- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

An Aptamer-based mRNA Affinity Purification Procedure (RaPID) for the Identification of Associated RNAs (RaPID-seq) and Proteins (RaPID-MS) in Yeast

Published: Vol 12, Iss 1, Jan 5, 2022 DOI: 10.21769/BioProtoc.4274 Views: 5399
Reviewed by: Julie WeidnerMarion HoggNicoletta Cordani
Original research article
The authors used this protocol in:

May 2021

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
RNA-RNA and RNA-protein interactions are involved in the regulation of gene expression. Here, we describe an updated and extended version of our RNA purification and protein identification (RaPID) protocol for the pulldown of aptamer-tagged mRNAs by affinity purification. The method takes advantage of the high affinity interaction between the MS2 RNA aptamer and the MS2 coat protein (MCP), as well as that between streptavidin-binding peptide (SBP) and streptavidin. Thus, it employs MCP-SBP fusions to affinity purify MS2-tagged target RNAs of interest over immobilized streptavidin. Purified aptamer-tagged mRNAs, along with any associated RNAs and proteins, are then sent for RNA sequencing (RaPID-seq) or mass spectrometry (RaPID-MS), which allows for the identification of bound cohort RNAs and proteins, respectively.
Background
RNA-binding proteins (RBPs) and RNA-associated proteins regulate every step in the gene expression pathway. There are also frequent occasions of regulation via RNA-RNA interactions (Guil and Esteller, 2015). Thus, identifying protein-RNA and RNA-RNA interactions is key to understanding RNA biology.
A wide list of methods is available to study RNA-protein interactions. These are classified further as RNA-centric methods or protein-centric methods. RNA-centric approaches characterize proteins bound to a specific RNA of interest, while RNAs bound to a specific protein are studied in protein-centric methods (Ramanathan et al., 2019). RNA-centric approaches use both in vitro and in vivo methods. The simplest method of RNA tagging and pulldown is either by 5′- or 3′-end RNA biotinylation (Zheng et al., 2016), or S1 aptamer tagging used for the in vitro pull down of an RNA of interest with immobilized streptavidin. In vivo methods include RNA affinity purification (RAP) (Hacisuleyman et al., 2014; McHugh and Guttman, 2018), tandem RNA isolation procedure (TRIP) (Matia-González et al., 2017), MS2 in vivo biotin-tagged RAP (MS2-BioTRAP) (Tsai et al., 2011), and CHART (Capture Hybridization Analysis of RNA Targets) (Simon et al., 2011). RAP has been used to study non-coding RNA, such as Xist (McHugh et al., 2015), whereas TRIP is used to study polyadenylated RNA (Matia-González et al., 2017). MS-BioTRAP employs the use of the MS2 aptamer system, in which the MS2 stem loop-tagged RNA and the MS2 coat protein are ectopically expressed (Tsai et al., 2011); however, their over-expression may not reflect the normal physiological level of RNA. CHART capture oligonucleotides are designed to specifically hybridize to the RNA of interest (Simon et al., 2011). In contrast to the RNA-centric approach, cross-linking immunoprecipitation (CLIP) is the most common protein-centric method used to characterize RNAs bound to a protein of interest (Ule et al., 2003; Licatalosi et al., 2008). While all potentially effective, the described methods have not been combined to study RNA-RNA and RNA-protein interactions together.
In addition to these methods, there are several transcriptome-wide approaches to study RNA-RNA and RNA-protein interactions in vivo and in vitro. Approaches like PARIS (psoralen analysis of RNA interactions and structures) (Lu et al., 2018), SPLASH (sequencing of psoralen crosslinked, ligated, and selected hybrids) (Aw et al., 2017), LIGR-seq (ligation of interacting RNA followed by high-throughput sequencing) (Sharma et al., 2016), and MARIO (mapping RNA interactome in vivo) (Nguyen et al., 2016) are used to study transcriptome-wide RNA-RNA interactions, but cannot be used to study the interactions of a specific transcript.
We developed RaPID (RNA purification and identification), which is an MS2 aptamer-based mRNA affinity purification technique (Slobodin and Gerst, 2010 and 2011). The method allows for the specific isolation of MS2 aptamer-labeled mRNAs from yeast or mammalian cells (Slobodin and Gerst, 2010 and 2011), and subsequent analysis of associated RNAs and proteins (Figure 1) using RNA seq and mass-spectrometry, respectively. The advantage of RaPID is the use of the high affinity interaction between the MS2 aptamer and MS2 coat protein (MCP), as well as that of the streptavidin-binding peptide (SBP), which is fused to MCP, to streptavidin beads (Slobodin and Gerst, 2010 and 2011). Hence, the method allows for the specific purification of MS2-tagged mRNAs and their associated proteins and RNAs from cells.
The RaPID-mass spectrometry (RaPID-MS) and RaPID RNA sequencing (RaPID-seq) protocols presented here are updated and extended versions of the protocols previously described by Slobodin and Gerst (2010 and 2011), and Haimovich et al. (2016), which were used to analyze RNA-protein and RNA-RNA interactions in yeast cells, respectively. Using these methods (see Figure 2), we identified both known (Slobodin and Gerst, 2010 and 2011) and novel (Zabezhinsky et al., 2016) RNA-protein interactions, as well as the phenomenon of mRNA multiplexing (Nair et al., 2021). Briefly, the endogenous yeast gene of interest is tagged with the MS2 aptamer (Haim et al., 2007; Haim-Vilmovsky and Gerst, 2009), and the cells transformed with a plasmid expressing MCP fused to both GFP and SBP (MCP-GFP-SBP). The cells are cultured under the required experimental conditions, following which MCP-GFP-SBP expression is induced and the cells fixed to preserve the ribonucleoprotein (RNP) complexes. Next, the cells are lysed and the RNP complexes precipitated by affinity purification using streptavidin-conjugated beads. The associated RNAs and proteins are then analyzed using RNA-seq and mass spectrometry, respectively.
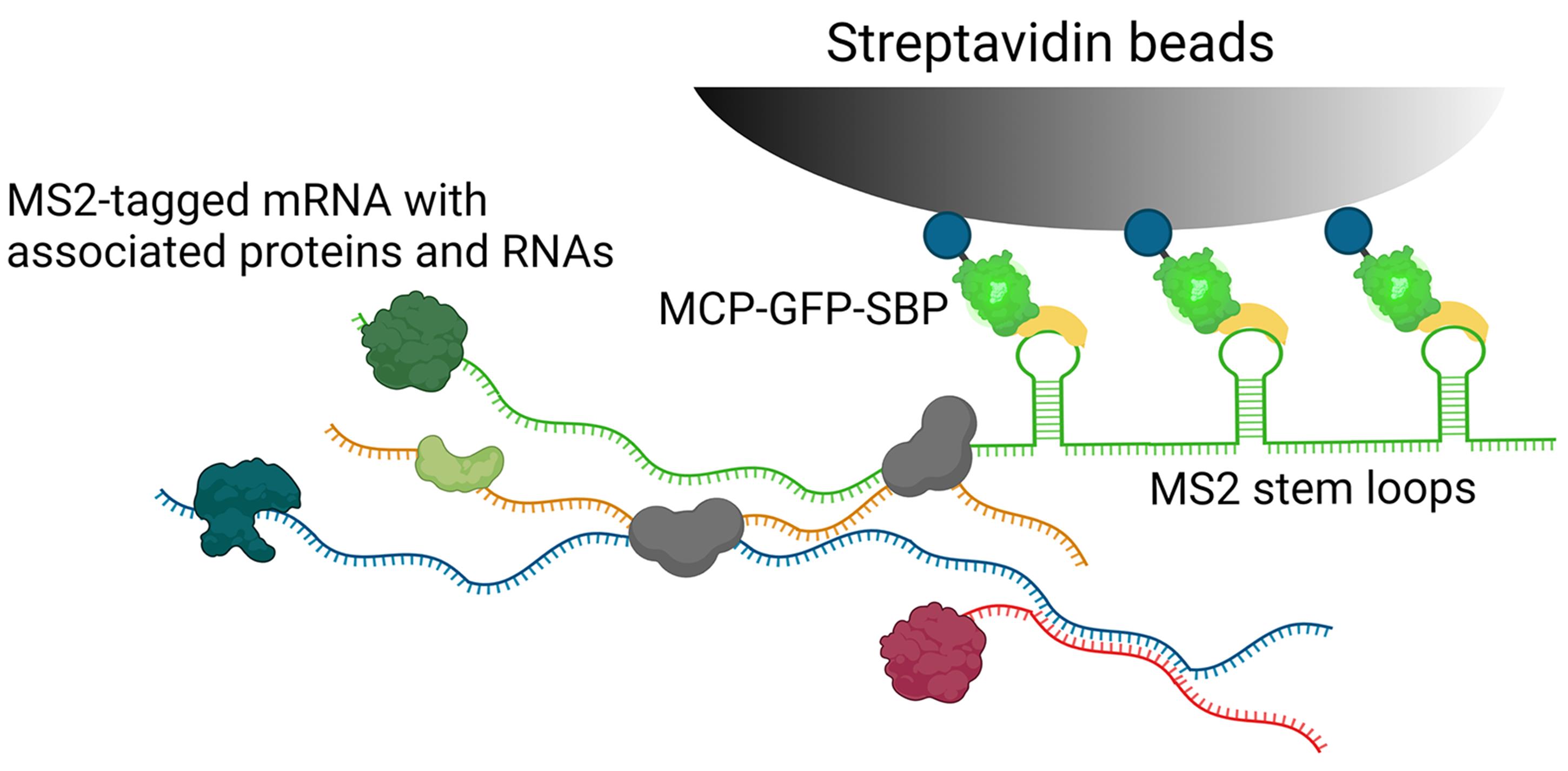
Figure 1. Schematic of the RaPID strategy. Cells co-expressing the MS2 aptamer-tagged RNA and MCP-GFP-SBP binding protein are fixed and lysed, as described in the text. Following which, the lysates are incubated with immobilized streptavidin beads and the MS2 aptamer-tagged mRNA along with associated untagged RNAs and RNA-binding proteins (RBPs) is precipitated. Protein and RNA extraction of the precipitates is then performed to identify RBPs by mass spectrometry and RNAs by RNA-seq or qRT-PCR.

Figure 2. Schematic flowchart of the RaPID-seq/mass-spectrometry procedure. Each major step is presented by graphical representation. A. Preparation of cells bearing the MS2 aptamer tagged mRNA and MCP-GFP-SBP. B. Cell culture. C. Induction of MCP-GFP-SBP protein expression. D. Crosslinking, harvesting, freezing of cells. E. Lysis, measurement of protein concentration. F. Add beads, incubation. G. Centrifugation, washes. H. Crosslink reversal, elution. I. Sample for RNA-seq. J. Library prep and sequencing. K. Analysis of gene expression. L. Sample for mass spectrometry. M. SDS-PAGE and silver staining. N. Mass spectrometry.
Materials and Reagents
Yeast strains
Wild-type strains used successfully with this procedure include BY4741 (EUROSCARF; MATa his3D1 leu2D0 met15D0 ura3D0), although any S. cerevisiae lab strain should suffice, provided it is mutated in the HIS3 and URA3 genes. Yeast can be stored at 4°C for up to 2 weeks on plates, or indefinitely when frozen at -80°C.
Reagents and disposables
17-gauge needle or 23-gauge needle (any vendor)
1.7 mL plastic tubes (autoclaved) (any vendor)
Sterile 15-mL polypropylene centrifuge tubes (any vendor)
Sterile Nuclease-free Barrier tips (10 µL, 200 µL, 1,000 µL) (any vendor)
Sterile 50 mL polypropylene centrifuge tubes (Greiner, catalog number: 227270)
Glass beads, 0.5 mm in diameter (Biospec products, catalog number: 11079-105)
Ultra-pure water (Biological Industries, catalog number: 01-866-1B)
Sterile double distilled water (DDW)
Lithium acetate dihydrate (LiOAc) (Sigma, catalog number: L6883)
Salmon sperm DNA (ssDNA) (Sigma, catalog number: D1626)
Ultrapure water (Molecular Biology Grade Water, nuclease free) (Sigma, catalog number: W4502)
Ethanol absolute (Bio Lab, catalog number: 05250521)
Formaldehyde 37% (Sigma, catalog number: F8775-25ML)
Glycine free base (Sigma, catalog number: G7126)
Liquid Nitrogen
Recombinant Rnasin Ribonuclease Inhibitor (Promega, catalog number: 20006332)
cOmpleteTM, Mini, EDTA-free Protease Inhibitor Cocktail ×25 (Roche, catalog number: 11836170001)
BCA protein assay kit (Pierce, catalog number: 23225)
Streptavidin-conjugated SepharoseTM beads (GE Healthcare, catalog number: 17-5113-01)
Yeast tRNA (Sigma, catalog number: R8508)
Avidin solution (Sigma, catalog number: A9275)
Bovine Serum albumin (BSA), lyophilized powder, ≥96% (Sigma, catalog number: A7906)
Biotin (Sigma, catalog number: B4501)
Dimethylsulfoxide (DMSO) (Sigma, catalog number: D8418)
MPC protein precipitation reagent (a component of the MasterPureTM Yeast RNA purification kit; Epicentre Biotechnologies, catalog number: MPY03100)
Glycogen 20 mg/mL solution (Fermentas, catalog number: R0561)
3 M sodium acetate (NaOAc) solution, pH 5.2 (Fermentas, catalog number: R1181)
qScript Flex cDNA Kit (Quantabio, catalog number: 95049-100-2)
DNaseI (Quantabio, catalog number: 95150-100)
N,N,N’,N’-Tetramethylethylenediamine (TEMED) (Sigma, catalog number: T9281-25ML)
30% acrylamide/Bis 29:1 (Bio-Rad, catalog number: 161-0156)
Silver Stain Kit (Pierce, catalog number: 24612)
TE buffer (see Recipes)
1 M LiOAc (see Recipes)
0.1 M LiOAc (see Recipes)
PEG solution (see Recipes)
ssDNA (see Recipes)
Selective medium (see Recipes)
Glycine solution (see Recipes)
Yeast lysis buffer (see Recipes)
Complete yeast lysis buffer (see Recipes)
Yeast tRNA (see Recipes)
Avidin solution (see Recipes)
Yeast tRNA (see Recipes)
BSA solution (see Recipes)
Washing buffer (see Recipes)
Biotin solution (see Recipes)
2× Cross-link reversal buffer (see Recipes)
5× Protein sample buffer (see Recipes)
Plasmids
pUG34-MS2-CP-GFP-SBP (aka pMCP-GFP-SBP) (Slobodin and Gerst, 2010)
pMS2-SL plasmid (Haim-Vilmovsky and Gerst, 2009)
Equipment
1 L flask
3 L Erlenmeyer flask
-80°C freezer
Sterile surgical blades for the excision of protein bands from gel (Jai Surgical Blades, catalog number: 400-11)
Orbital shaker incubator (MRC, TOU-120)
Pipette aid (recommended: S1 pipet filler, Thermo Fisher Scientific, catalog number: 9501)
(Optional) Vacuum trap
Chemical (fume) hood
Ultrospec 10 Cell Density Meter (Biochrom, catalog number: 634-0882)
Rotator/Mixer Intelli-Mixer (Thomas Scientific, catalog number: RM-2M)
Benchtop refrigerated centrifuge (Eppendorf, catalog number: 5810 R)
Benchtop refrigerated microcentrifuge (Eppendorf, catalog number: 5417 R)
Centrifuge (refrigerated) suitable for large volumes (Sorvall, RC5C plus with SLA3000 rotor) or equivalent centrifuge
Bottles for Sorvall centrifuge, 500 mL (e.g., Thermo, Nalgene, catalog number 10430613)
Digital Disruptor Genie (Scientific Industries, catalog number: SI-DD38 120V). This should be pre-cooled in a cold room prior to the experiment.
Standard equipment for sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) experiments
Sterile box capable of accommodating acrylamide gels
Software
Proteome Discoverer (version 1.4)
Procedure
MS2 tagging and growing the yeast cell culture
The MS2 aptamer is a short RNA stem-loop (SL) structure originating from the MS2 bacteriophage. MS2 tagging, i.e., inserting 12-24 repeats of the MS2-SL, allows the visualization and pulldown of endogenously tagged mRNAs using MCP fused to a fluorescent protein, like GFP (Figure 2). Addition of the SBP moiety allows for the pulldown of MCP-GFP using immobilized streptavidin. However, to identify nonspecific signals during the pull-down experiments several controls are needed, such as untagged cells and cells tagged with a control mRNA.
Tag the gene of interest with MS2 aptamer a multiple binding sites for the RNA-binding MS2 coat protein (MCP), using a PCR-based genomic-tagging strategy and homologous recombination as previously described (Haim et al., 2007; Haim-Vilmovsky and Gerst, 2009). See also Note 1.
Transform the strain having the gene tagged with MS2 aptamer with plasmids expressing the MCP-GFP-SBP using standard yeast LiOAc transformation (Gietz and Schiestl, 2007), and plate on SC-His synthetic media agarose plate.
Pick a single colony that has been transformed with MS2-CP-GFP-SBP containing the HIS3 selection marker, inoculate it into a 50-mL test tube with 7-8 mL of selective medium (SC-His), and grow in a shaking incubator at 30°C for 6-10 h.
Dilute to 500 mL culture for an overnight culture. For cells at mid-logarithmic phase, the final OD600 the next morning should be between 0.4-0.8. Cultures at different OD600 values can be used if other cell states are required, but further steps may need modification according to the amount of cells obtained. To calculate the starter culture volume to take, follow these steps:
Measure the cell number or OD600 of a culture and determine the doubling time using the formula below. For the wild-type strain (BY4741) used in this experiment, the doubling time was 2 h.
[log10 (Nt/N0)]/log10(2) = g
g – number of generations
N0 – # of cells or OD600 at start
Nt – # of cells or OD600 at the end
t = time cultured
d – doubling time (d=t/g)
Determine the amount of starter culture to add to a larger, overnight culture using this formula:
N0 – OD600 at which to start
Nt – OD600 that you want at the end
g – number of generations the culture will go through before harvesting
N0 = Nt/2g
g = t/d
After overnight growth, measure absorbance of the culture and, if needed, keep growing the culture at least up to OD600 = 0.4. Collect the cells by centrifugation at 1,100 × g for 5 min at 4°C, using a SLA3000-type rotor in a Sorvall centrifuge, and discard the growth medium.
Resuspend the cell pellet by gentle pipetting (do not vortex) in 200 mL of fresh growth medium lacking methionine, transfer to 1 L flask, and incubate at 30°C for 45 min with shaking to induce the expression of the MS2-CP-GFP-SBP. See also Note 2.
Fix the cells with formaldehyde by directly adding 270 μL of formaldehyde to a final concentration of 0.05%, and incubate at room temperature (RT) with slow shaking for 10 min.
Terminate the cross-linking reaction by adding 25 mL of glycine solution to a final concentration of 0.125 M and incubate for an additional 2 min.
Centrifuge the cells, discard the supernatant according to chemical waste procedures at your institute, and resuspend the cells using a pipette (do not vortex) in 10 mL of PBS in a 50 mL tube.
Centrifuge the cells at 2,500 × g for 5 min at 4°C.
Discard the PBS completely and keep the tube inverted for 1-2 min on tissue paper to remove residual PBS.
Quickly freeze the cell pellet using liquid nitrogen. For storage, transfer the cells to a -80°C freezer. The cells may be stored for prolonged periods under these conditions.
Yeast Cell Lysis
Note: All the steps mentioned below should be performed on ice.
Place the frozen cell pellet on ice and thaw by adding 4 mL of complete yeast lysis buffer.
Once the cells are thawed, transfer 0.7 mL into 1.5 mL microcentrifuge tubes prefilled with a volume of 0.5 mL of acid-prewashed and oven-baked glass beads.
Vortex the cells using the disruptor for 20 min at 4°C at maximum speed. Turn off the shaker and let stand for 5 min to prevent excessive heating, then turn on for an additional 20 min.
Centrifuge microfuge tubes in a bench-top centrifuge precooled to 4°C at 1,000 × g for 2 min. Transfer the supernatant into fresh microfuge tubes, and centrifuge again at 15,000 × g for 10 min.
Collect the supernatants from each sample (i.e., each individual culture) into disposable 15 mL test tubes. Around 4 mL of supernatant is obtained after centrifugation. Measure the protein concentration with the BCA protein assay kit according to the manufacturer’s instructions.
Transfer the total amount of protein extract desired for RaPID-seq/MS (typically 80-100 mg per pulldown are used for an RaPID-seq or RaPID-MS experiment, whereas 5-10 mg are used for simple pulldowns for qRT-PCR or Western analysis) to a fresh 15 mL tube, add the avidin solution (10 μg of avidin per 1 mg of protein extract), and incubate at 4°C for 1 h with constant rotation/shaking. This step is done to block any biotinylated protein from binding to the streptavidin beads. See Notes 3 and 4 regarding protein measurements and the required amounts for each type of experiment.
Separate the amounts desired for the isolation of RNA and protein into different tubes. In addition, place aside two tubes, each with 1% of initial amount of total extract. One tube is used for “input RNA” (proceed to Step 14 for RNA isolation), and can be used to normalize for gene expression when performing either qRT-PCR or Nanostring experiments (see Note 5). The other tube will be used for “input protein” (proceed to Step 15). These samples are kept on ice and processed in parallel to the eluate, or can be kept frozen at -80°C until used.
In parallel, aliquot 30 µL of streptavidin-conjugated beads. Wash the beads twice by adding 1 mL of ice-cold PBS; once in lysis buffer, swirl (do not vortex) 2-3 times, and centrifuge at 1,000 × g. Block the beads for 1 h at 4°C with 2% BSA and tRNA (0.1 mg per 100 μL of beads) prepared in lysis buffer. After 1 h, centrifuge and wash once with lysis buffer.
Discard the buffer from the washed beads, add the avidin-blocked total cell extract to the beads along with 0.1 mg of yeast tRNA, and incubate at 4°C for 2 h to overnight with constant rotation.
Centrifuge the tubes at 1,000 × g for 2 min and remove the supernatant. Transfer the beads to 1.7 mL tubes and wash them three times with lysis buffer and twice with washing buffer, with centrifugation (as above) between washes.
Perform a last wash in ice-cold PBS and remove the excess buffer.
Add 150 μL of the biotin elution solution to the beads, and incubate for 1 h at 4°C with rotation, for elution of the mRNA-protein/RNA complexes from the beads.
Centrifuge at 1,000 × g for 2 min, transfer the eluate into a fresh microfuge tube, centrifuge once again, and transfer the eluate into a new tube to assure that no beads were carried over.
To reverse crosslink the fraction destined for RNA isolation, add an equal volume of the cross-link reversal buffer to the eluate and incubate at 70°C for 45 min.
To reverse crosslink the fraction destined for protein isolation, add an appropriate volume of the 5× protein sample buffer to reach the 1× concentration, and incubate at 70°C for 45 min.
De-crosslinked RNA and protein samples can be kept at -20°C.
Data analysis
Analysis of precipitated RNA
After reverse crosslinking, add 175 μL of MPC protein precipitation reagent to each 300 μL of eluate and vortex vigorously for 10 s.
Centrifuge for 10 min at 4°C at ≥10,000 × g.
Carefully transfer the supernatant into a new microfuge tube. Add glycogen (80 μg/mL final concentration) and NaOAc (0.3 M final concentration), and vortex thoroughly for 20 s. Add an equal volume of isopropanol, mix by inverting the tubes ~20 times, and incubate overnight at -20°C.
Centrifuge the tubes (12,000 × g for 10 min at 4°C). The pellet containing the RNA should be visible. Carefully discard the supernatant.
Wash the pellet with 70% ice-cold ethanol. Centrifuge for 5 min at 4°C at ≥10,000 × g. Carefully discard the supernatant.
Heat the tube in a dry bath at 50°C for 10 min with an open cap, to evaporate residual ethanol. Do not over-dry, since this will make the pellet hard to dissolve.
Dissolve the pellet in 30 μL of Ultra-pure water. Heat at 50°C for 5-10 min to achieve complete solubility. Store the dissolved RNA in -20°C or -80°C freezers for long term storage.
Construct the library for RNA-seq as previously described (Levin et al., 2010). Oligo(dT) selection is optional, depending upon whether polyA+ RNAs alone are desired or not.
Reverse transcribe RNA with SuperScript III (Invitrogen) at 55°C, and amplify the cDNA with Herculase (Stratagene) in the presence of 5% DMSO for 16 cycles of PCR, followed by a cleanup with 1.8× volumes of AMPure beads (Beckman-Coulter, IN).
Sequence the library to a depth of ~107 reads using standard methodologies (e.g., we used an Illumina HiSeq2000 sequencer with paired-end 76 base reads; Nair et al., 2020).
Data analysis of RaPID RNA seq
Estimate the gene expression levels from the RNA-Seq data using Kallisto (Bray et al., 2016) and targeting the Saccharomyces cerevisiae (S. cerevisiae) reference transcriptome (derived from the Saccharomyces Genome Database and leveraging the Saccharomyces cerevisiae S288C genome version R64-2-1). An example of the read counts acquired for various RNAs for different MS2-tagged mRNAs is provided in Figure 3A.
Note: Consider the 100 bases downstream of the annotated 3' UTR of each gene as part of the mRNA, in order to cover possible unannotated 3’UTR regions.
Identify the differentially expressed genes. We used edgeR (Robinson et al., 2009), with the dispersion parameter manually set to 0.1. Those genes reported as at least four-fold differentially expressed (FDR < 0.001) were retained as significantly differentially expressed. After log transformation of the data [log2(TPM +1)], the gene expression values from the control sample are subtracted from the experimental samples. Expression quantitation, differential expression, and plotting of heat maps were facilitated through use of the transcriptome analysis modules integrated into the Trinity software suite (Haas et al., 2013). It is difficult to determine in advance how many RNAs are enriched with a specific MS2-tagged bait mRNA, since it depends on the MS2-tagged mRNA, the yeast strain used (e.g., MATa or MATα, or a specific deletion strain), and the growth conditions employed. From our experience, we have found up to ten interacting RNAs identified by RaPID-seq. It is important to validate these results by other methods (e.g., co-smFISH, RaPID pulldowns followed by qRT-PCR). We successfully identified and validated such RNA-RNA interactions for various mRNAs (e.g., encoding mating factors, heat-shock proteins, MAP kinases etc.).
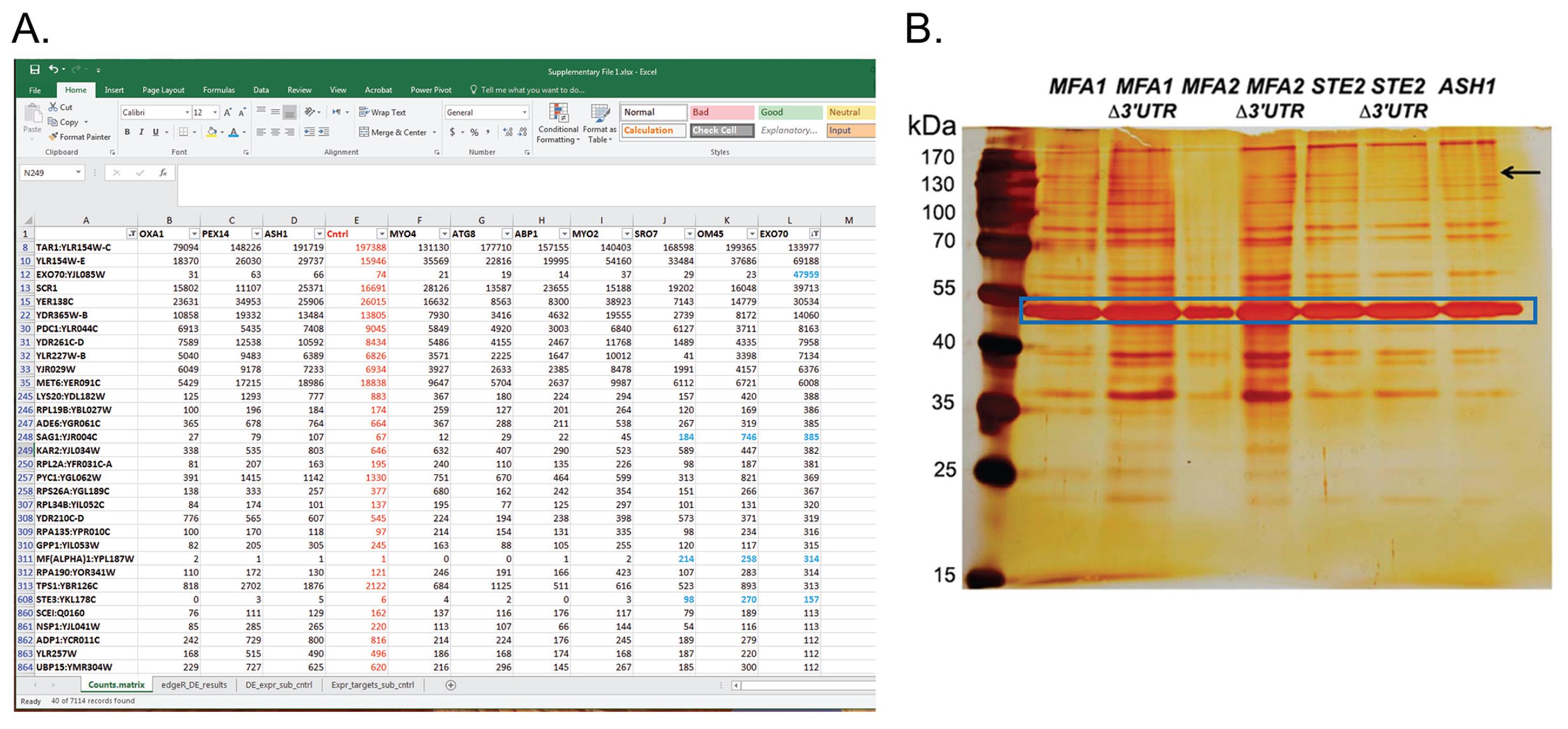
Figure 3. Examples of read counts from RaPID-seq with various MS2-tagged mRNAs and a silver stain gel for RaPID-MS. (A) Shown is a screenshot of the “Counts.matrix” excel sheet from Supplementary File 1 from Nair et al. (2021). It shows the number of reads assigned to each gene (gene names are in column A) for various MS2-tagged target mRNAs (column headers). Cntrl – control untagged strain. rRNA, tRNA and snRNAs were filtered out. The sheet (as shown) is sorted from largest to smallest for EXO70 mRNA (column L). Blue labeled cells show mating-related genes that are enriched in RaPID-seq pulldowns of EXO70, SRO7 and OM45. Note that the MS2-tagged EXO70 target mRNA is highly enriched for its own RNA. Many RNAs are common to all strains, including the untagged control cells, and are therefore considered as background. Only a few chosen genes are shown out of ~7,100 genes identified. (B) Silver stain of SDS-PAGE gel for RaPID-MS (reproduced from Nair et al., 2021). Yeast strains expressing MS2 aptamer-tagged MFA1, MFA2, and STE2 either with or without their 3’UTRs, along with ASH1 were grown and processed for RaPID-MS. The arrow indicates a band of ~150 kDa that exists in the STE2 lane (and all MFA1 and MFA2 lanes), but not in the ASH1 or STE2 ∆3’UTR lane. This band was cut from the gel and sent for MS. Blue square – indicates the band of MCP-GFP-SBP. kDa – kilodaltons.
Analysis of precipitated protein
To identify unknown proteins, there are two alternatives. The first is to separate the proteins on a medium-sized 1.5 mm thick gel (e.g., 20 cm × 15 cm, Bio-Rad) 9% SDS-PAGE gel. Use a comb that will allow loading an entire sample volume per lane (about 180 µL).
Perform silver staining using a Silver Stain Kit (Pierce) according to manufacturer’s instructions. It is fully compatible with subsequent mass-spectrometry analysis.
Analyze the staining results. If specific bands are detected (see Figure 3B), excise them with a sterile blade and send to mass-spectrometry for identification.
The second possibility is to send the entire sample for mass spectrometry.
We recommend discussing protein sample preparation and submission, peptide measurements, and data analysis with personnel of the selected proteomics facility prior to the experiment. In Section D we provide some details to allow a basis for discussion.
Data analysis of RaPID-MS
Reversed-phase nano-liquid chromatography-electrospray ionization tandem mass spectrometry and data analysis was done at the Israel National Center for Personalized Medicine (Weizmann Institute, Rehovot, Israel). The gel slice was processed and trypsinized at the core facility. A nanoUPLC (10 μm tip; New Objective; Woburn, MA, USA) coupled to a quadrupole orbitrap mass spectrometer (Q Exactive HF) using a FlexIon nanospray apparatus (Proxeon) was used. The parameters used are: MS1 resolution was set to 120,000 (at m/z 200), mass range of m/z 300-1650, AGC of 3e6 and maximum injection time was set to 60 ms. MS2 resolution was set to 30,000, quadrupole isolation m/z 1.7, AGC of 1e5, dynamic exclusion of 20 s, and maximum injection time of 60 ms. Data is processed at the core facility using Proteome Discoverer (version 1.4), with two search algorithms – SequestHT and Mascot, using the UniprotKB yeast protein database. A list of common lab contaminants is added and the following variable modifications as well: Carbamidomethyl on C, Oxidation on M, and N-terminal acetylation. Data is filtered for maximum 1% false discovery rate.
By using RaPID-MS, we successfully identified and validated novel proteins that interact with the MS2-tagged mRNA, and its associated mRNAs (from RaPID-seq) (Slobodin and Gerst, 2010; Zabezhinsky et al., 2016; Nair et al., 2021). Although in each case we focused on a single excised band and on a single identified protein for further exploration, this protocol can potentially yield multiple RNA interacting proteins to study.
Discussion
To validate RaPID RNA-seq and RaPID-MS results it is recommended to perform subsequent RaPID experiments using qRT-PCR (RaPID-qPCR) and western blotting, respectively. It is also important to include in these follow-ups both aptamer-tagged and non-tagged strains expressing the MS2 coat protein. This allows for measuring the quantitative enrichment of mRNA and protein from the tagged strains versus non-tagged strains, and is indicative of specificity in the pulldown of RNA and protein, respectively. It is also highly advisable to use a proper negative control (e.g., an unrelated tagged strain), to normalize gene expression using the input sample, and to validate the results with additional experiments. For example, RaPID-seq results indicating RNA-RNA interactions can be validated using single-molecule fluorescence in situ hybridization (smFISH) experiments, using dual sets of labeled probes that can detect potential interaction partners. This protocol does not distinguish RNA-RNA interaction via base paring or through mutual protein binding. Protocols using psoralen crosslinking [e.g., PARIS (psoralen analysis of RNA interactions and structures) (Lu et al., 2018) or SPLASH (sequencing of psoralen crosslinked, ligated, and selected hybrids) (Aw et al., 2017)] could be complementary to RaPID for this purpose. To validate RaPID-MS results, we recommend performing the reciprocal experiment – i.e., to affinity purify the protein and test whether the mRNA is co-precipitated with the protein.
The uniqueness of RaPID is its ability to precipitate a single RNA species from the thousands expressed, due to the high affinity between the MS2 aptamer and MCP (Slobodin and Gerst, 2010 and 2011). While this non-biased affinity purification approach is potentially applicable to any aptamer-binding component pair (e.g., PP7, S1m) (Larson et al., 2011; Leppek and Stoecklin, 2014), RaPID has already proven effective in precipitating known and novel protein-RNA, as well as RNA-RNA, interactions, using single target mRNAs as bait (Slobodin and Gerst, 2010; Zabezhinsky et al., 2016; Nair et al., 2021). Our protocol has been used on a variety of mRNAs in yeast, some of which were expressed at low levels (e.g., UGO1; <5 copies per cell) and some highly expressed (e.g., HSP104; >100 copies per cell after heat-shock), working well in all cases. Thus, the protocol should be suitable for most mRNAs. Nevertheless, we recommend the use of live imaging with the MS2 system or single-molecule FISH, to estimate general transcript abundance and to scale up the protocol for rare transcripts. Finally, we note that RaPID dovetails well with m-TAG, an MS2 aptamer genome-tagging approach originally developed to allow for the localization of endogenously expressed messages in the yeast (Haim-Vilmovsky and Gerst, 2009). Together, the two approaches can be useful towards developing transcriptome-wide mRNA interaction maps for yeast.
Notes
In the current protocol, we have used the version 3 (V3) MS2 aptamer for mRNA pulldown experiments, as originally developed for m-TAG (Haim et al., 2007). However, there are other versions of the MS2 aptamer developed by Singer group. For example, the V6 MS2 aptamer (Tutucci et al., 2018) may be advantageous since it does not show the accumulation of 3’UTR decay products that V3 may sometimes yield, particularly upon gene overexpression using a non-native promoter and/or under conditions that induce P-body formation (Haimovich et al., 2016). We recommend using a 12 or 24 SL cassette, since it is also useful for single molecule mRNA imaging. Shorter cassettes will be very difficult to image, but may work for RaPID. However, having more repeats increases the chance of the mRNA binding to the streptavidin beads. We do not have experience using less than 12 SL repeats.
Induction of the binding protein does not necessarily allow for visualization of endogenously-expressed RNP granules, since the MCP-GFP SBP fusion has only one GFP (unlike in m-TAG, where MCP is fused to three GFPs). We performed a 45 min induction step, which was sufficient. Prolonged induction (i.e., >90 min in our yeast strains) can lead to excessive expression of the MCP-GFP-SBP and, possibly, to a higher level of background of precipitated proteins using RaPID. To optimize RaPID for other strains/growth conditions, users can perform RaPID using different induction times and compare the affinity purified RNA and proteins from the mRNA of interest with the background obtained from pulldowns using control strains or from different conditions.
The amount of the protein extract needed for performing RaPID-seq and/or RaPID-mass spectrometry in yeast is typically between 80-100 mg. Between 5-10 mg of total protein extract is sufficient for the identification of precipitated proteins using western analysis, or for the analysis of RNA by qRT-PCR. Thus, the culture volume of yeast can vary, depending upon the desired downstream application, as well as the efficiency of lysis steps.
For each RaPID reaction, an equal amount of cell extract is recommended. Hence, the concentration of total cellular protein should be accurately measured. The lysates used for this procedure are usually concentrated; we dilute them 1:10 and 1:100 in order to accurately determine the protein concentration. However, starting with an equal amount of cells for each reaction is also advisable.
For normalization of gene expression for qRT-PCR, equal amounts of RNA from input samples of tagged and untagged or control strains are taken for cDNA preparation using the cDNA kit, according to manufacturer’s instructions. qRT-PCR is performed using gene specific primers. Fold enrichment is calculated using the following formula:
Fold enrichment = 2–[(Ct tagged RaPID – Ct tagged input) – (Ct untagged RaPID – Ct untagged input)]
Ct refers to cycle threshold.
Recipes
Tris-ethylenediaminetetraacetic acid (EDTA) buffer (TE)
10 mM Tris-HCl and 1 mM EDTA in double distilled (or fully deionized) water (DDW)
pH 7.5
1 M LiOAc
Lithium acetate 10.2% (wt/vol) in TE
Filter-sterilize and store at RT for up to 1 year.
0.1 M LiOAc
Mix 0.1 volumes of 1 M LiOAc (pH 7.5) with 0.9 volumes of TE.
Prepare the solution under sterile conditions and store at RT for up to 1 year.
50% (wt/vol) polyethylene glycol (PEG) 3350
Dissolve 250 g of PEG 3350 in 300 mL of TE buffer, while stirring and warming to 50°C.
Fill to 500 mL, and filter sterilize.
Salmon sperm DNA (ssDNA)
The DNA is dissolved in ultrapure water at a concentration of 10 mg/mL.
The solution will need to be stirred for at least 2-4 h at RT to dissolve the DNA. Shearing the DNA will help to reduce the viscosity by passing the DNA solution rapidly 12 times through a 17-gauge needle or once through a 23-gauge needle.
Selective medium
Synthetic complete (SC) were mixed essentially according to the protocol of Rose et al. (1990).
In brief, 1 L of synthetic medium is prepared by mixing 7 g of synthetic dry mix (mix composed of 294 g of yeast nitrogen base with ammonium sulfate and without amino acid; 0.3 g each of arginine, cysteine, and proline; 0.45 g each of isoleucine, lysine, and tyrosine; 0.75 g each of glutamic acid, phenylalanine, and serine; 1.0 g each of aspartate, threonine, and valine) with 850 mL DDW, adding 350 µL of 10 N NaOH, followed by stirring and autoclaving in a large (i.e., 3 liters) Erlenmeyer flask. After cooling the autoclaved mixture to 55°C, add 100 mL of either prewarmed 20% glucose (wt/vol) along with 10 mL of a sterile-filtered 100 amino acid stock solution (see below). The 100 amino acid stock is composed of up to six amino acids/bases (e.g., adenine, histidine, leucine, methionine, tryptophan, and uracil) for the preparation of specific selective media. For example, SC medium contains all six, whereas synthetic medium lacking histidine (SC-H) would contain all except histidine. The 100 amino-acid stock solution is prepared by first adding 0.4 g of each amino acid/base required to 150 mL of DDW, followed by the addition of 3 mL of concentrated HCl while stirring, developing the volume up to 200 mL, and then sterile filtering. Liquid media can be stored at RT for up to 2 months. The 100 amino-acid stock is stored up to 1 year at 4°C, whereas the synthetic dry mix can be stored up to 1 year at RT.
! CAUTION: NaOH and HCl are very corrosive and can cause severe burns. May cause serious permanent eye damage. Very harmful upon ingestion. Harmful by skin contact or by inhalation. Use safety glasses, adequate ventilation, and neoprene or PVC gloves!
Glycine solution
1 M glycine solution at pH 7.0
Yeast lysis buffer
20 mM Tris-HCl at pH 7.5
150 mM NaCl
0.5% (vol/vol) Triton X-100
1.8 mM MgCl2
Prepared using RNase-free water
Complete yeast lysis buffer
Supplement Yeast lysis buffer with the following reagents (to be freshly added before use):
1× proteinase inhibitor cocktail
1 mM dithiothreitol (DTT)
80 U/mL RNAsin® ribonuclease inhibitor
Avidin solution
1 mg/mL prepared in PBS
Store at -20°C
Yeast tRNA
10 mg/mL solution in nuclease free water
Store at -20°C
BSA solution
4% solution in PBS
Store at 4°C
Prepared from BSA fraction V
Washing buffer
20 mM Tris-HCl at pH 7.5
300 mM NaCl
0.5% (vol/vol) of NP-40
Biotin solution
0.2 M stock solution in dimethylsulfoxide (DMSO)
Store at 4°C for up to two weeks.
For elution, prepare a 6 mM solution in PBS prewarmed to 37°C.
This solution should be prepared directly before use and be well mixed.
2× Cross-link reversal buffer
100 mM Tris-HCl at pH 7.0
10 mM EDTA
20 mM DTT
2% (wt/v) of sodium dodecylsulfate (SDS)
5× Protein sample buffer
400 mM Tris-HCl at pH 6.8
50% (v/v) of glycerol
10% (wt/v) SDS
0.5% (wt/v) Bromophenol blue
Store at RT.
Before use, add fresh 0.5 M DTT or 5% (v/v) of β-mercaptoethanol.
Warning! DTT and β-mercaptoethanol may be harmful upon inhalation or skin contact. Use hood while preparing buffers containing these agents.
Acknowledgments
This work was supported by grants to J.E.G. from the Takiff Family Foundation and Jeanne and Joseph Nissim Center for Life Sciences (Weizmann Institute of Science), and the Israel Science Foundation (#578/18). R.R.N. was supported by a VATAT Fellowship for Postdoctoral Fellows from China and India (Israel Council of Higher Education). J.E.G. holds the Besen-Brender Chair in Microbiology and Parasitology (Weizmann Institute of Science).
This protocol was described in: Nair, R. R., Zabezhinsky, D., Gelin-Licht, R., Haas, B. J., Dyhr, M. C., Sperber, H. S., Nusbaum, C. and Gerst, J. E. (2021). Multiplexed mRNA assembly into ribonucleoprotein particles plays an operon-like role in the control of yeast cell physiology. eLife 10: e66050.
Competing interests
The authors declare they have no competing financial or non-financial interests.
References
- Aw, J. G. A., Shen, Y., Nagarajan, N. and Wan, Y. (2017). Mapping RNA-RNA Interactions Globally Using Biotinylated Psoralen. J Vis Exp (123): 55255.
- Bray, N. L., Pimentel, H., Melsted, P. and Pachter, L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34(5): 525-527.
- Gietz, R. D. and Schiestl, R. H. (2007). High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2(1): 31-34.
- Guil, S. and Esteller, M. (2015). RNA-RNA interactions in gene regulation: the coding and noncoding players. Trends Biochem Sci 40(5): 248-256.
- Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., Couger, M. B., Eccles, D., Li, B., Lieber, M., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8(8): 1494-1512.
- Hacisuleyman, E., Goff, L. A., Trapnell, C., Williams, A., Henao-Mejia, J., Sun, L., McClanahan, P., Hendrickson, D. G., Sauvageau, M., Kelley, D. R., et al. (2014). Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat Struct Mol Biol 21(2): 198-206.
- Haim, L., Zipor, G., Aronov, S., Gerst, J.E. (2007). A genomic integration method to visualize localization of endogenous mRNAs in living yeast. Nat Methods 4(5): 409-412.
- Haim-Vilmovsky, L. and Gerst, J. E. (2009). m-TAG: a PCR-based genomic integration method to visualize the localization of specific endogenous mRNAs in vivo in yeast. Nat Protoc 4(9): 1274-1284.
- Larson, D. R., Zenklusen, D., Wu, B., Chao, J. A. and Singer, R. H. (2011). Real-time observation of transcription initiation and elongation on an endogenous yeast gene. Science 332(6028): 475-478.
- Leppek, K. and Stoecklin, G. (2014). An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Res 42(2): e13.
- Licatalosi, D. D., Mele, A., Fak, J. J., Ule, J., Kayikci, M., Chi, S. W., Clark, T. A., Schweitzer, A. C., Blume, J.E., et al. (2008). HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456: 464-469.
- Lu, Z., Gong, J. and Zhang, Q. C. (2018). PARIS: Psoralen Analysis of RNA Interactions and Structures with High Throughput and Resolution. Methods Mol Biol 1649: 59-84.
- Matia-González, A. M., Iadevaia, V. and Gerber, A. P. (2017). A versatile tandem RNA isolation procedure to capture in vivo formed mRNA-protein complexes. Methods 118-119: 93-100.
- McHugh, C. A., Chen, C. K., Chow, A., Surka, C. F., Tran, C., McDonel, P., Pandya-Jones, A., Blanco, M., Burghard, C., Moradian, A., et al. (2015). The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521(7551): 232-236.
- McHugh, C. A. and Guttman, M. (2018). RAP-MS: A Method to Identify Proteins that Interact Directly with a Specific RNA Molecule in Cells. Methods Mol Biol 1649: 473-488.
- Nair, R. R., Zabezhinsky, D., Gelin-Licht, R., Haas, B. J., Dyhr, M. C., Sperber, H. S., Nusbaum, C. and Gerst, J. E. (2021). Multiplexed mRNA assembly into ribonucleoprotein particles plays an operon-like role in the control of yeast cell physiology. Elife 10 : e66050.
- Nguyen, T. C., Cao, X., Yu, P., Xiao, S., Lu, J., Biase, F. H., Sridhar, B., Huang, N., Zhang, K. and Zhong, S. (2016). Mapping RNA-RNA interactome and RNA structure in vivo by MARIO. Nat Commun 7: 12023.
- Ramanathan, M., Porter, D. F. and Khavari, P. A. (2019). Methods to study RNA-protein interactions. Nat Methods 16(3): 225-234.
- Robinson, M. D., McCarthy, D. J. and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1): 139-140.
- Rose, M. D., Winston, F. and Hieter, P. (1990). Methods in Yeast Genetics, A Laboratory Course Manual. In: Biochemistry and Molecular Biology Education. Cold Spring Harbor Laboratory Press. New York.
- Sharma, E., Sterne-Weiler, T., O'Hanlon, D. and Blencowe, B. J. (2016). Global Mapping of Human RNA-RNA Interactions. Mol Cell 62(4): 618-626.
- Simon, M. D., Wang, C. I., Kharchenko, P. V., West, J. A., Chapman, B. A., Alekseyenko, A. A., Borowsky, M. L., Kuroda, M. I. and Kingston, R. E. (2011). The genomic binding sites of a noncoding RNA. Proc Natl Acad Sci USA 108(51): 20497-20502.
- Slobodin, B. and Gerst, J. E. (2010). A novel mRNA affinity purification technique for the identification of interacting proteins and transcripts in ribonucleoprotein complexes. RNA 16(11): 2277-2290.
- Slobodin, B. and Gerst, J. E. (2011). RaPID: an aptamer-based mRNA affinity purification technique for the identification of RNA and protein factors present in ribonucleoprotein complexes. Methods Mol Biol 714: 387-406.
- Tsai, B. P., Wang, X., Huang, L. and Waterman, M. L. (2011). Quantitative profiling of in vivo-assembled RNA-protein complexes using a novel integrated proteomic approach. Mol Cell Proteomics 10(4): M110 007385.
- Ule, J., Jensen, K. B., Ruggiu, M., Mele, A., Ule, A. and Darnell, R. B. (2003). CLIP identifies Nova-regulated RNA networks in the brain. Science 302(5648): 1212-1215.
- Zabezhinsky, D., Slobodin, B., Rapaport, D. and Gerst, J. E. (2016). An Essential Role for COPI in mRNA Localization to Mitochondria and Mitochondrial Function. Cell Rep 15(3): 540-549.
- Zheng, X., Cho, S., Moon, H., Loh, T. J., Jang, H. N. and Shen, H. (2016). Detecting RNA-Protein Interaction Using End-Labeled Biotinylated RNA Oligonucleotides and Immunoblotting. Methods Mol Biol 1421: 35-44.
Article Information
Copyright
![]() Nair et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Nair et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Nair, R. R., Haimovich, G. and Gerst, J. E. (2022). An Aptamer-based mRNA Affinity Purification Procedure (RaPID) for the Identification of Associated RNAs (RaPID-seq) and Proteins (RaPID-MS) in Yeast. Bio-protocol 12(1): e4274. DOI: 10.21769/BioProtoc.4274.
- Nair, R. R., Zabezhinsky, D., Gelin-Licht, R., Haas, B. J., Dyhr, M. C., Sperber, H. S., Nusbaum, C. and Gerst, J. E. (2021). Multiplexed mRNA assembly into ribonucleoprotein particles plays an operon-like role in the control of yeast cell physiology. Elife 10 : e66050.
Category
Molecular Biology > RNA > RNA purification > Affinity purification
Molecular Biology > RNA > RNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.