, enforced by the master regulator FOXP3 (Katz et al., 2018). More recently, we discovered that cellular metabolic processes, including glycolysis and fatty acid synthesis, can influence the apoptotic susceptibility of effector T cells (Larsen et al., 2017; Voss et al., 2017 and 2019). Despite these mechanistic insights into RICD of terminally-differentiated (i.e., “late-stage”) effector T cells, it remains unclear how early activated T cells undergoing clonal expansion (i.e., “early stage”) are spared from premature RICD in the presence of abundant antigen and IL-2. We recently described how transient FOXP3 expression can protect newly-activated human conventional T cells from RICD by promoting basal autophagy and suppressing p53 (Voss et al., 2021). The protocol described herein is based on our recent paper describing how TIM-3, a co-signaling protein often linked to CD8 T cell exhaustion, can influence subtle and temporal changes in RICD sensitivity (Lake et al., 2021).
Our original in vitro RICD protocol for human late-stage effector T cells was described previously (Katz and Snow, 2013). Here, we expand on a modified protocol to measure RICD sensitivity at different stages of the T cell response, including the interrogation of specific target proteins like TIM-3 to elucidate their role in tuning RICD sensitivity. Our method offers a quick and easy way to model human T cell activation and RICD induction in an in vitro setting, which may be coupled with other apoptotic readouts such as Annexin V staining, caspase activation, and mitochondrial depolarization (Wlodkowic et al., 2011). Clinicians and research scientists may utilize this procedure to evaluate and manipulate RICD sensitivity over time in healthy donor T cells, or test for abnormal RICD in patients with lymphoproliferative disease, or other primary immune regulation disorders (PIRDs). Through this approach, our method has proven highly valuable in delineating myriad molecular mechanisms that govern T cell homeostasis via RICD.
Materials and Reagents
50 mL conical tubes (sterile) (Genesee, catalog number: 28-106)
15 mL conical tubes (sterile) (Genesee, catalog number: 28-103)
9” Glass Pasteur pipettes (Daigger, catalog number: EF20402C)
10 mL serological pipettes (Genesee, catalog number: 12-104)
5 mL serological pipettes (Genesee, catalog number: 12-102)
6 well tissue culture plates (Diagger, catalog number: EF24881)
Vented tissue culture flasks (Genesee, catalog number: 25-209)
96 well round bottom plates (Genesee, catalog number: 25-109)
1.1 mL polypropylene tubes (USA Scientific, catalog number: 1412-1000)
5 mL round bottom tubes for flow cytometry (VWR, catalog number: 60819-728)
16 mL sterile polystyrene culture tubes (Genesee, catalog number: 21-129)
Mouse IgG1 Isotype Control SAFIRE (Bio-gems, 44212-25)
Whole blood or “buffy coat” (i.e., PBMC enriched fraction of whole blood post centrifugation)
Ficoll-Paque PLUS (GE Healthcare, catalog number: 17-1440-03)
1× Phosphate-buffered saline (PBS) (VWR, catalog number: 95042-486)
ACK Lysis Buffer (VWR, catalog number: 10128-806)
RPMI-1640 w/ Glutamine (VWR, catalog number: 95042-508)
Fetal Bovine Serum (Milipore Sigma, catalog number: F0926-500)
Penicillin-Streptomycin (Thermo Fisher, catalog number: 15140163)
Trypan blue 4 g/L (VWR, catalog number: K940-100ML)
Immunocult CD3/CD28/CD2 T Cell Activator (Stem Cell Technologies, catalog number: 10970)
Anti-human CD3 antibody, clone HIT3a NA/LE (BD Biosciences, catalog number: 555336)
Anti-human CD28 antibody (BD Biosciences, catalog number: 555725)
Anti-CD3 antibody OKT3 (Bio-gems, catalog number: 01521-05)
Recombinant human IL-2 (Peprotech, catalog number: 200-02)
Propidium iodide (Thermo Fisher, catalog number: P1304MP)
Amaxa P3 Primary Cell 4D-Nucelofactor X Kit L (Lonza, V4XP-3024) (included: Lonza P3 buffer, Lonza pipettes, and Lonza cuvettes)
RoboSepTM Buffer (Stem Cell Technologies, catalog number: 20104)
Human CD8+ T cell Isolation Kit (Stem Cell Technologies, catalog number: 17953)
TIM-3 siRNA (Thermo Fisher, catalog number: 4392420) (stored at -20°C, aliquoted)
Nonspecific siRNAcontrol (Thermo Fisher, catalog number: 12935300) (stored at -20°C, aliquoted)
TIM-3 blocking monoclonal antibody, clone F38-2E2 (Biolegend, catalog number: 35004)
Recombinant human Tim-3-FC Chimera (R&D Systems, catalog number: 2365-TM-050)
200 Proof Ethanol (Decon Labs Inc., catalog number: 2716)
Ultra-pure water (university provided)
70% ethanol (see Recipes)
Complete RPMI Media (see Recipes)
Antibiotic-free RPMI Media (see Recipes)
Recombinant human IL-2 solution (rIL-2) (see Recipes)
siRNA aliquots (see Recipes)
PI solution (see Recipes)
Equipment
Centrifuge Sorvall Legend X1XTR (Thermo Fisher, catalog number: 75004539)
Class II A2 biosafety cabinet (for sterile workflow)
Accuri C6 Flow Cytometer (Becton Dickenson)
Vortex-Genie 2 (Scientific Industries Inc., catalog number: SI-0236)
4D NucleofectorTM Core Unit and 4D NucleofectorTM X Unit (Lonza, catalog number: AAF-1002B and AAF-1002X) (included: Lonza cuvette holder)
“The Big Easy” EasySepTM Magnet (Stem Cell Technologies, catalog number: 18001)
Pipette Aid (Genesee, catalog number: 33-901)
Shel Lab 37°C water bath (Sheldon Manufacturing Inc., catalog number: SWB15)
HeracellTM 150i 5% CO2 Incubator (Thermo Fisher, catalog number: 50116048)
Cell counter or hemocytometer
Software
Accuri C6 Flow Cytometer Software (Becton Dickenson)
FlowJo (Becton Dickenson, https://www.flowjo.com/)
Procedure
Isolation of Peripheral Blood Mononuclear Cells Using Ficoll
*To avoid confusion, sample(s) in the form of buffy coat or whole blood specimen tube (e.g., ACD tube) from one patient will be called a “donor”. Processing steps are described for a single buffy coat donor of 25 mL. However, we have used this scalable protocol to process up to 15 unique donors (8–30 mL whole blood) simultaneously. All steps are performed in a sterile biosafety cabinet (BSC), except for centrifugations (closed tubes). Isolation of PBMCs can be accelerated through other technologies [e.g., SepMate tubes (StemCell Technologies) or CPT tubes (Becton Dickinson)]. However, in our experience the Ficoll protocol described herein provides the best purification and yield by comparison.
Prepare and sterilize materials with 70% ethanol for use in the sterile BSC: 9” glass pipette tips, Ficoll Plus, 50 mL conical centrifuge tubes, 1× PBS, ACK Lysis Buffer, and donor samples. Place complete RPMI media in a water bath at 37°C.
Pour 25 mL of donor sample into a labeled 50 mL centrifuge tube.
Dilute donor sample 1:1 with 1× sterile PBS: pour PBS into the 50 mL centrifuge tube until the 50 mL mark is reached.
Cap the tube and invert 4–6 times to mix sample and PBS.
Pour half of the PBS/blood mixture into another labelled 50 mL centrifuge tube so that each contains 25 mL.
Place one glass Pasteur pipette into each 50 mL centrifuge tube so that the tip of the pipette is touching the base of the tube.
Distribute 13 mL of Ficoll Plus into each 50 mL tube through the Pasteur pipette using a 10 mL serological pipette tip and Pipette Aid (pipette by adding 3 ml aliquots of Ficoll at a time, repeat until 13 mL is added), creating a clear distinct Ficoll bottom layer under the sample/PBS mixture top layer as depicted in Figure 2 and Video 1.
Note: Filling the Pasteur pipettes with Ficoll gradient can be slow and tricky. The 9” Pasteur pipettes fill exactly 3 mL at a time, but drain more slowly to achieve an undisturbed Ficoll layer at the bottom of each tube. Additionally, caution should be taken to gently rest the serological pipette tip on the Pasteur pipette as to not break the Pasteur pipette. When 13 mL have been drained to the base of the 50 mL centrifuge tube, the Pasteur pipette should be gently dragged up the side of the 50 mL tube and slowly removed, as to not disturb the gradient and allow any residual Ficoll in the pipette to drain to the base.
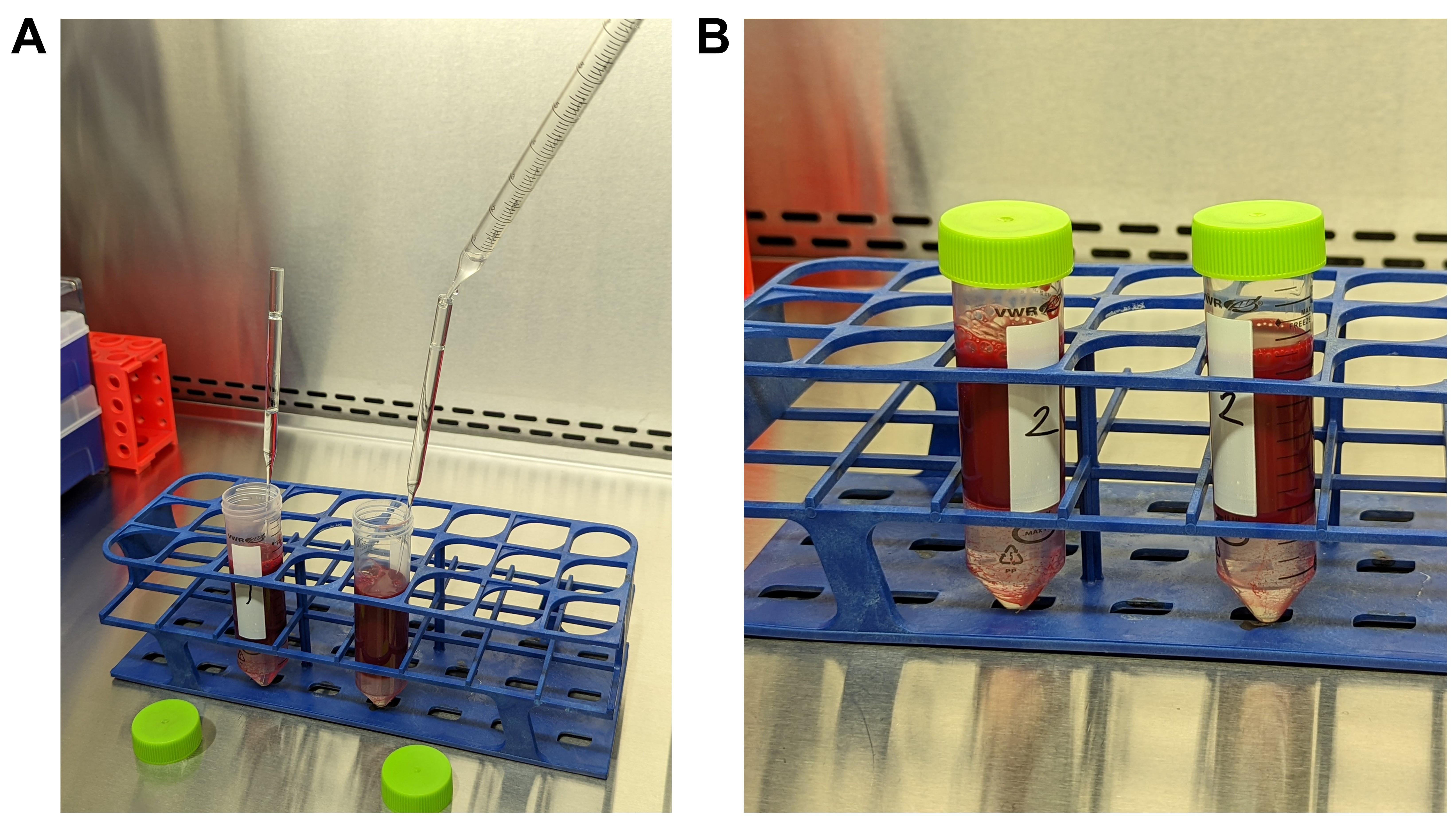
Figure 2. Addition of Ficoll for PBMC isolation. A. The set-up for Ficoll distribution is depicted. Two 50 mL centrifuge tubes have equal portions of the sample/PBS mixture. The Pasteur pipette is placed in the mixture so the tip rests in the conical portion of the tube. A 10 mL serological pipette is filled with Ficoll and gently rested on the opening of the Pasteur pipette, slowly depositing 3 mL of Ficoll or until the pipette is full. The process of refilling the serological pipette, and distributing the Ficoll in increments no greater than 3 mL, occurs until 13 mL is present in each centrifuge tube. B. The final product of Steps A7 and A8 should resemble this panel. The Pasteur pipette was removed slowly as to not disturb the Ficoll layered on the bottom. There is only a small presence of RBC in the Ficoll. The solutions are now able to be slowly transferred to the centrifuge. Video 1. Ficoll technique for isolation of PBMC from human buffy coats.
Video 1. Ficoll technique for isolation of PBMC from human buffy coats.After slowly removing the Pasteur pipette, cap the centrifuge tubes gently so as to not disturb the Ficoll layer at the bottom.
Gently place the 50 mL centrifuge tubes in the centrifuge (swinging bucket rotor, balanced on each side) and spin at 2,000 RPM at room temperature for 20 min (full speed acceleration, no deceleration).
Note: Rapid deceleration and braking can disrupt the Ficoll gradient; therefore, the centrifuge rotors should decelerate naturally with no brake.
While tubes are spinning, label another set of sterile 50 mL centrifuge tubes.
Carefully retrieve tubes and bring back to the BSC to uncap.
Note: Post-centrifugation and separation on the Ficoll gradient, the PBMC layer (i.e., buffy coat) is extremely delicate to shaking and disturbances. Carry PBMC 50 mL tubes in a rack to and from the centrifuge, and avoid inadvertent mixing of layers in the gradient when retrieving the PBMC (i.e., do not expel liquid from the pipette).
Using a 5 mL serological pipette tip, gently collect the white layer of PBMC sitting at the plasma:Ficoll gradient interface and place it into a new 50 mL centrifuge tube, as shown in Figure 3. Minimize the volume retrieved, avoiding the Ficoll and erythrocyte/granulocyte pellet.
Note: Ficoll gradients produce four distinct layers that may be retained for different uses depending on experimental set-up and interest. From top to bottom, the order is as following: plasma/PBS, PBMC, Ficoll, erythrocyte/granulocyte pellet. Occasionally, erythrocytes may remain in the PBMC layer and cause a light red discoloration. The ACK Lysis Buffer is used to eliminate these residual erythrocytes. Avoid transferring any part of the pellet or “chunks” of erythrocytes in the PBMC layer, by pipetting carefully.

Figure 3. Isolation of the PBMC layer after Ficoll separation. A. After the Ficoll gradient is formed via centrifugation, the white PBMC layer will become apparent (shown by the red arrow). The PBMC layer should have few RBCs. B. Removal of the white PBMC layer may be achieved by using the 5 mL serological pipette tip and delicately placing the tip on top of the PBMC layer, slowly removing it while avoiding the serum (above) and Ficoll (below). Angling the tube may help.Repeat Step A12 if there are residual PBMCs visible at the interface, and place the resulting into the same centrifuge tube (total volume <8–10 mL per tube). Discard original tubes with Ficoll unless you are also collecting plasma (top) and/or granulocytes (pellet) for additional use.
Fill the new PBMC tube with 1× sterile PBS until the 50 mL line is reached.
Centrifuge the tube at 1,400 × g at room temperature for 10 min (full speed acceleration, and deceleration from this point forward).
During this spin, label another set of 50 mL centrifuge tubes in the BSC to create two sets of centrifuge tubes per donor/sample.
Attempting to not disturb the pellet, pour half of the contents of the recently centrifuged tube into the newly labelled one (approximately 25 mL each tube). Fill both up to the 50 mL mark with 1× PBS.
Repeat Step A14.
Note: Due to the greater density of Ficoll, excess Ficoll removed with the PBMC layer can prohibit the proper sedimentation of PBMCs when centrifuging during PBS wash steps (A12–15). If the wash still appears cloudy after centrifugation, decant supernatant into a separate tube, dilute further in PBS, and repeat centrifugation to maximize PBMC recovery.
Aspirate the supernatant from each 50 mL centrifuge tube without disturbing the PBMC pellet, which may appear white, pink or red depending on the presence of residual erythrocytes.
Resuspend the pellet(s) in a total 5 mL of ACK Lysis Buffer. Incubate at room temperature for 5 min.
Note: If there is still a red tint or streak present in the PBMC pellets after ACK lysis, this step may be repeated. However, further incubations can decrease PBMC recovery.
Fill the 50 mL centrifuge tube to the 50 mL mark with 1× PBS.
Centrifuge the tube at 1,400 × g at room temperature for 10 min.
Aspirate off supernatant and resuspend the white PBMC pellet in 10 mL of complete RPMI media.
Count PBMC in media using a hemocytometer or automated cell counter, diluting as needed to acquire an accurate cell count. Use a 1:1 dilution of Trypan Blue to discriminate live (white/clear) vs. dead cells (blue).
Notes:
When isolating cells from a buffy coat of 20–25 mL, we generally recover 150–900 million PBMC (average ~400–500 million). Therefore, a 50 mL dilution of all PBMC is too concentrated for accurate cell counting. Hence if buffy coats are the primary source of PBMC, we suggest further diluting a 10 μL aliquot of this solution 1:10. PBMC recovery from whole blood is generally lower (1 × 106–2 × 106 cells/mL blood). Residual erythrocytes may still be seen in the hemocytometer; if excessive, an additional RBC lysis may be required.
PBMC may also be stimulated and cultured as a whole, kept between 1 × 106–2 × 106 cells/mL in complete RPMI media supplement with an appropriate rIL-2 concentration and used for RICD assays at the desired post-activation date.
Purification of resting CD8+ T cells from PBMC
*The protocol provided is nearly identical to what is described within the instruction manual of Stem Cell Technologies human CD8+ T cell Isolation Kit (https://www.stemcell.com/easysep-human-cd8-t-cell-isolation-kit.html#section-related-products), assuming an average input of 500 million PBMC. We have also isolated CD4+ T cells, naïve cells, and other T cell populations to test for RICD sensitivity.
Note: On average, we use 250–500 million PBMC to isolate 15–40 million CD8+ T cells in 5–10 mL of RoboSepTM Buffer. Stem Cell Technologies EasySepTM Buffer or PBS may also be used (especially when diluting).
Prepare the BSC with required materials: EasySepTM magnet, 16 mL cell culture tube, and RoboSepTM Buffer.
Centrifuge PBMCs at 1,400 × g at room temperature for 5 min.
Aspirate the media supernatant and resuspend pellet in 5 mL of RoboSepTM Buffer (concentration = 5 × 107 PBMC/mL).
Transfer cell suspension to a 16 mL sterile culture tube.
Add 50 μL/mL of CD8+ T cell isolation cocktail to the tube (total = 500 μL). Pipette up and down to mix.
Incubate at room temperature for 5 min.
Vortex RapidSpheresTM for 30 s during the Step 6 incubation.
Add 50 μL/mL of RapidSpheresTM to the solution (total = 500 μL). Pipette up and down to mix.
Incubate at room temperature for 3 min.
Place the solution tube uncapped into the EasySepTM magnet. Leave at room temperature for 3 min.
Without removing or disturbing the tube in the magnet, carefully decant the contents (i.e., isolated CD8+ T cells) into a new 15 mL conical tube.
Centrifuge the isolated CD8+ T cells at 90 × g at room temperature for 10 min.
Resuspend the CD8+ T cell pellet in 10 mL complete RPMI media and count cells (see Step A24).
Activating, modifying, and culturing CD8+ T cells
*The stimulating agent used in our experiments is typically Immunocult Human CD3/CD28/CD2 T Cell Activator, referenced in the following steps. However, other stimulating agents are discussed in the notes.
Activating and culturing of unmodified CD8+ T cells
Adjust CD8+ T cells to 2 × 106 cells/mL in complete RPMI media.
Add the activating agent Immunocult at 25 μL/mL. Using a serological pipette, gently mix by pipetting up and down, and transfer to a vented tissue culture flask.
Note: Our lab uses Immunocult anti-CD3/CD28/CD2 activating reagent to robustly stimulate CD4+ and CD8+ T cells. However, we have used many different activation stimuli, ranging from bead cross-linked or plate-bound anti-CD3/CD28, anti-CD3/CD28 Dynabeads, homemade magnetic beads, or PMA/Ionomycin, a method for bypassing the TCR engagement to stimulate T cells. Soluble anti-CD3/CD28 antibodies (Abs) do not provide adequate stimulation of purified T cells in the absence of Fc receptor bearing accessory cells, and should only be used when stimulating T cells in unfractionated PBMC (Katz and Snow, 2013).
Place the tissue culture flask in 5% CO2 incubator at 37°C.
Count the cells using trypan blue 24 h later. Add complete RPMI media to the mixture to achieve a cell concentration of 1 × 106 cells/mL.
Continue culturing until day 3, sustaining cells at a concentration between 1 × 106–2 × 106 cells/mL.
Note: Monitoring of cells after activation is important to ensure proper activation. The presence of CD69 on day 1 and CD25 on days 2–3 may be observed through flow cytometry. Cells that are not properly blasting, aka not increasing in size and not clumping, can give rise to skewed RICD results.
On day 3 post-activation, count the cells with Trypan blue. Start warming complete RPMI media (in a 37°C water bath) and thaw recombinant IL-2 (rIL-2) on ice (individual aliquot = 3 × 105 U/mL).
Note: IL-2 is an important addition for T cell proliferation due to the upregulation of the IL-2 receptor (CD25) 2–3 days after activation. Rapid expansion will occur up to days 7–10, requiring media color and cell density to be monitored every day. If whole PBMC populations were activated, IL-2 addition will result in accumulations of largely CD4+ and CD8+ T cells.
Transfer CD8+ T cells to appropriate sterile tubes and centrifuge at 1,400 × g at room temperature for 5 min.
Aspirate supernatant and resuspend cells in 5 mL of 1× PBS. Fill the remainder of the centrifuge tube.
Repeat Step C1g.
Aspirate supernatant and resuspend cells in complete RPMI media at 1 × 106 cells/mL supplemented with 100 U/mL of rIL2 (1:3,000 dilution from stock).
Count cells with trypan blue 24 h later in preparation for the RICD assay (day 4: early stage), or continue culturing to generate late-stage effector T cells for subsequent RICD assays (e.g., day 14) by keeping cells between 1 × 106–2 × 106 cells/mL in complete RPMI media + 100 U/mL rIL-2. Move cell suspension to additional/larger vented tissue culture flasks as necessary.
Activating and culturing modified CD8+ T cells
* Herein we describe our protocol for siRNA transfection in human primary CD8+ T cells with a control [non-specific (NS) siRNA] vs.TIM-3 specific siRNA in a single donor. This protocol is also scalable for more donors and/or conditions involving additional siRNAs, or other modifications such as CRISPR (Seki et al., 2018). All siRNAs used were maintained at 50 μM and frozen in 10 μL aliquots (consult providing company for usable siRNA concentrations). We typically transfect 1 × 107 million CD8+ T cells per condition, but we have achieved suitable knockdown results with fewer cells (5–8 million).
Place CD8+ T cells at 2 × 106 cells/mL in antibiotic free media (RPMI + 10% FCS) in a vented tissue culture flask in the incubator at 37°C for 30 min.
Note: CD8+ T cells are relatively stable in the 37°C incubator prior to activation. We have kept unstimulated CD8+ T cells for at least 3 days resting in the incubator prior to activation with no large changes in activation potential and phenotype. PBMC can tolerate overnight, especially T cells, but the possible adherence of these mononuclear cells to the flasks will limit recoverability.
During this time, prepare and sterilize materials in the BSC for siRNA nucleofection: 2 Lonza cuvettes, 2 Lonza pipettes, and a Lonza cuvette holder. Thaw siRNA aliquots on ice (500 pmol each).
Prepare a 6-well cell culture dish with 3 mL of antibiotic free RPMI media per condition per donor: one condition for control, one condition for targeted knockdown. Place in the 5% CO2 incubator at 37°C until ready.
Centrifuge 2 × 107 CD8+ T cells at 90 × g at room temperature for 10 min.
During the centrifugation, place the Lonza P3 buffer on the bench top to warm to room temperature.
Place the cell samples in the hood along with the Lonza P3 buffer, cell culture plate, and thawed aliquots for NS and TIM-3 siRNA.
Aspirate the media from the CD8+ T cell pellet and resuspend in P3 buffer at 90 μL per 1 × 107 cells, gently pipetting up and down once or twice.
Transfer 90 μL of P3 cell/buffer solution to a tube containing a 10 μL aliquot of NS siRNA and immediately transfer the 100 µL volume directly into the Lonza cuvette.
Repeat for TIM-3 siRNA (or siRNA specific for another target gene).
Place each cuvette in the Lonza 4D Nucleofector and nucleofect under the following conditions: Buffer: buffer – P3, Program: – Human, unstim HF. Once a successful nucleofection program is completed, return the cuvettes to the hood.
Using the Lonza transfer pipettes, in one motion, pick up ~100–200 μL culture media from the appropriate well in the cell culture plate, add it dropwise into the cuvette, retrieve the entire solution of media/P3 buffer and return dropwise to the same well in the cell culture plate. While this step is time imperative, it is equally imperative to gently handle the cells to avoid breakage and death. Repeat for additional cuvettes in separate wells as needed.
Continue at Step C1b.
Plating T cells for RICD Assay
* The assay is run at 1 × 106 cells/mL. Each condition will require 1.2 × 106 cells and 1.2 mL of media; extra cells (1.3 × 106–1.4 × 106 cells in the corresponding volume) are extremely helpful to ensure an adequate quantity of solution, regardless of pipetting error, etc. A final concentration of 100 ng/mL of anti-CD3 OKT3 for restimulating the T cells is assumed here. Pharmacological inhibitors or blocking antibodies can be used and dosed 1 h before restimulation. TIM-3 blocking antibodies, IgG1 isotype controls, and chimeric proteins were used at 10 μg/mL.
Count CD8+ T cells with trypan blue.
Collect required CD8+ T cells from each condition and centrifuge settings at 1,400 × g at room temperature for 5 min.
During the spin, prepare enough warmed complete RPMI media supplemented with 100 U/mL IL-2 and bring required materials into the BSC: 96-well round bottom tissue culture plate, supplemented media, and anti-CD3 (OKT3 mAb).
Aspirate off media and resuspend pelleted CD8+ T cells in 600 μL of media.
Add 100 μL of the cell suspension into each well of the 96-well plate as shown in Figure 4 (6 wells total). The assay runs each condition in triplicate, either unstimulated or restimulated with OKT3.
Note: Using a range of OKT3 restimulating concentrations (~1–1,000) ng/mL can be helpful to generate a dose-dependent RICD response, observed as an increase in % cell loss by PI staining 24 h later. We suggest observing the dose curve in unmodified activated CD8+ T cells and then moving forward in experimentation with the selected concentrations.
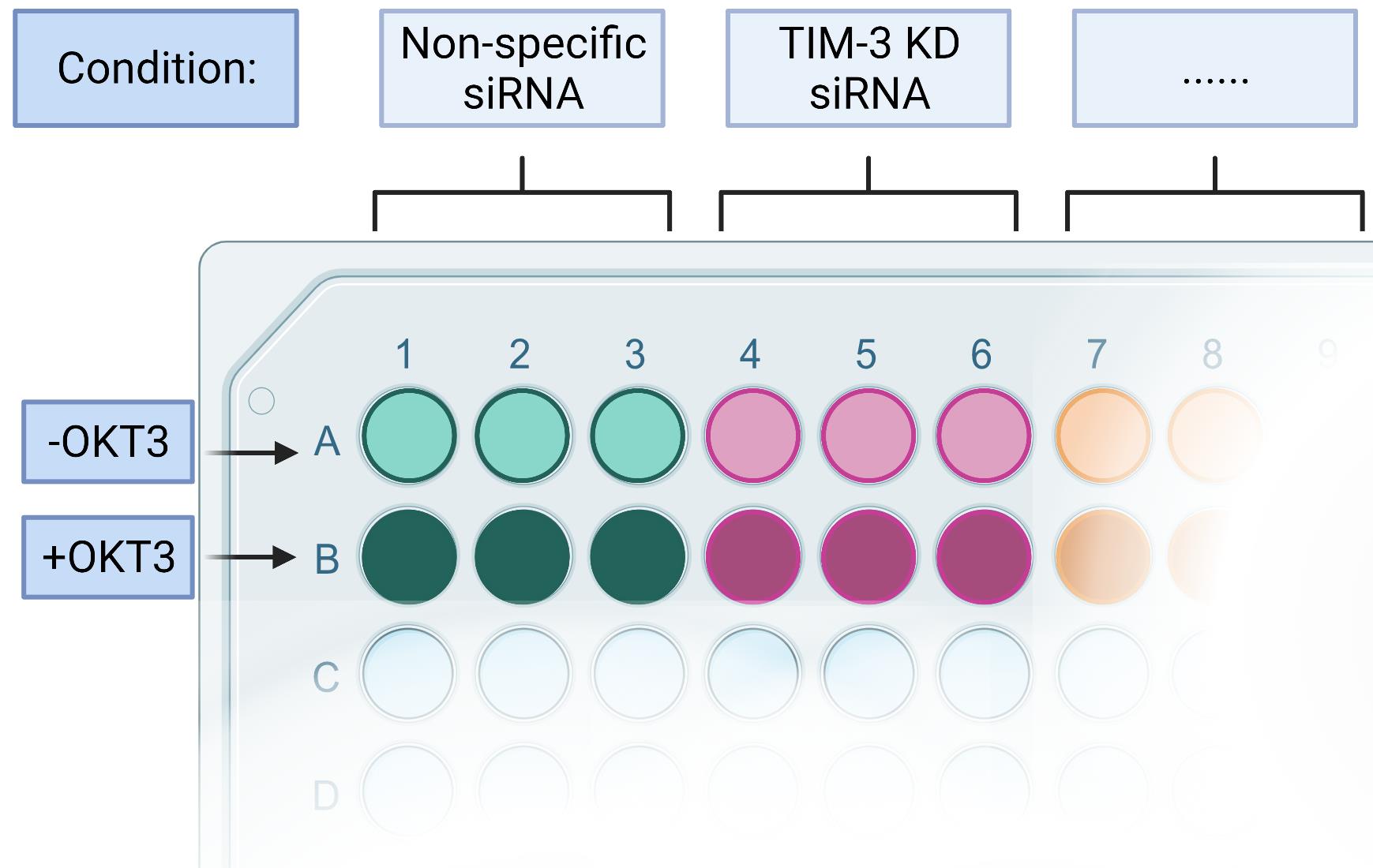
Figure 4. RICD assay set-up in a 96-well plate. The RICD assay should be formulated in a way where two types of controls are present. A control condition, consisting of either unaltered cells or control treatment (such as transfected non-specific siRNA), and a control triplicate where OKT3 is not added. Each condition, control or experimental, will consist of six wells: three containing OKT3 and three without it. Any number of conditions can be added to the plate. Figure created using Biorender.com.Note: We did not include the full 96-well plate in order to conserve space and be able to clearly identify applicable/utilized wells.
Repeat for each condition of CD8+ T cells.
Dilute OKT3 in complete RPMI media to a 200 ng/mL concentration.
Add 100 μL of OKT3 solution to the appropriate wells (100 ng/mL final concentration). For unstimulated control wells, add 100 μL of complete RPMI media.
Place the 96 well plate in the 5% CO2 incubator at 37°C for 24 h.
Note: PI+ cells typically appear by 18 h post-restimulation, but we run the assay around 24 h later without any large differences in cell loss.
Reading and Quantifying RICD Assay
Turn on and initialize the Accuri C6 flow cytometer (or flow cytometer of choice).
Using a 1 µg/mL solution of propidium iodide (PI), add 10 μL to each well in the 96-well plate on the benchtop.
Note: Propidium iodide is excited at 488 nm and emits around ~636 nm maximum, with a broad emission spectrum that can bleed into other channels (FL-3). This should be considered if other stains/fluorochromes are used simultaneously.
Thoroughly mix the wells with a 200 μL pipette. A 12 tip multichannel pipette is preferred for efficiency.
Transfer all wells into corresponding 1.1 mL polypropylene tubes.
Prepare Accuri acquisition template and gating strategy as follow:
Note: Plot 1 used in our analysis helps to make sure we visualize the appropriate size of cells and gauge cell viability based on this. SSC is side scatter and usually used to interpret internal complexity (shape and granularity) of a cell. FSC is forward scatter and useful for interpreting cell size. Together these parameters can help to show relative viability and purity of the cell sample for quality control. This plot is also useful for discerning different cell populations in PBMC. Plot 2 graphs positive PI staining from dead cells versus cell size, and is where our quantification statistics will be taken from. PI staining is not bimodal, as PI-positive staining can have a gradient of mean fluorescence intensity (MFI) increase. A third plot, FSC-H vs. FSC-A, may be utilized to exclude cell duplicates prior to data analysis.
Threshold: events >1,000,000 FSH-H
Dot Plot 1: FSC vs. SSC
Dot Plot 2: PSC vs. FL-2 (PI)
Place each 1.1 mL tube into a round bottom 5 mL flow cytometry sample tube. Briefly vortex each sample (1–2 s) before placing it on the Accuri C6 SIP.
Acquire sample data for 5 s on the slowest flow rate.
Locate cells and gate appropriately on the live CD8+ T cell population in Plot 2. Examples are shown in Figure 3.
Repeat run of the first sample to collect ~15,000 events according to the live cell gate in Plot 2. Record the number of seconds for this acquisition.
Set the C6 to acquire data from each subsequent sample for the set amount of constant time determined in the previous step. Time is the key measurement that will remain the same throughout the duration of a condition, but may change between conditions. This allows for reproducibility and experimental continuity.
Note: Using our example, we have two conditions: non-specific siRNA control cells and TIM-3 KD cells. Each condition will have six wells each, three without anti-CD3 and three with. The time to reach approximately 15,000 events in the live cell gate in the FL-2 vs. FSC plot will be recorded using the 1st non-specific siRNA sample in the triplicate without anti-CD3. This time is now used to acquire live cell events in the remainder two wells without anti-CD3 and the triplicate with anti-CD3. The next condition will repeat the timing process and does not need to be the same as the condition prior. However, large changes in time between conditions (>20 s) may signify inaccurate cell concentrations and skew results.
Repeat Steps E6-E10 for each condition of the CD8+ T cells.
Data analysis
RICD analysis has been previously described and published (Katz and Snow, 2013). Analysis involves calculating the percentage loss of viable cells in the restimulated conditions compared to the unrestimulated conditions, providing the most accurate assessment of cell death. Events recorded can be pulled from the Accuri C6 program from gates that were established during data acquisition, or from gates redrawn and established using another analysis program such as FlowJo (Figure 5). Calculations are as follows:
AVG-OKT3 = Average of triplicate events recorded in the viable FL-2 gate without OKT3
AVG+OKT3 = Average of triplicate events recorded in the viable FL-2 gate with OKT3
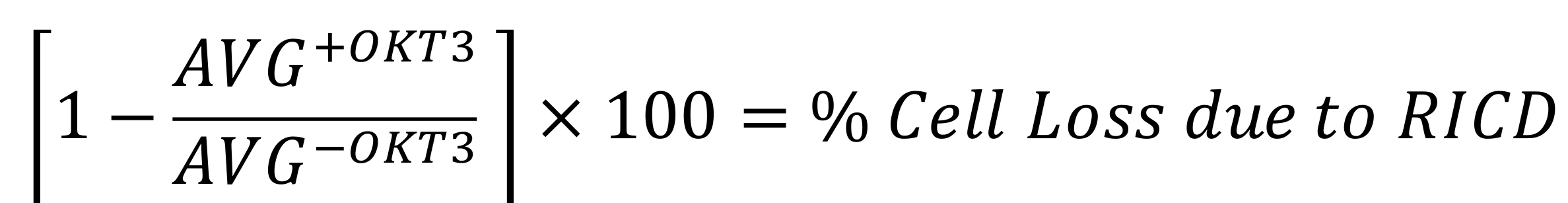
If there is a large variance in events recorded within the same triplicate, we will generally discard the experiment. This can happen due to pipetting errors, donor variation, or material/reagent changes. OKT3, the anti-CD3 Ab clone, induces a variable amount of apoptosis in T cells depending on the donor and what day post-activation the RICD assay is set up. Early stage cells typically experience between 10–25% cell death, whereas late stage cells may vary as much as 40–80% in cell loss. The benefit of counting viable cells within a specified period of time is the ability to account for cells that have died via apoptosis and disintegrated, not able to be detected by the flow cytometer. This method provides greater sensitivity and reproducibility over looking at percentage of positive cells or MFI changes.
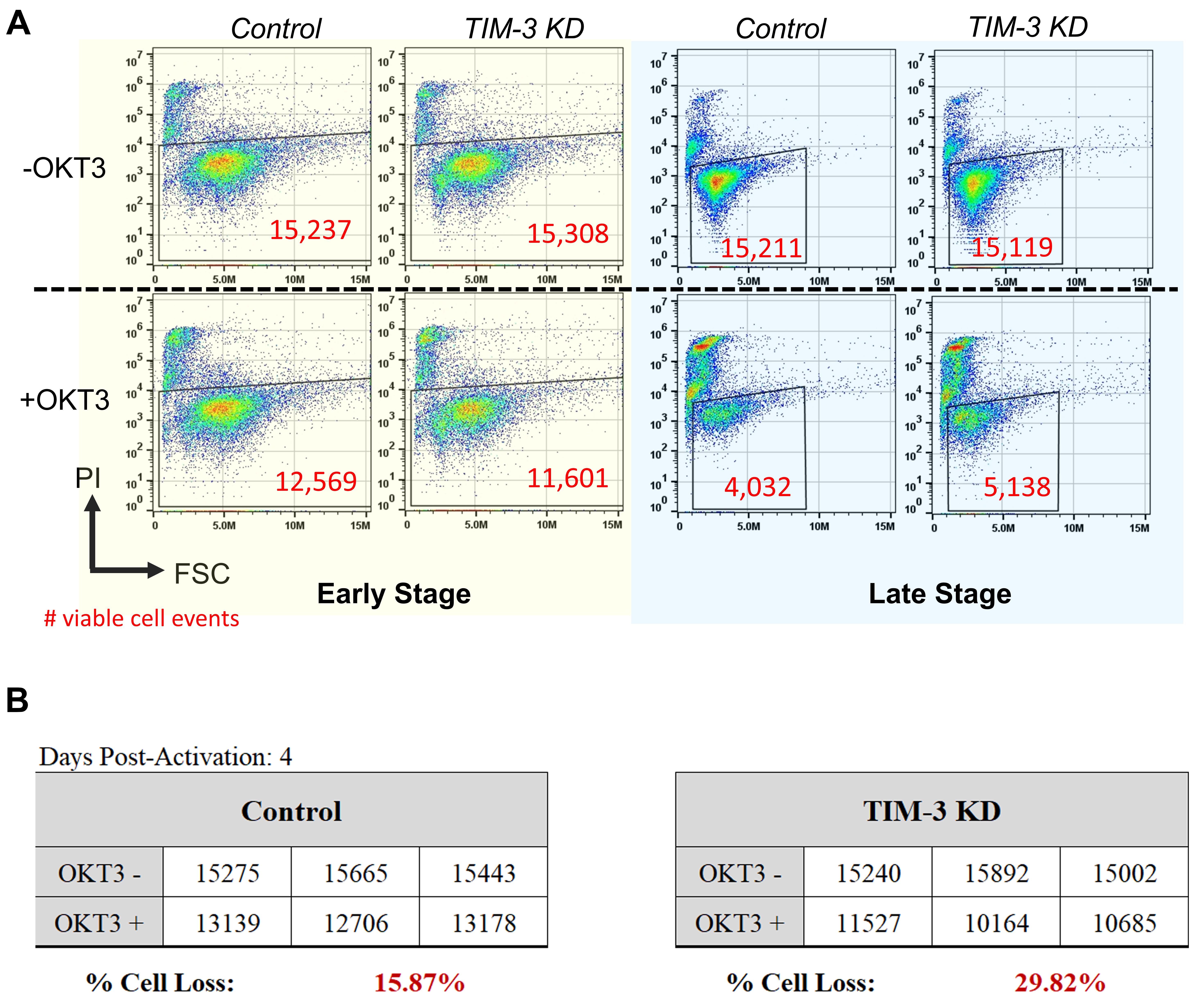
Figure 5. Flow cytometry output and RICD calculation. Illustrated here is our data analysis workflow from flow cytometry to RICD calculation using viable cell events. A. In the dot plots FSC vs. PI (FL-2), example images are present for early vs. late stage CD8 T cells (instead of the triplicate images usually acquired). The larger decrease in viable cell numbers can be appreciated in late stage populations, both for control and TIM-3 KD, due to increased RICD sensitivity at this time point. The decrease in viable cell events in +OKT3 compared to the corresponding -OKT3 plot can also be observed. Similar to what was published, loss of TIM-3 early stage exacerbated RICD while loss of TIM-3 late stage was protective towards RICD. B. A typical event record and calculation of percentage cell loss using the formula provided above with example RICD sensitivity seen at day 4 post-activation between NS (non-specific siRNA) control cells and TIM-3 KD.
Recipes
70% ethanol
700 mL of 200 Proof EtOH
300 mL of ultra pure water
Complete RPMI Media
500 mL of RPMI-1640 media w/Glutamine (2 nM)
10% heat-inactivated fetal bovine serum
1% Pen Strep
Antibiotic-free RPMI Media
Complete RPMI Media without Pen Strep
Recombinant human IL-2 solution (rIL-2)
Briefly centrifuge the parent vial (quick spin) to ensure all lyophilized IL-2 is at the bottom of the vial. Store on ice until step 2.
Prepare a fresh batch of 100 mM acetic acid. Sterile filter ~5 mL into a conical tube using a 0.45 μm syringe filter.
Add 2 mL of 100 mM acetic acid to the parent vial and pipet up and down to dissolve/mix. Current IL-2 concentration = 0.5 mg/ml. Place on ice.
Prepare ~40 mL of fresh 1× PBS + 0.1% BSA – sterilize using 0.45 μm syringe filter into a 50 mL conical tube.
Transfer 2 mL of IL-2 to a separate 50 mL conical tube containing 31.3 mL of sterile 1× PBS + 0.1% BSA. Final IL-2 concentration = 0.3 mg/mL (~3,000 U/mL).
Aliquot ~0.5 mL of diluted IL-2 into sterile labeled 1.5 mL microtubes. Store aliquots at -80°C.
siRNA aliquots
Reconstitute lyophilized siRNA material with nuclease-free water to 50 µM. Separate into 10 µL aliquots.
PI solution
Dilute Thermo Fisher Propidium Iodide to 1 µg/mL in double distilled water. Protect from light.
Acknowledgments
This work was funded by grants from the National Institutes of Health (R01GM05821, R35GM139619 to A.L.S) and Uniformed Services University, where this research and protocol design was performed. We also would like to thank the blood donor volunteers at the National Institutes of Health and Dr. Michael Leonardo for access to buffy coats. Additionally, we thank our lab members Bradley Bauman, Gina Dabbah, and Dr. Batsukh Dorjbal for their support, critiques, and assistance in this work. The original research utilizing this protocol was published in the following journal article: Lake et al. (2021).
Competing interests
No conflicts of interest to be declared.
Ethics
Anonymous, de-identified buffy coat and blood samples are obtained with informed consent from healthy volunteers at the National Institutes of Health Blood Blank (provided through Dr. Michael Leonardo).
References
- Ashwell, J. D., Cunningham, R. E., Noguchi, P. D. and Hernandez, D. (1987a). Cell growth cycle block of T cell hybridomas upon activation with antigen. J Exp Med 165(1): 173-194.
- Ashwell, J. D., Longo, D. L. and Bridges, S. H. (1987b). T-cell tumor elimination as a result of T-cell receptor-mediated activation. Science 237(4810): 61-64.
- Boehme, S. A. and Lenardo, M. J. (1993). Propriocidal apoptosis of mature T lymphocytes occurs at S phase of the cell cycle. Eur J Immunol 23(7): 1552-1560.
- Brunner, T., Mogil, R. J., LaFace, D., Yoo, N. J., Mahboubi, A., Echeverri, F., Martin, S. J., Force, W. R., Lynch, D. H., Ware, C. F. and et al. (1995). Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature 373(6513): 441-444.
- Dhein, J., Walczak, H., Baumler, C., Debatin, K. M. and Krammer, P. H. (1995). Autocrine T-cell suicide mediated by APO-1/(Fas/CD95). Nature 373(6513): 438-441.
- Katz, G. and Snow, A. L. (2013). Fluorescence-activated cell sorting-based quantitation of T cell receptor restimulation-induced cell death in activated, primary human T cells. Methods Mol Biol 979: 15-23.
- Katz, G., Krummey, S. M., Larsen, S. E., Stinson, J. R. and Snow, A. L. (2014). SAP facilitates recruitment and activation of LCK at NTB-A receptors during restimulation-induced cell death. J Immunol 192(9): 4202-4209.
- Katz, G., Voss, K., Yan, T. F., Kim, Y. C., Kortum, R. L., Scott, D. W. and Snow, A. L. (2018). FOXP3 renders activated human regulatory T cells resistant to restimulation-induced cell death by suppressing SAP expression. Cell Immunol 327: 54-61.
- Lenardo, M., Chan, K. M., Hornung, F., McFarland, H., Siegel, R., Wang, J. and Zheng, L. (1999). Mature T lymphocyte apoptosis--immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol 17: 221-253.
- Larsen, S. E., Bilenkin, A., Tarasenko, T. N., Arjunaraja, S., Stinson, J. R., McGuire, P. J. and Snow, A. L. (2017). Sensitivity to Restimulation-Induced Cell Death Is Linked to Glycolytic Metabolism in Human T Cells. J Immunol 198(1): 147-155.
- Lake, C. M., Voss, K., Bauman, B. M., Pohida, K., Jiang, T., Dveksler, G. and Snow, A. L. (2021). TIM-3 drives temporal differences in restimulation-induced cell death sensitivity in effector CD8+ T cells in conjunction with CEACAM1. Cell Death Dis 12(4): 400.
- Mateo, V., Menager, M., de Saint-Basile, G., Stolzenberg, M. C., Roquelaure, B., Andre, N., Florkin, B., le Deist, F., Picard, C., Fischer, A., et al. (2007). Perforin-dependent apoptosis functionally compensates Fas deficiency in activation-induced cell death of human T lymphocytes. Blood 110(13): 4285-4292.
- Nagy, N. and Klein, E. (2010). Deficiency of the proapoptotic SAP function in X-linked lymphoproliferative disease aggravates Epstein-Barr virus (EBV) induced mononucleosis and promotes lymphoma development. Immunol Lett 130(1-2): 13-18.
- Nourse, J. P., Jones, K., Dua, U., Runnegar, N., Looke, D., Schmidt, C., Tey, S. K., Kennedy, G. and Gandhi, M. K. (2010). Fulminant infectious mononucleosis and recurrent Epstein-Barr virus reactivation in an adolescent. Clin Infect Dis 50(6): e34-37.
- Ruffo, E., Malacarne, V., Larsen, S. E., Das, R., Patrussi, L., Wulfing, C., Biskup, C., Kapnick, S. M., Verbist, K., Tedrick, P., et al. (2016). Inhibition of diacylglycerol kinase alpha restores restimulation-induced cell death and reduces immunopathology in XLP-1. Sci Transl Med 8(321): 321ra327.
- Snow, A. L., Oliveira, J. B., Zheng, L., Dale, J. K., Fleisher, T. A. and Lenardo, M. J. (2008). Critical role for BIM in T cell receptor restimulation-induced death. Biol Direct 3: 34.
- Snow, A. L., Marsh, R. A., Krummey, S. M., Roehrs, P., Young, L. R., Zhang, K., van Hoff, J., Dhar, D., Nichols, K. E., Filipovich, A. H., et al. (2009). Restimulation-induced apoptosis of T cells is impaired in patients with X-linked lymphoproliferative disease caused by SAP deficiency. J Clin Invest 119(10): 2976-2989.
- Snow, A. L., Pandiyan, P., Zheng, L., Krummey, S. M. and Lenardo, M. J. (2010). The power and the promise of restimulation-induced cell death in human immune diseases. Immunol Rev 236: 68-82.
- Seki, A. and Rutz, S. (2018). Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J Exp Med 215(3): 985-997.
- Voss, K., Larsen, S. E. and Snow, A. L. (2017). Metabolic reprogramming and apoptosis sensitivity: Defining the contours of a T cell response. Cancer Lett 408: 190-196.
- Voss, K., Luthers, C. R., Pohida, K. and Snow, A. L. (2019). Fatty Acid Synthase Contributes to Restimulation-Induced Cell Death of Human CD4 T Cells. Front Mol Biosci 6: 106.
- Voss, K., Lake, C., Luthers, C. R., Lott, N. M., Dorjbal, B., Arjunaraja, S., Bauman, B. M., Soltis, A. R., Sukumar, G., Dalgard, C. L., et al. (2021). FOXP3 protects conventional human T cells from premature restimulation-induced cell death. Cell Mol Immunol 18(1): 194-205.
- Wlodkowic, D., Telford, W., Skommer, J. and Darzynkiewicz, Z. (2011). Apoptosis and beyond: cytometry in studies of programmed cell death. Methods Cell Biol 103: 55-98.
- Zhang, N., Hartig, H., Dzhagalov, I., Draper, D. and He, Y. W. (2005). The role of apoptosis in the development and function of T lymphocytes. Cell Res 15(10): 749-769.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Pohida, K., Lake, C. M., Yee, D. and Snow, A. L. (2022). Restimulation-Induced Cell Death (RICD): Methods for Modeling, Investigating, and Quantifying RICD Sensitivity in Primary Human T Cells via Flow Cytometric Analysis. Bio-protocol 12(4): e4326. DOI: 10.21769/BioProtoc.4326.
Category
Immunology > Immune cell function > Lymphocyte
Immunology > Immune cell isolation > Leukocyte
Cell Biology > Cell viability > Cell death
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.