- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Extraction and Electrophoretic Analysis of Bacterial Lipopolysaccharides and Outer Membrane Proteins
Published: Vol 11, Iss 24, Dec 20, 2021 DOI: 10.21769/BioProtoc.4263 Views: 6156
Reviewed by: Alexandros AlexandratosLionel SchiavolinAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2021
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Lipopolysaccharides (LPS) (or lipooligosaccharides [LOS], which lack the O-antigen side chains characteristic of LPS), and outer membrane proteins (OMP) are major cell-surface molecules in the outer membrane (OM) of gram-negative bacteria. The LPS is responsible for causing endotoxic shock in infected hosts and, in conjunction with some OMPs, provides protection to the bacterium against host innate immune defenses and attachment to host cells. Electrophoretic analysis can provide valuable information regarding the size, number, and variability of LPS/LOS and OMP components between bacterial strains and mutants, which aids in understanding the basic biology and virulence factors of a particular species. Furthermore, highly purified extracts are normally not required if only electrophoretic analysis is to be done, and various methods have been established for such procedures. Here, we review ameliorated procedures for fast and convenient extraction of LPS/LOS and protein-enriched outer membranes (PEOM) for optimal electrophoretic resolution. Specifically, we will describe the phenol-based micro-method for LPS/LOS extraction, a differential extraction procedure with sodium lauryl sarcosinate for PEOM, and gel preparation for electrophoretic analysis of LPS/LOS samples in detail.
Graphic abstract:

Workflow for the preparation and analysis of LPS/LOS and PEOM.
Background
The outer membrane (OM) of Gram-negative bacteria is a unique lipid bilayer with two asymmetric membrane leaflets. While the inner leaflet of the OM consists of conventional phospholipids, the outer leaflet predominately contains the lipid A component of lipopolysaccharides (LPS) or lipooligosaccharides (LOS) (Silhavy et al., 2010). LPS molecules typically contain three domains: a hydrophobic membrane anchor known as lipid A (the endotoxic component of LPS), a non-repeating “core” oligosaccharide, and a variable repeating chain of glycoses that make up the O-antigen. LOSs lack the repeating O-antigen of LPS, the core oligosaccharide often branches off different heptose residues, and may phase vary in composition. Furthermore, the terminal oligosaccharide of some species’ LOS can mimic host cell surface oligosaccharides and/or is decorated with sialic acid (Tsai, 2001). LOSs are often present in bacteria that reside predominately on host mucosal surfaces (Preston et al., 1996). The proteins of the OM (OMP) represent about 50% of the OM mass, and can be divided into integral membrane proteins and lipoproteins. Unlike the integral IM proteins spanning the membrane in the form of hydrophobic α-helices, integral OM proteins have β sheets that fold into cylinders (Bos et al., 2007). Collectively, LPS/LOS and OMP contribute to the OM’s ability to serve as a selective permeability barrier that prevents the entry of harmful substances and allows the influx of nutrient molecules (Nikaido, 2003). Due to their important roles as virulence factors, antigenic factors, and targets for molecular typing, these components are of great interest and have been examined in many biomedical studies (Cloeckaert et al., 1996; Inzana et al., 1997; Giordano et al., 2020) for their roles as adhesins, (endo)toxins, protective barriers to host immunity, and enzymatic proteins.
For detailed studies of the composition of these macromolecules embedded in the OM, it is imperative to prepare highly purified materials and to differentiate subtle changes with sensitive screening methods. LPS/LOS has been successfully extracted with a variety of reagents, including pyridine (Goebel et al., 1945), trichloroacetic acid (Ribi et al., 1961), EDTA (Leive, 1965), phenol (Westphal and Jann, 1965), water (Roberts et al., 1967), ether (Galanos et al., 1969), sodium dodecyl sulfate (SDS) (Darveau and Hancock, 1983), butanol (Morrison and Leive, 1975), chloroform-methanol (Raetz et al., 1985), and methanol (Nurminen and Vaara, 1996). A commonly used method is a variation of the phenol-water extraction procedure (Johnson et al., 1976), which applies EDTA and enzymatic treatment prior to harsh chemical extraction. However, micro versions of the phenol-water protocol (Inzana, 1983) or treatment with proteinase K (Hitchcock and Brown, 1983) are adequate when highly purified materials are not required, such as for electrophoretic gel analysis (Figure 1).
One critical step in the isolation of protein-enriched outer membranes (PEOM) is to separate the OM from the inner (cytoplasmic) membrane (IM) and from proteins in the periplasmic space. Due to differences in the densities of the OM and IM, techniques such as sucrose density gradient centrifugation have been developed for the separation of OM and IM of different bacterial species (Miura and Mizushima, 1968; Osborn et al., 1972; Hancock and Carey, 1979; Loeb et al., 1981). Assays for enzymes specific to the IM (succinic dehydrogenase) can be used to confirm the OM is not contaminated with non-OM proteins (Loeb et al., 1981). However, the IM and OM also differ in their susceptibility to solubilization by detergents. Therefore, a simpler and more rapid approach to isolating PEOM is to use differential extraction with detergents, such as sodium lauryl sarcosinate (Filip et al., 1973; Barenkamp et al., 1981) or Triton X-100 (Schnaitman, 1971; Loeb et al., 1981). Differential gradient centrifugation is ideal for the initial characterization of OM proteins, but extraction with detergent is more time-efficient, sufficient for PEOM preparations, and appropriate for general qualitative and quantitative electrophoretic analysis (Filip et al., 1973; Murakami et al., 2002; Hobb et al., 2009). Different detergents and specific procedures are likely to be more effective for different bacterial species. For example, compared to Triton X-100, we have found sodium lauryl sarcosinate to be more effective for the extraction of Histophilus somni PEOM (Figure 2), which can be determined by comparison to PEOM isolated by sucrose density gradients.
The most common and cost-effective analytical method for examination of polysaccharides and proteins is SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Previously, we have reported that optimal gel resolution of LPS/LOS can be achieved by using a bilayer stacking gel and high-quality reagents (Inzana and Apicella, 1999). Other methods have also been described that improve the electrophoretic resolution of LPS/LOS, such as replacing glycine with tricine in the cathode running buffer (Lesse et al., 1990). However, we have not noticed any improvement in resolution using tricine in the cathode buffer compared to the use of two stacking gels. When combined with staining techniques such as silver staining for LPS/LOS (Tsai and Frasch, 1982), and Coomassie blue (Diezel et al., 1972) or silver staining (Merril et al., 1981) for proteins, SDS-PAGE analyses can provide sensitive and relatively rapid information about changes in the structure of LPS/LOS or the expression of OMP. Here, we present protocols for rapid extraction of LPS/LOS and PEOM preparations for electrophoretic analysis. One modification of the protocol previously described for electrophoretic analysis of LPS/LOS (Inzana and Apicella, 1999), is the use of the solubilization buffer described by Tsai and Frasch (1982), which incorporates sucrose in place of glycerol and a lower concentration of 2-mercaptoethanol. These protocols should be applicable to most Gram-negative bacteria.
Materials and Reagents
Notes:
Acrylamide and bisacrylamide are highly neurotoxic. Phenol, sodium lauryl sarcosinate, SDS, ammonium persulfate, TEMED, bromophenol blue, β-mercaptoethanol, and glacial acetic acid are irritants to the eyes, skin, or respiratory tract. Avoid eye/skin contact and inhalation. Handle these compounds while wearing personal protective equipment.
Ethanol, glacial acetic acid, and TEMED are highly flammable. Phenol is combustible. Use these chemicals in a chemical fume hood.
Microcentrifuge tubes (1.5-2-ml) (Thermo Fisher Scientific, catalog number: 50-751-5024 or 07-200-210)
Conical centrifuge tubes (15- or 50-ml) (Thermo Fisher Scientific, catalog number: 14-959-49B, 06-443-21)
Ultra-centrifuge tubes (open-top, thinwall, ultra-clear) and adapters (16 mm diameter) (Thermo Fisher Scientific, catalog numbers: NC9570324, NC1532017)
Glass vials (1-fluid dram or smaller size) (Thermo Fisher Scientific, DWK Life Sciences WheatonTM, catalog number: 06-408B)
One-milliliter syringe (Thermo Fisher Scientific, Air-TiteTM, catalog number: 14-817-173)
Needles (18, 20 and 22 gauge) (Thermo Fisher Scientific, catalog numbers: 14-826-5G, 14-826-5C, 14-826-5A)
Plastic tubing (1/32" I.D. × 3/32" O.D. × 1/32" Wall; Thermo Fisher Scientific, catalog number: 14-387-347)
Acid-washed glass plates (18 cm × 16 cm; Thermo Fisher Scientific, HoeferTM, catalog number: 03-500-204)
Columbia broth (Thermo Fisher Scientific, BD DifcoTM, catalog number: DF0944-17-0), or any medium suitable for growth of the bacteria to be used
Columbia blood agar, 5% sheep, 15 × 100 mm plate (Hardy Diagnostics, catalog number: A16), or any agar plate suitable for growth of the bacteria to be used
High-grade distilled water, such as from Milli-Q Integral water purification system
HPLC-grade water (VWR, catalog number: 87003-652)
Sodium chloride (NaCl) (Thermo Fisher Scientific, J.T. BakerTM, catalog number: 02-004-045)
Sodium phosphate monobasic (NaH2PO4) (Sigma-Aldrich, catalog number: S3139-250G)
Sodium phosphate dibasic heptahydrate (Na2HPO4·7H2O) (Thermo Fisher Scientific, catalog number: S373-500)
Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M8266-100G)
Trizma® base (Sigma-Aldrich, catalog number: T1503-1KG)
HEPES (Sigma-Aldrich, catalog number: H3375-250G)
Liquid phenol (90%) (Thermo Fisher Scientific, catalog number: A931I-4)
Ethanol (Thermo Fisher Scientific, catalog number: BP2818500)
Protease inhibitor (Thermo Fisher Scientific, PierceTM, catalog number: PIA32955)
DNase I (10,000-25,000 units/mg) (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18-047-019)
RNase A (≥5000 U/mg protein) (Thermo Fisher Scientific, catalog number: FEREN0531)
Sodium lauryl sarcosinate (Sigma-Aldrich, catalog number: L5777-100G)
BCA (bicinchoninic acid) Protein Assay Kit (Thermo Fisher Scientific, PierceTM, catalog number: PI23227)
Acrylamide (VWR, catalog number: VWRV0341-500G)
N,N′-Methylene-bisacrylamide (VWR, catalog number: VWRV0172-100G)
Urea (VWR chemicals BDH, catalog number: BDH4602-500G)
Tetramethylethylenediamine (TEMED) (Sigma-Aldrich, catalog number: T9281-25ML)
Sodium dodecyl sulfate (SDS) (Sigma-Aldrich, catalog number: L3771-1KG)
Glycine (Sigma-Aldrich, catalog number: G7403-1KG)
Ammonium persulfate (Sigma-Aldrich, catalog number: A3678-25G)
Sucrose (Sigma-Aldrich, catalog number: S0389)
Glycerol (Sigma-Aldrich, catalog number: G5516-500ML)
2-Mercaptoethanol (Sigma-Aldrich, catalog number: M3148-100ML)
Bromophenol blue (Sigma-Aldrich, catalog number: B8026-5G)
4-12% Bis-Tris gels (Thermo Fisher Scientific, catalog number: NP0336BOX)
Protein staining solution (Thermo Fisher Scientific, PageBlueTM, catalog number: PI24620)
Glacial acetic acid (Thermo Fisher Scientific, catalog number: A38-212)
Periodic acid (Sigma-Aldrich, catalog number: P0430-25G)
Silver nitrate (Sigma-Aldrich, catalog number: 209139)
Calcium chloride dihydrate (CaCl2·2H2O) (Thermo Fisher Scientific, catalog number: C79-500)
Ammonium hydroxide solution (NH4OH) (Sigma-Aldrich, catalog number: 338818-100ML)
Sodium Hydroxide (NaOH) (Thermo Fisher Scientific, catalog number: S318-1)
Citric Acid monohydrate (Sigma-Aldrich, catalog number: C1909-500G)
Formalin (37% formaldehyde) (Sigma-Aldrich, catalog number: 252549)
Phosphate buffered saline (PBS, see Recipes)
5 M NaCl (see Recipes)
2 M MgCl2 (see Recipes)
Tris buffers (see Recipes)
HEPES buffer (see Recipes)
Acrylamide (30%) stock (see Recipes)
9 M Urea solution (see Recipes)
SDS solution (10%, see Recipes)
10× Tris/glycine running buffer (see Recipes)
1× SDS Tris/glycine running buffer (see Recipes)
5% Ammonium persulfate solution (see Recipes)
2× Solubilization buffer (see Recipes)
Fixative (see Recipes)
Oxidizing solution (see Recipes)
Silver nitrate solution (20%, see Recipes)
Silver stain reagent (see Recipes)
Developer solution (see Recipes)
Equipment
Milli-Q Integral water purification system (MilliporeSigma, catalog number: ZIQ7010T0)
Colorimeter CO7000 (VWR, catalog number: WAPR80-3000-42)
Incubator shaker (Thermo Fisher Scientific, HoeferTM, catalog number: SHKE5000 S)
Orbital/rocking shaker (Thermo Fisher Scientific, catalog number: 11-676-681)
Hot plate with magnetic stiring capability (Thermo Fisher Scientific, catalog number: SP88850200)
Sonicator (Qsonica, catalog number: Q500-110)
Fume hood (Labconco, catalog number: 100600040)
Centrifuges and rotors for handling 0.5-50-ml phenol-resistant centrifuge tubes or bottles [e.g., Beckman Avanti J-E centrifuge (Beckman Coulter, catalog number: A20699) with JA-20 (Beckman Coulter, catalog number: 334831) and JLA-10.500 rotors (Beckman Coulter, catalog number: 369681); Sorvall Legend XTR centrifuge (Thermo Scientific, catalog number: 75210061) with TX-750 rotor (Thermo Scientific, catalog number: 75003180), F13-14x50cy rotor (Thermo Scientific, catalog number: 75003661), round buckets (Thermo Scientific, catalog number: 75003608), and conical adapters (Thermo Scientific, catalog number: 75003639)]
Microcentrifuge with a temperature-control system (Eppendorf, catalog number: 5401000137)
Ultracentrifuge and rotors (e.g., Optima XE-90 with Type 70.1 Ti rotor, Beckman Coulter, catalog numbers: A94471, 342184)
Bottles (250-ml) and adapters for Beckman Avanti J-E centrifuge (Thermo Fisher Scientific, catalog number: NC9304619 and NC1476800)
Nephelo culture flasks (Thermo Fisher Scientific, catalog numbers: 50-194-5583 for 300-ml and 50-194-5576 for 500-ml)
Filtering flasks with side arm (vacuum flask) (Thermo Fisher Scientific, KIMAXTM, catalog number: 10-181B)
Erlenmeyer Flasks (Thermo Fisher Scientific, PYREXTM, catalog number: 10-090C)
Micro stir bars (1/16 × 5/16 inch) (Thermo Fisher Scientific, catalog number: 14-512-150)
Spacers (0.75 mm thick, 16 cm long, 2 cm wide; Thermo Fisher Scientific, HoeferTM, catalog number: 03-500-209)
Gel casting stand (Thermo Fisher Scientific, HoeferTM, catalog number: 03-500-247)
Gel casting frames (Thermo Fisher Scientific, HoeferTM, catalog number: 03-500-241)
Gel combs (15 Wells, 0.75 mm thick; Thermo Fisher Scientific, HoeferTM, catalog number: 03-500-188)
Polyacrylamide vertical gel electrophoresis system (for LOS/LPS: Hoefer, HoeferTM SE 600, catalog number: SE600-15-1.5) (for PEOM: Thermo Fisher Scientific, InvitrogenTM, catalog number: EI0002)
Power supply that is capable of loads from 5 mA and adjustable in 1 mA steps (Bio-Rad, PowerPacTM, catalog number: 1645056)
Acid-washed glass dishes or trays (20.1 × 20.1 × 5.5 cm; Thermo Fisher Scientific, catalog number: 15-242B)
Gel imaging system (Bio-Rad, ChemiDocTM, catalog number: 17001402)
Procedure
Bacterial culture
Note: The culture procedure should be optimized for the bacterium used in the study. The final growth phase for LPS and PEOM extraction should be determined based on the purpose of the study. In general, bacteria grown to at least mid-log phase are preferable for the extraction of LPS and PEOM.
Prepare a starter culture at an OD600 of 0.8 by inoculating the bacteria freshly isolated from an agar plate into 5 ml of suitable broth in a Nephelo culture flask for LPS extraction, or 25 ml of broth for extraction of PEOM.
Dilute the starter culture 1:20 into 5 ml or 200 ml of fresh broth for LPS extraction or PEOM extraction, respectively.
Incubate at 37°C at 180 rpm in an incubator shaker for 3 to 4 h, or until the bacteria reach mid-log phase (approximately 109 CFU/ml for Histophilus somni and some Pasteurellaceae bacteria).
Transfer 2 ml of the culture to a microcentrifuge tube and proceed to section B for LPS extraction, or utilize the 200 ml of culture for PEOM extraction (section C).
Extraction of LPS/LOS using the hot phenol-water microextraction method (Inzana, 1983)
Centrifuge 2 ml of the above bacterial suspension in a microcentrifuge tube at 10,000 × g for 5 min at 4°C.
Discard the supernatant and wash the pellet once with 1 ml of PBS.
Resuspend the washed cells in 300 μl of distilled water and transfer the suspension to a 1-fluid dram glass vial (or similar) containing a micro stir bar (1/16 × 5/16 inch).
Add an equal volume of 90% phenol (pre-warmed to 65-70°C is preferable, but not required) to the bacterial suspension.
Place the vials in a beaker of water on a hot plate/stir plate set to 65-70°C. The water should be no higher than half the length of the vials, and multiple vials can be secured together with a rubber band to prevent tipping.
Stir the mixture vigorously at 65-70°C for 15 min.
Chill the mixture in the vial on ice only enough to cool to about 15°C and transfer the mixture to a 1.5-ml microcentrifuge tube.
Centrifuge at 10,000 × g for 10 min at 15°C.
Carefully transfer the supernatant to a 15-ml conical centrifuge tube and set the tube aside.
Transfer the phenol phase back into the glass vial.
Add 300 μl of water to re-extract the phenol phase and repeat Steps B5 to B9.
Pool the aqueous phases in the 15-ml conical centrifuge tube.
Adjust the sodium concentration of collected aqueous phases to ~0.5 M by adding one tenth volume of 5 M NaCl.
Add 5-10 volumes of cold (-20°C) 95% ethanol to precipitate the LPS and mix the solution thoroughly.
Maintain the mixture at -20°C for at least 6 h or overnight.
Centrifuge the mixture at 2,000 × g for 10 min at 4°C.
Aspirate the ethanol under vacuum with a pasteur pipette until the tube is dry. Tip the tube to the side with the pellet side up so you do not have to get the pipette tip too close to the pellet.
Suspend the opaque pellet in 100 μl of distilled water.
Transfer the 100 μl of suspension to a 1.5-ml microcentrifuge tube.
Repeat Steps B14 to B18 for a second round of precipitation with ethanol.
Suspend the precipitate with 50 μl of distilled water.
Store the sample at -20°C. Samples may be stored as aliquots to avoid repeated freeze-thawing.
Extraction of protein-enriched outer membranes (PEOM)
Note: All steps for extraction of PEOM should be performed at 4°C with protease inhibitor.
Centrifuge the 200 ml of bacterial suspension in a 250-ml bottle at 10,000 × g at 4°C for 15 min.
Wash the pellet twice in 30 ml of 0.05 M Tris buffer, pH 7.8, containing 2 mM MgCl2 (freeze pellets at -20°C if stopping here).
Resuspend the pellet in 25 ml of 10 mM HEPES buffer, pH 7.4, containing protease inhibitor (follow the manufacture’s instruction for the working concentration), and 20 μg/ml each of DNase I and RNase A.
Sonicate the suspension in a 50-ml beaker of ice for 12 bursts of 15 s each, with at least one minute of incubation on ice between each burst (keep below 10°C). Alternatively, two passages through a French Press can be used to lyse the cells (8,000 lbs/in2).
Transfer the resultant sample into 50-ml conical tubes.
Centrifuge at 12,000 × g for 5 min to remove unbroken cells.
Transfer the supernatant to ultra-centrifuge tubes.
Centrifuge the supernatant at 255,000 × g for 60 min (this yields a total membrane preparation including the IM, peptidoglycan, and OM).
Resuspend the pellet in 1 ml of 10 mM HEPES buffer.
Homogenize the pellet suspension with a 1-ml syringe using an 18-G, then 20-G, then 22-G needle until thoroughly resuspended. Covering the end of each needle with tubing will prevent scratching the tube (as shown in the graphic abstract). Take care to avoid sudsing.
Place aside 0.1 ml of the suspension in a 1.5-ml microcentrifuge tube and freeze at -20°C (do this only once).
Transfer the remaining homogenized suspension into ultracentrifuge tubes (~10-ml size).
Fill to the top of the tube with 10 mM HEPES buffer containing 2% sodium lauryl sarcosinate. Seal the tubes and tip back-and-forth gently to thoroughly mix.
Maintain the tubes at room temperature for 30 min without shaking.
Centrifuge at 255,000 × g for 60 min to obtain PEOM.
Remove the supernatant and overlay the pellet with 1 ml of 10 mM HEPES buffer.
Repeat Steps C9 to C14, but do not remove 0.1 ml of the suspension again.
Resuspend the final pellet in approximately 100-200 μl of distilled water.
Dilute the sample from Step C18 to a protein concentration of 0.5-3 mg/ml, based on the result of BCA protein assay performed following the manufacturer's instructions.
Aliquot the sample and store at -20°C for future use.
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) for LPS/LOS
Note: For optimal resolution of LOS/LPS, use high quality reagents throughout the procedure and prepare fresh if indicated.
Preparation of 14% separating gel (20 ml for one 18 cm × 16 cm gel with 0.75 mm spacers)
Mix 9.3 ml of 30% acrylamide stock, 6.4 ml of 9 M urea, 4 ml of 1.88 M Tris buffer (pH 8.8), and 0.2 ml of 10% SDS in a vacuum flask, gently mix, and degas for 10 min.
Add 0.15 ml ammonium persulfate solution (5%) and 5 μl of TEMED.
Gently mix the solution and immediately dispense the separating gel solution between the glass plates to 4 cm below the top of the plate using a syringe or pipette.
Overlay the gel with 0.1% SDS to a depth of a few millimeters.
Allow the gel to polymerize for at least 1 h.
Rinse and blot the top of the gel with filter paper after polymerization.
Preparation of 3% stacking gel A (10 ml for one 18 × 16 cm gel with 0.75 mm spacers)
Mix 1 ml of 30% acrylamide stock, 1 ml of 1.25 M Tris buffer (pH 6.8), 0.1 ml of 10% SDS, and 7.8 ml of HPLC-grade water in a vacuum flask and degas for 10 min.
Add 0.1 ml ammonium persulfate solution (5%) and 5 μl of TEMED.
Gently mix the solution and immediately dispense the stacking gel solution between the glass plates to where the bottom of the well comb will be. The bottom of the well comb teeth should just touch the top of the gel after polymerization.
Allow the stacking gel to polymerize for ~2 h.
Preparation of 3% stacking gel B (10 ml for one 18 × 16 cm gel with 0.75 mm spacers)
Mix 1 ml of 30% acrylamide stock, 1 ml of 10× Tris/glycine running buffer lacking SDS, 0.1 ml 10% SDS, and 7.8 ml of HPLC-grade water.
Place solution in a vacuum flask and degas for 10 min.
Insert the comb chosen for the run about half-way between the glass plates.
After degassing, add 0.1 ml ammonium persulfate solution (5%) and 5 μl of TEMED to the acrylamide solution.
Gently mix the solution and immediately dispense the stacking gel solution between the glass plates.
The teeth of the comb should just touch the top of stacking gel A when fully inserted.
Allow the stacking gel to polymerize for ~2 h.
The comb can be removed, the wells filled with 1× running buffer, and the gel stored overnight at 4°C after wrapping in plastic wrap.
Alternatively, only stacking gel A can be used, but resolution of the bands will be reduced.
Preparation of LPS/LOS samples
Add 1-10 µg (depending on the number of bands) of purified LPS/LOS sample or 5 μl of the micro-extract (prepared as above), and dilute to a volume of 10 µl with HPLC-grade water.
Note: If the LPS/LOS gel is overloaded or the development is allowed to proceed for too long, the bands will be obscured, and a smear will form. Normally, 10-12 μg of LPS or 2-3 μg of LOS per well is ideal. If 5 μl of extracted sample is too much or too little, the volume for each sample should be adjusted for a second gel run. Over-development can be prevented or reversed somewhat by addition of 5% acetic acid, but this will also make the bands yellow.
Add an equal volume of 2× solubilization buffer.
Boil the samples for 5 min by using a floating pad to secure the samples and placing in a beaker of water on a hot plate set to 100°C.
Electrophoresis
Remove the comb and rinse the sample wells with 1× SDS Tris/glycine running buffer, and then fill with the same buffer.
Add samples carefully, allowing them to sink to the bottom, and not spilling over into other wells.
Place gels into the electrophoresis chamber.
Fill the upper (first) and then lower reservoir with cold (4°C) 1× SDS Tris/glycine running buffer. Two liters is the minimum, 4 L is preferred.
Run the gel at 9 mAmp/gel constant current through the stacking gels, then at 12 mAmp/gel through the separating gel. The total run time will be about 5 h.
When the dye front reaches the bottom of the separating gel, stop the electrophoresis.
SDS-PAGE for PEOMs
Preparation of protein samples
Add the PEOM sample to the desired concentration (1-10 µg depending on the number of bands) and dilute to a maximum volume of 7.5 µl (5 μl is preferred) with HPLC-grade water.
Add an equal volume of 2× solubilization buffer.
Boil the samples for 10 min by using a floating pad to secure the samples and placing in a beaker of water on a hot plate set to 100°C.
Electrophoresis
Remove the comb from a commercial 4-12% Bis-Tris gel and rinse the sample wells with distilled water.
Place gels into the electrophoresis chamber.
Fill the upper buffer chamber and the wells with 1× SDS-Tris/glycine running buffer.
Add samples allowing them to sink to the bottom.
Fill the lower buffer chamber with 1× SDS-Tris/glycine running buffer.
Run the gel at 180 V constant voltage.
When the dye front reaches the bottom of the gel, stop the electrophoresis.
Silver staining for LPS/LOS gels
Remove the gel from between the glass plates.
Fix the gel in a glass dish with 200 ml of fixative overnight at room temperature covered with plastic wrap; shaking is not required.
Discard the fixative.
Oxidize the gel in 200 ml of oxidizing solution (made fresh) in the dark by covering the dish with aluminum foil to minimize light exposure.
Rotate the dish rapidly (~70 rpm) for 5 min.
Discard the oxidizing solution and wash the gel 3 times by rotating slowly with 1 L of distilled water for 15 min each.
Prepare the silver stain reagent during the last wash.
Dissolve 1 g of silver nitrate into 5 ml of distilled water.
In a separate 500-ml flask, add 2 ml of NH4OH to 28 ml of 0.1 M NaOH to make the NaOH/NH4OH solution.
While gently shaking, slowly add all the silver nitrate solution dropwise to the NaOH/NH4OH solution. A brown precipitate forms and then becomes clear.
Add 115 ml of distilled water. (At this point, the reagent should be perfectly clear. If the solution remains brown, do not use it, and make up again with fresh NH4OH.)
Discard the last wash, add the silver stain reagent to the gel, and shake rapidly (~70 rpm) for 10 min.
Pour the silver stain reagent into a flask and precipitate with a small amount (large pinch) of CaCl2 before disposing.
Wash the gel 3 times with 1 L of distilled water for 10 min each.
Discard the last wash and add 200 ml of the developer solution.
Stop the reaction just before optimal development by rinsing the gel in cold water. If over-development occurs, the gel can be destained with 5% acetic acid, but the bands may yellow.
Store cold, but photograph or scan as soon as possible.
Coomassie blue staining for protein gels
Remove the gel from the cassette and place in a clean tray.
Wash the gel three times for 5 min each using distilled water with gentle agitation.
Discard the last wash.
Add a sufficient volume of the Page Blue protein staining solution to cover the gel.
Incubate at room temperature for 60 min with gentle agitation.
Discard the staining solution and rinse the gel two times with distilled water.
Wash the gel three times for 5 min each using distilled water with gentle agitation (or wash more times until the background becomes clear).
Store cold until ready to photograph.
Data analysis
Profiles of LOSs (lanes 1-4) and LPSs (lanes 5-8) of various genera (Inzana and Apicella, 1999).
“Using the reagents described with a single stacking gel, an acceptable profile of LPSs and LOSs was obtained (Figure 1A). However, when a separate comb gel (stacking gel B) containing the same buffer as the upper reservoir buffer was used, the profiles were much sharper, particularly the lower molecular weight LOS bands (Figure 1B)”. © 1999 WILEY-VCH Verlag GmbH, Weinheim, Fed. Rep. of Germany.
“The number of bands that could be resolved for Haemophilus and Neisseria species could more accurately be discerned following electrophoresis through the bilayer stacking gel (Figure 1B, lanes 1-4)”. © 1999 WILEY-VCH Verlag GmbH, Weinheim, Fed. Rep. of Germany.
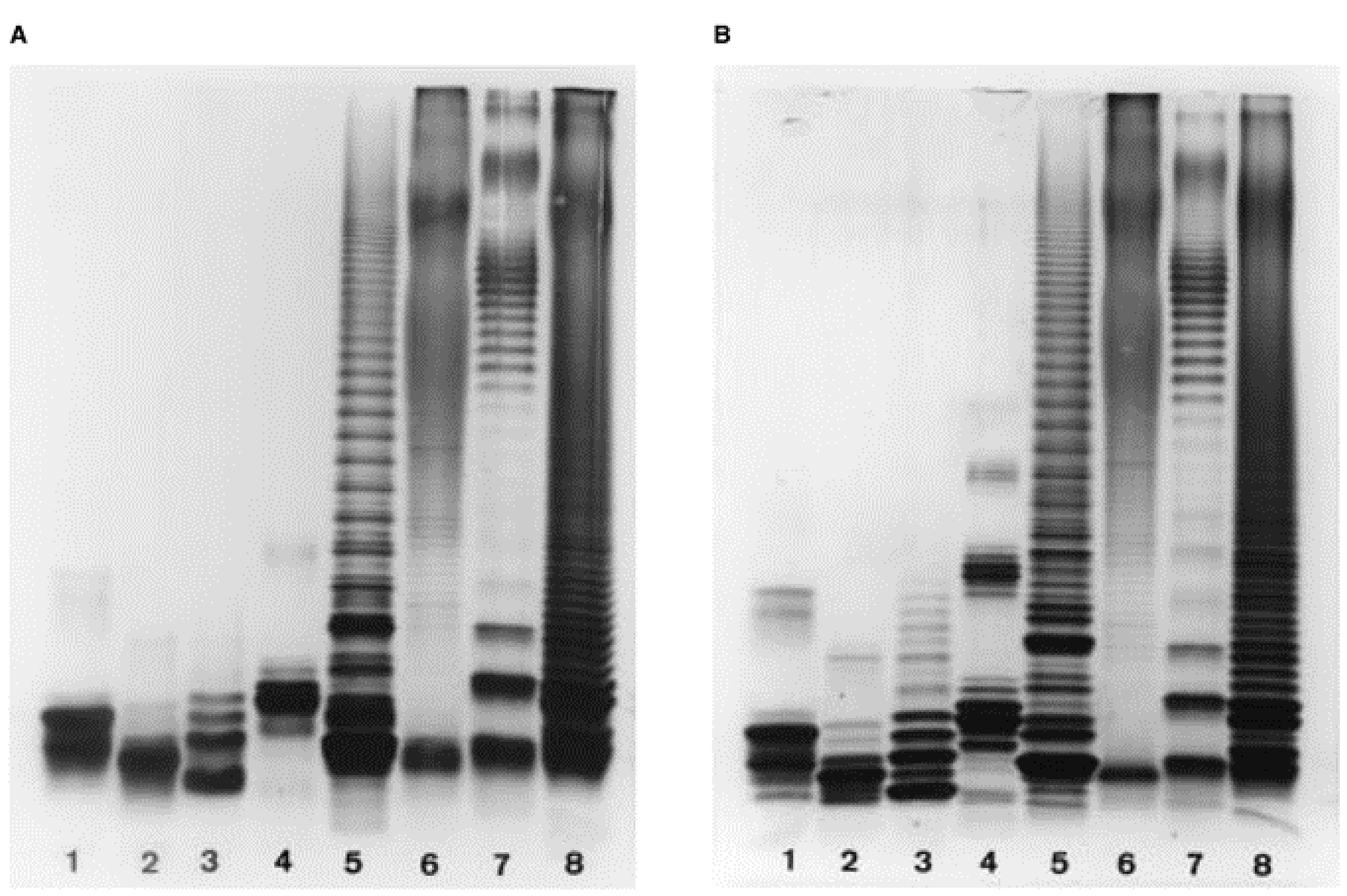
Figure 1. Electrophoretic profiles of LOSs (lanes 1-4) and LPSs (lanes 5-8) of various genera following conventional SDS-PAGE using (A) a single stacking gel or (B) two stacking gels. Lanes, bacteria from which LOS/LPS samples were isolated, and sample concentration: (1) Haemophilus influenzae type b, 1.5 μg; (2) Histophilus somni, 2.2 μg; (3) Neisseria gonorrhoeae, 1.0 μg; (4) Pasteurella multocida, 1.5 μg; (5) Actinobacillus pleuropneumoniae type 7, 7.5 μg; (6) Brucella abortus, 12 μg; (7) Escherichia coli, 12 μg; (8) Salmonella typhimurium, 8 μg. Reproduced with permission from (Inzana and Apicella, 1999) © 1999 WILEY-VCH Verlag GmbH, Weinheim, Fed. Rep. of Germany.
Electrophoretic profiles of OMPs from Histophilus somni strain (Figure 2).
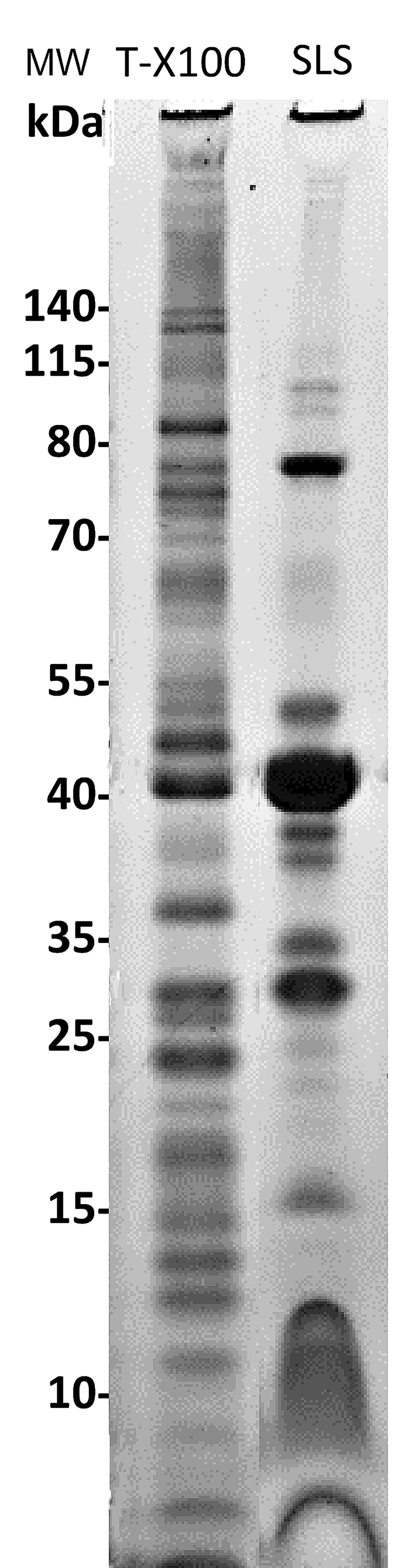
Figure 2. Outer membrane protein (OMP) electrophoretic profiles from Histophilus somni strain 2336 following cell lysis and extraction with Triton-X-100 (T-X100) or sodium lauryl sarcosine (SLS). The T-X100 extract contains many proteins associated with the inner membrane, and the SLS extract is enriched for well-known major OMPs, such as the ~40 kDa protein. However, lower molecular size proteins (<15 kDa) were not well resolved using SLS. Molecular size markers (MW) are to the left. Four μg of protein were added/lane indicating there is about 10 ng of protein/band.Phosphate buffered saline (PBS)
8.5 g NaCl (150 mM)
0.23 g NaH2PO4 (1.9 mM)
2.17 g Na2HPO4·7H2O (8.1 mM)
Add distilled water up to 1 L, adjust pH to 7.4.
5 M NaCl
29.22 g NaCl
Add distilled water up to 100 ml.
2 M MgCl2
19.04 g MgCl2
Add distilled water up to 100 ml.
Tris buffer (0.05 M, pH 7.8) containing 2 mM MgCl2
0.61 g Trizma® base
100 μl of 2 M MgCl2 (add before use)
Add distilled water up to 100 ml, adjust pH to 7.8, store at 4°C.
Tris buffer (1.25 M, pH 6.8)
15.14 g Trizma® base
Add distilled water up to 100 ml, adjust pH to 6.8, store at 4°C.
Tris buffer (1.88 M, pH 8.8)
22.77 g Trizma® base
Add distilled water up to 100 ml, adjust pH to 8.8, store at 4°C.
HEPES buffer (10 mM, pH 7.4)
0.24 g HEPES
Add distilled water up to 100 ml, adjust pH to 7.4.
Acrylamide stock (30% T, 0.8% C)
29.2 g acrylamide
0.8 g bis-acrylamide (C, cross-linker)
Add distilled water up to 100 ml. Filter stock solution, wrap in aluminum foil, and store at 4°C.
Urea solution (9 M)
54.05 g urea
Add distilled water up to 100 ml and store at room temperature.
10% SDS solution
10 g SDS
Add distilled water up to 100 ml and store at room temperature.
10× Tris/glycine running buffer
30.29 g Trizma® base (0.25 M)
144.13 g glycine (1.92 M)
Add distilled water up to 1 L, store at room temperature.
1× SDS-Tris/glycine running buffer (4 L)
400 ml of 10× Tris/glycine running buffer
40 ml of 10% SDS solution
Add distilled water up to 4 L, store at 4°C.
5% Ammonium persulfate solution
50 mg ammonium persulfate
Add distilled water up to 1 ml (prepare fresh for each experiment).
2× solubilization buffer (100 ml)
For LPS/LOS gels:
10 ml of 1 M Tris-HCl buffer, pH 6.8 (0.1 M)
2 g SDS (2%)
20 g sucrose (20%)
0.5 ml of 1% bromophenol blue (0.005%)
Add distilled water up to 100 ml.
Just before use, add 10 μl of 2-mercaptoethanol to 990 μl of 2× solubilization buffer.
For OMP gels:
12.5 ml of 1 M Tris-HCl buffer, pH 6.8 (0.125 M)
4 g SDS (4%)
20 ml glycerol (20%)
0.5 ml of 1% bromophenol blue (0.005%)
Add distilled water up to 100 ml.
Just before use, add 100 μl of 2-mercaptoethanol to 0.9 ml of 2× solubilization buffer.
Fixative (200 ml)
10 ml glacial acetic acid (final concentration 5%)
80 ml ethanol (final concentration 40%)
110 ml distilled water
Oxidizing solution
1.4 g periodic acid (final concentration 0.7%)
200 ml of the fixative
20% Silver nitrate solution
1 g silver nitrate
Add distilled water up to 5 ml.
Silver stain reagent (see Procedure section E for details of preparation)
28 ml of 0.1 N NaOH
2 ml of NH4OH solution
5 ml of 20% silver nitrate (add silver nitrate dropwise)
115 ml of distilled water
Developer solution
50 mg of citric acid
0.5 ml of 37% formaldehyde (formalin)
Add distilled water up to 1 L.
- Barenkamp, S. J., Munson, R. S., Jr. and Granoff, D. M. (1981). Subtyping isolates of Haemophilus influenzae type b by outer-membrane protein profiles. J Infect Dis 143(5): 668-676.
- Bos, M. P., Robert, V. and Tommassen, J. (2007). Biogenesis of the gram-negative bacterial outer membrane. Annu Rev Microbiol 61: 191-214.
- Cloeckaert, A., Verger, J. M., Grayon, M. and Vizcaíno, N. (1996). Molecular and immunological characterization of the major outer membrane proteins of Brucella. FEMS Microbiol Lett 145(1): 1-8.
- Darveau, R. P. and Hancock, R. E. (1983). Procedure for isolation of bacterial lipopolysaccharides from both smooth and rough Pseudomonas aeruginosa and Salmonella typhimurium strains. J Bacteriol 155(2): 831-838.
- Diezel, W., Kopperschläger, G. and Hofmann, E. (1972). An improved procedure for protein staining in polyacrylamide gels with a new type of Coomassie Brilliant Blue. Anal Biochem 48(2): 617-620.
- Filip, C., Fletcher, G., Wulff, J. L. and Earhart, C. F. (1973). Solubilization of the cytoplasmic membrane of Escherichia coli by the ionic detergent sodium-lauryl sarcosinate. J Bacteriol 115(3): 717-722.
- Galanos, C., Lüderitz, O. and Westphal, O. (1969). A new method for the extraction of R lipopolysaccharides. Eur J Biochem 9(2): 245-249.
- Giordano, N. P., Cian, M. B. and Dalebroux, Z. D. (2020). Outer Membrane Lipid Secretion and the Innate Immune Response to Gram-Negative Bacteria. Infect Immun 88(7).
- Goebel, W.F., Binkley, F. and Perlman, E., (1945). Studies on the flexner group of dysentery bacilli: I. The specific antigens of Shigella paradysenteriae (Flexner). J Exp Med 81(4): 315-330.
- Hancock, R. E. and Carey, A. M. (1979). Outer membrane of Pseudomonas aeruginosa: heat- 2-mercaptoethanol-modifiable proteins. J Bacteriol 140(3): 902-910.
- Hitchcock, P. J. and Brown, T. M. (1983). Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J Bacteriol 154(1): 269-277.
- Hobb, R. I., Fields, J. A., Burns, C. M. and Thompson, S. A. (2009). Evaluation of procedures for outer membrane isolation from Campylobacter jejuni. Microbiology (Reading) 155(Pt 3): 979-988.
- Inzana, T. J. (1983). Electrophoretic heterogeneity and interstrain variation of the lipopolysaccharide of Haemophilus influenzae. J Infect Dis 148(3): 492-499.
- Inzana, T. J. and Apicella, M. A. (1999). Use of a bilayer stacking gel to improve resolution of lipopolysaccharides and lipooligosaccharides in polyacrylamide gels. Electrophoresis 20(3): 462-465.
- Inzana, T. J., Hensley, J., McQuiston, J., Lesse, A. J., Campagnari, A. A., Boyle, S. M. and Apicella, M. A. (1997). Phase variation and conservation of lipooligosaccharide epitopes in Haemophilus somnus. Infect Immun 65(11): 4675-4681.
- Johnson, K. G., Perry, M. B. and McDonald, I. J. (1976). Studies of the cellular and free lipopolysaccharides form Neisseria canis and N. subflava. Can J Microbiol 22(2): 189-196.
- Leive, L. (1965). Release of lipopolysaccharide by EDTA treatment of E. coli. Biochem Biophys Res Commun 21(4): 290-296.
- Loeb, M. R. and Smith, D. H. (1980). Outer membrane protein composition in disease isolates of Haemophilus influenzae: pathogenic and epidemiological implications. Infect Immun 30(3): 709-717.
- Loeb, M. R., Zachary, A. L. and Smith, D. H. (1981). Isolation and partial characterization of outer and inner membranes from encapsulated Haemophilus influenzae type b. J Bacteriol 145(1): 596-604.
- Lesse, A. J., Campagnari, A. A., Bittner, W. E. and Apicella, M. A. (1990). Increased resolution of lipopolysaccharides and lipooligosaccharides utilizing tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis. J Immunol Methods 126(1): 109-117.
- Merril, C. R., Goldman, D., Sedman, S. A. and Ebert, M. H. (1981). Ultrasensitive stain for proteins in polyacrylamide gels shows regional variation in cerebrospinal fluid proteins. Science 211(4489): 1437-1438.
- Miura, T. and Mizushima, S. (1968). Separation by density gradient centrifugation of two types of membranes from spheroplast membrane of Escherichia coli K12. Biochim Biophys Acta 150(1): 159-161.
- Morrison, D. C. and Leive, L. (1975). Fractions of lipopolysaccharide from Escherichia coli O111:B4 prepared by two extraction procedures. J Biol Chem 250(8): 2911-2919.
- Murakami, Y., Imai, M., Nakamura, H. and Yoshimura, F. (2002). Separation of the outer membrane and identification of major outer membrane proteins from Porphyromonas gingivalis. Eur J Oral Sci 110(2): 157-162.
- Nikaido, H. (2003). Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67(4): 593-656.
- Nurminen, M. and Vaara, M. (1996). Methanol extracts LPS from deep rough bacteria. Biochem Biophys Res Commun 219(2): 441-444.
- Osborn, M. J., Gander, J. E., Parisi, E. and Carson, J. (1972). Mechanism of assembly of the outer membrane of Salmonella typhimurium. Isolation and characterization of cytoplasmic and outer membrane. J Biol Chem 247(12): 3962-3972.
- Preston, A., Mandrell, R. E., Gibson, B. W. and Apicella, M. A. (1996). The lipooligosaccharides of pathogenic gram-negative bacteria. Crit Rev Microbiol 22(3): 139-180.
- Raetz, C. R., Purcell, S., Meyer, M. V., Qureshi, N. and Takayama, K. (1985). Isolation and characterization of eight lipid A precursors from a 3-deoxy-D-manno-octylosonic acid-deficient mutant of Salmonella typhimurium. J Biol Chem 260(30): 16080-16088.
- Ribi, E., Haskins, W. T., Landy, M. and Milner, K. C. (1961). Preparation and host-reactive properties of endotoxin with low content of nitrogen and lipid. J Exp Med 114(5): 647-663.
- Roberts, N. A., Gray, G. W. and Wilkinson, S. G. (1967). Release of lipopolysaccharide during the preparation of cell walls of Pseudomonas aeruginosa. Biochim Biophys Acta 135(5): 1068-1071.
- Schnaitman, C. A. (1971). Solubilization of the cytoplasmic membrane of Escherichia coli by Triton X-100. J Bacteriol 108(1): 545-552.
- Silhavy, T. J., Kahne, D. and Walker, S. (2010). The bacterial cell envelope. Cold Spring Harb Perspect Biol 2(5): a000414.
- Tsai, C. M. (2001). Molecular mimicry of host structures by lipooligosaccharides of Neisseria meningitidis: characterization of sialylated and nonsialylated lacto-N-neotetraose (Galβ1-4GlcNAcβ1-3Galβ1-4Glc) structures in lipooligosaccharides using monoclonal antibodies and specific lectins. Adv Exp Med Biol 491: 525-542.
- Tsai, C. M. and Frasch, C. E. (1982). A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Anal Biochem 119(1): 115-119.
- Westphal, O. and Jann, K. (1965). Bacterial lipopolysaccharides extraction with phenol-water and further applications of the procedure. Methods Carbohydr Chem 5: 83-91.
Recipes
Acknowledgments
Funding for work from the author’s laboratory was provided by USDA-NIFA, NIH, and Virginia Polytechnic Institute and State University College of Veterinary Medicine. The original work on the LPS/LOS protocols was derived from Tsai and Frasch (1982), Inzana and Apicella (1999).
The original work on preparation and analysis of PEOM protocols was derived from Loeb and Smith (1980), Barenkamp et al. (1981) and Loeb et al. (1981).
Competing interests
The authors have no financial or non-financial competing interests.
References
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Lee, Y. J. and Inzana, T. J. (2021). Extraction and Electrophoretic Analysis of Bacterial Lipopolysaccharides and Outer Membrane Proteins. Bio-protocol 11(24): e4263. DOI: 10.21769/BioProtoc.4263.
Category
Microbiology > Microbial biochemistry > Carbohydrate
Microbiology > Microbial proteomics > Membrane proteins
Biochemistry > Carbohydrate > Polysaccharide
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.