- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Label-free Quantification of Direct Protein-protein Interactions with Backscattering Interferometry

Published: Vol 11, Iss 24, Dec 20, 2021 DOI: 10.21769/BioProtoc.4256 Views: 3572
Reviewed by: Manjula MummadisettiPooja VermaBhanu Jagilinki
Original research article
The authors used this protocol in:

Oct 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The functional performance of a cell depends on how macromolecules, in particular proteins, come together in a precise orientation, how they assemble into protein complexes and interact with each other. In order to study protein-protein interactions at a molecular level, a variety of methods to investigate these binding processes yield affinity constants and/or the identification of binding regions. There are several well-established biophysical techniques for biomolecular interaction studies, such as fluorescence spectroscopy and surface plasmon resonance. Although these techniques have been proven to be efficient, they either need labeling or immobilization of one interaction partner. Backscattering interferometry (BSI) is a label-free detection method, which allows label- and immobilization-free interaction analysis under physiologically relevant conditions with high sensitivity and in small volumes. We used BSI to measure the interaction of the neuronal calcium sensor recoverin with its target G protein-coupled receptor kinase 1 (GRK1) as a model system. Increasing concentrations of purified recoverin were mixed with a specific concentration of a GRK1 fusion protein. In this protocol, we provide a full description of the instrumental setup, data acquisition, and evaluation. Equilibrium dissociation constants of recoverin-GRK1 interaction determined by the BSI instrumental setup are in full agreement with affinity constants obtained by different methods as described in the literature.
Background
Molecular binding interactions are based on driving forces set by hydrogen bonding, hydrophobic and electrostatic interactions, and van der Waals and London dispersion forces. These interactions are fundamental in cellular processes and have led to several significant discoveries in the fields of biochemistry, proteomics, chemistry, and biophysics (Archakov et al., 2003). The struggle to find label-free approaches for interaction studies has remained a major goal in the last three decades. Techniques such as NMR studies (Ames et al., 1994), mass spectrometry (Anderson et al., 2014), surface plasmon resonance (Dell’Orco and Koch, 2016), biolayer interferometry (Kumar et al., 2016), and backscattering interferometry (Bornhop et al., 2007 and 2016; Sulmann et al., 2017) are currently among the most applied methods. Backscattering interferometry (BSI) is a relatively new method and is known for its high sensitivity, immobilization-free, and label-free quantification to study protein-protein or protein ligand interactions. BSI has a refractive index (RI) detector, which measures protein-protein interactions and can also be used to study protein-ligand interactions (Bornhop et al., 2016). More recently, BSI has been successfully used in drug discovery studies (Mizuno et al., 2019), as well as protein-small molecule and protein-ion interactions (Kammer et al., 2018). The current detection limit of the BSI signal is ΔRI = 10–6. ΔRI is defined as the relative difference in refractive index between two samples having nearly identical RI values. For example, comparing RI values of 1.391312 with 1.391318 yields a ΔRI = 6 × 10-6 (Bornhop et al., 2007).
Recoverin is a neuronal calcium sensor that interacts with G protein-coupled receptor kinase 1 (GRK1, rhodopsin kinase) in a calcium dependent manner. The interplay of recoverin and GRK1 is important for primary visual processes, as it controls the phosphorylation of rhodopsin (Gorodovikova et al., 1994; Klenchin et al., 1995). Phosphorylation of rhodopsin and several feedback loops are important control steps in photoresponse recovery (Koch and Dell’Orco, 2015). Previous work revealed that the N-terminal amphipathic α-helix of GRK1 comprising the first 25 amino acids is essential for the interaction, as it comes into contact with a hydrophobic pocket in recoverin (Figure 1) (Ames et al., 2006; Higgins et al., 2006). Recoverin-GRK1 complex formation is under the control of calcium. At high saturating calcium concentrations, recoverin exposes its covalently attached myristoyl-group, opening the hydrophobic pocket for the N-terminus of GRK1. In this state, GRK1 is inhibited and unable to phosphorylate rhodopsin. This process is reversed when the calcium concentration drops (Komolov et al., 2009; Zernii et al., 2011). More recently,Abbas et al. (2019) reported that the interaction between recoverin and GRK1 involves a network of switching electrostatic interactions in recoverin by using BSI. Thus, Figure 1 represents the optimal structural configuration of the recoverin-GRK1 complex. By employing a combinatorial approach using BSI and various other techniques, we have further investigated the complex formation using wild-type and mutant forms of recoverin. Results from surface plasmon resonance, isothermal titration calorimetry, tryptophan fluorescence spectroscopy, CD spectroscopy, and molecular dynamics simulations were all in agreement with our BSI studies. The calcium-dependent interaction of recoverin with its target region in GRK1 (peptide encompassing 25 amino acids of the N-terminus) is well suited as a benchmark example for establishing BSI in any laboratory.
In this protocol, we employ BSI to study biomolecular interactions between purified recoverin and the N-terminus of GRK1 containing the first 25 amino acids N-terminally tagged with glutathione-S-transferase (GST-NRK25). We provide a step-by-step protocol including the instrument setup, the detection of signals, designing a binding experiment, and data analysis.
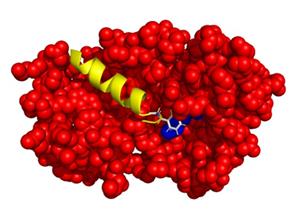
Figure 1. Possible interaction between Recoverin-GRK1. Recoverin is represented by red spheres. The blue spheres represent the C-terminal region of recoverin that interacts with GRK1. GRK1 consists of 558 amino acids and the interaction domain (M1-S25), shown as a yellow coloured helix, is present in the hydrophobic pocket of the protein recoverin.
Materials and Reagents
Eppendorf tubes (SARSTEDT)
Pipette tips 10 µl (Sartorius)
Pipette tips 200 µl (Sartorius)
Pipette tips 1,000 µl (Sartorius)
Pipette set (SARSTEDT, Nümbrecht)
Falcon tubes 50 ml (DARSTEDT AG and CO.KG, catalog number: 9042211)
Photometer cuvettes (Ratiolabs, catalog number: 2712120)
Glass beaker (Schott Duran)
Disposable vinyl gloves (Semper guard)
Lens cleaning wipe papers (Thorlabs, Inc.)
Ethanol (Sigma-Aldrich, catalog number: STBJ2652)
HCl (VWR chemicals 25% solution, catalog number: 20257-296)
Acetone 99.5% (Sigma-Aldrich, catalog number: STBJ1946)
KOH (Roth, catalog number: 6751.1)
HEPES (Roth, catalog number: 9105.2)
Magnesium chloride (MgCl2) (VWR Chemicals, catalog number: 25108.295)
Calcium chloride (CaCl2) (Roth, catalog number: 5239.1)
Potassium chloride (KCl) (Apllicem, catalog number: 7447.40.7)
Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: STB117768)
Bradford reagent (Bio-Rad, catalog number: 500-00069)
BSI buffer (see Recipes)
Equipment
Caramba Druckluftspray for cleaning electronic devices with air pressure (Böttcher AG, Germany)
Head light (Energizer)
Sample labeling marker (STAEDTLER permanent Marker pen)
Lyophylizer (Alpha 1-2 LD plus, Martin Christ Gefriertrocknungsanlagen GmbH, catalog number: 101521)
Vortex (Vortex Genie 2) (Sigma-Aldrich, catalog number: Z258423-1EA)
Sonicator (Bandelin Sonorex)
-20°C freezer (Liebherr)
Photometer (Food ALYT OMNILAB, catalog number: 122197)
Toolbox (Newport)
Microfluidic chip (Micronit-Vanderbilt)
Backscattering Intrument, built according to Bornhop et al. (2007 and 2016) and Kammer et al. (2018) .
Software
Standard FFT program 9.0 Alphas LV82 simple with saving frames
BSI SCSR analysis program-2012 (Bornhop lab at Vanderbilt University, USA)
Procedure
Schematic structure of device setup and components
The BSI system consists of a microfluidic chip, a HeNe laser, a mirror, and a CCD camera. These components are positioned and connected as illustrated in Figure 2. The position of each component and its distance to other components is crucial for the BSI setup, allowing the recording of the signals originating from protein-protein or protein-ligand interactions. Fixed positions are indicated by red boxes. It is important to keep the components in these positions at the right distances, whereas the positions of the blue boxes are optional and can be flexibly adjusted.
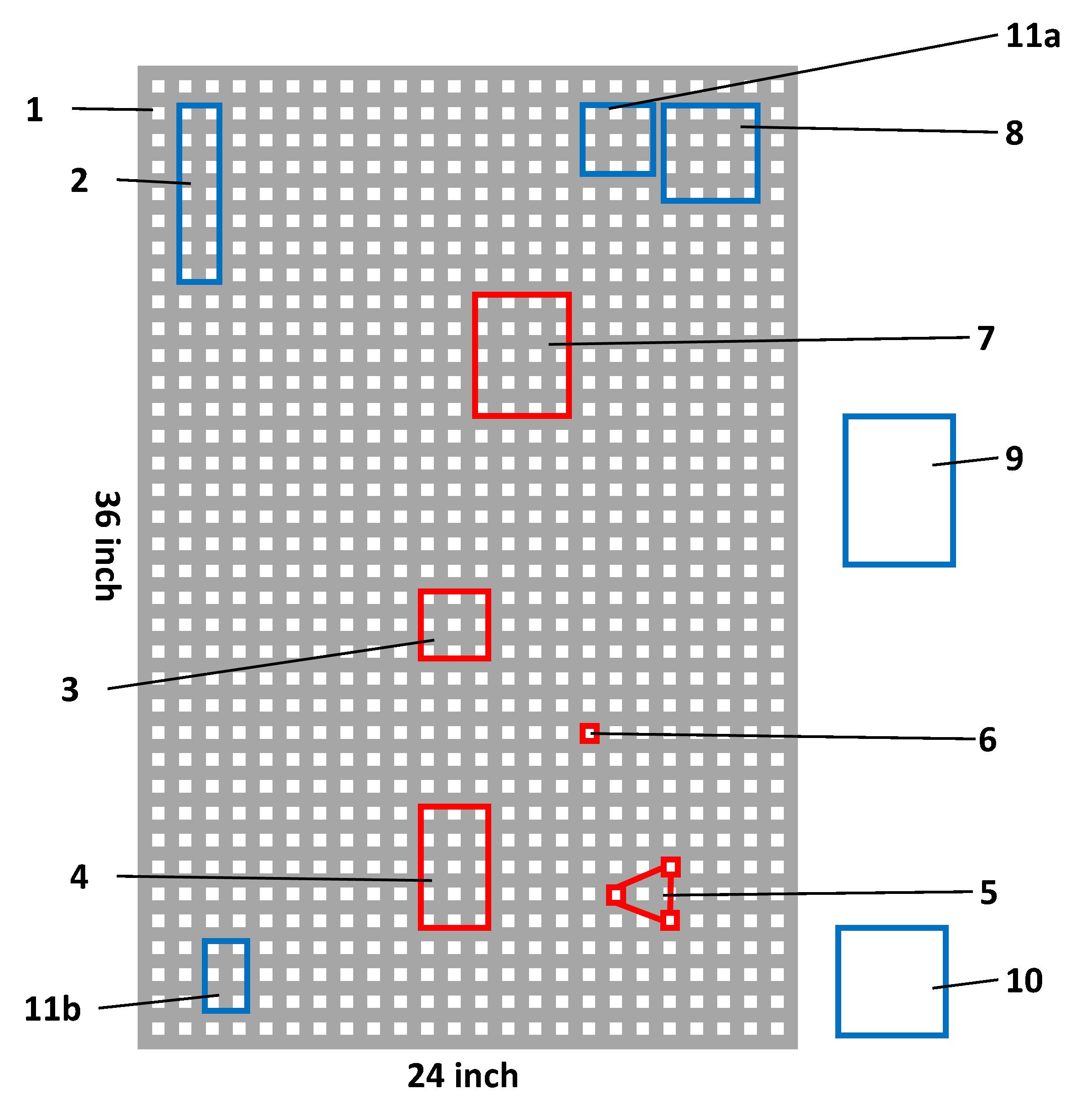
Figure 2. Schematic structure of BSI instrument setup. Boxes in red and blue represent the position of each device component representing fixed and flexible positions, respectively. Numbers correspond to device components as listed below.Components shown in Figures 2 and 3:
1. The table is 24 × 36 inches.
2. Laser
3. The point where laser leaves the cable
4. Chip holder
5. Mirror and brackets
6. Suction device
7. CCD camera
8. Power source for the laser
9. Temperature controller
10. Vacuum pump for the suction device
11a. Switch A to turn on the laser
11b. Switch B that controls the opening and closing of channels in the chip

Figure 3. Photograph of the actual setup for the BSI instrument displaying various components at their exact position. Numbers in yellow indicate the setup position as shown in Figure 2. Figure 3 shows the actual setup for the BSI instrument displaying the exact position and distances for each component.Device preparation
In order to start sample measurements, device assembling and preparation are important and should be done very accurately to avoid any erroneous signals that do not originate from biomolecular interactions.
Turn on the laser at least three hours before starting the experiment. During these three hours, either prepare the device or prepare the samples. It is recommended to prepare the device a day before doing the actual experiments. If possible, also prepare your samples a day before.
The chip holder is temperature sensitive which allows choosing the temperature for the experiments. Turn on the temperature device, which is usually set to 25°C. It will take a few minutes until the temperature reaches 25°C (shown in Figure 4).

Figure 4. Temperature controller. A temperature controller is used to set the temperature for the microfluidic chip. Experiments can be performed at any desired temperatures.Chip cleaning: The chip for BSI is a special microfluidic device that contains five flow channels (Figure 5). Chip cleaning is crucial as the laser beam directly shines on the chip surface and reflects back to the mirror. High concentrations of an alkaline or acid solution (1 M KOH or 1 M HCl) can be used to clean the chip. Only a few microliters of these solutions are recommended to use at once, followed by 70% ethanol and cleaning with an excess of water. The chip should never be touched by bare hands, always use gloves to hold the chip, and use lens cleaning paper towels to wipe and swipe the chip.
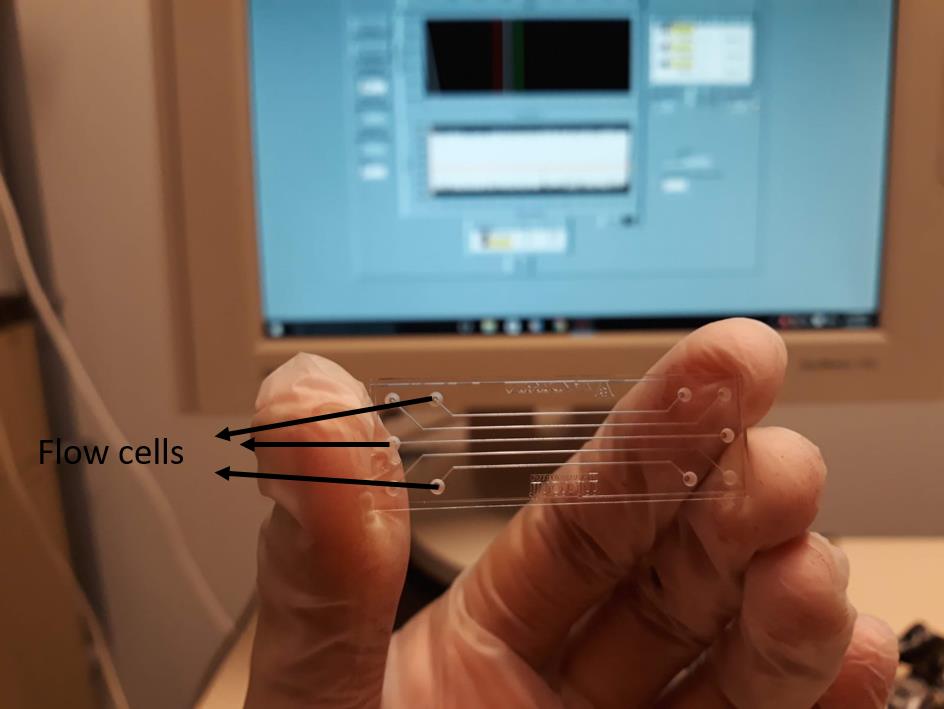
Figure 5. Chip for BSI. The chip is shown after cleaning. It has five flow cells and each of them can be used for measurements.Chip drying: There are two ways to dry the cleaned chip: manually, by blowing in some air onto the chip surface with a piece of paper (Do not blow air from the mouth. A suction device can suck excessive water left inside flow channels), or using commercial dryers (e.g., Druckluftspray from Caramba).
After drying the chip, place the chip inside chip holder. Fix the chip inside the chip holder with extreme care. There are two stages of chip fixing (Figure 6A). In the initial stage, place the chip inside the holder and tighten the metal screws. Second, special small screws made of plastic are used to fix the chip itself, so it does not move. The laser beam should directly point to the selected flow cell. To do so, you can adjust the laser signal by using adjusting screws shown in Figure 6B.

Figure 6. Microfluidic chip device setup. (A) Fixing of the chip inside the holder. The silver screws are fixed first followed by fixing the red plastic screws. Handle with extreme care as they touch the chip directly. (B) The chip holder indicating where the chip and adjusting screws for laser positioning are placed.Connect the flow cell to the suction vacuum pump right after fixing the chip in the chip holder. A small-sized plastic cable is connected to the suction vacuum pump, which can be turned ON/OFF using a regulatory switch, marked as 11b (shown in Figures 2 and 3). A small red tube (Figure 7) is directly placed inside the opening of one of the flow cells of your choice. Fix it firmly. Take a 10 µl pipette filled with water and place the pipette tip inside the opening of the flow cell. Check whether the water from the pipette is sucked out by the vacuum tube that was attached. If not, adjust the positions and/or try other channels until it works. Connecting the flow cell to the vacuum pump can be the most challenging task for a successful BSI setup. So, care and patience are of the utmost importance for this step. Figure 7 shows the successful connection of the flow cell to the vacuum pump.

Figure 7. Vacuum pump connection to the microfluidic chip device. The connecting tube shows the connection to the chip flow cell inside the chip.Maintain the pump pressure by adjusting the position of the connecting cable to the flow cell in the chip. Sometimes the pump sucks the samples from the flow cell very fast and sometimes too slow. Find a balanced pressure by trying gently.
To set the signals, fill the flow cell with the buffer that you will use to prepare your samples. For signal measurement, the laser beams onto the mirror (yellow arrow in Figure 8) and is directly reflected to the flow cell (yellow arrow in Figure 8), where it is reflected back to the mirror and finally to the CCD camera (red arrows in Figure 8). Molecular interactions occurring in protein samples loaded in the flow cell influence the reflective angle of the laser beam. Signals monitored by the CCD camera are displayed in the form of fringe patterns, which are further processed by the BSI software. An example of a fringe pattern is shown in Figure 9.

Figure 8. Indication of laser path. The laser source is where the finger pointed to. A laser beam (indicated by a yellow arrow) shines onto a mirror (white round-shaped device), where it is reflected and shines onto the chip. After reflecting back to the mirror (path shown in red). it finally reaches the CCD camera, which is covered with a piece of paper in this image.
Figure 9. Fringe patterns from CCD camera. The red dots represent the fringe pattern, which is displayed on the CCD camera.Fringe pattern readout: BSI software is used to readout fringe patterns, which change depending on the changes of reflecting conditions in the flow cell originating from biomolecular interactions. Signal differences can be detected down to nanomolar protein concentrations.
Selection of fringes: fringes are represented in the form of oscillating peaks (Figure 10) and the desired peaks are manually selected. Each peak corresponds to one point in the fringe pattern. Following aspects should be considered while selecting fringes.
The amplitude of the peaks should possibly be equal and peaks with a smaller amplitude (less than 2000 intensity units, Figure 10, upper part) should be excluded from further processing. Suitable peaks of high contrast exhibit sharp edges, are Gaussian-shaped, equally spaced, and are less noisy (Bornhop et al., 2016). Intensities should be equal (see Fringe selection in Figure 10, upper part).
If possible, five peaks should be selected, and these peaks will represent your data points during measurement.
The middle black window in Figure 10 indicates the Fourier transformation. Set the margins as indicated by red, green, and white lanes in Figure 10 (display in the middle).
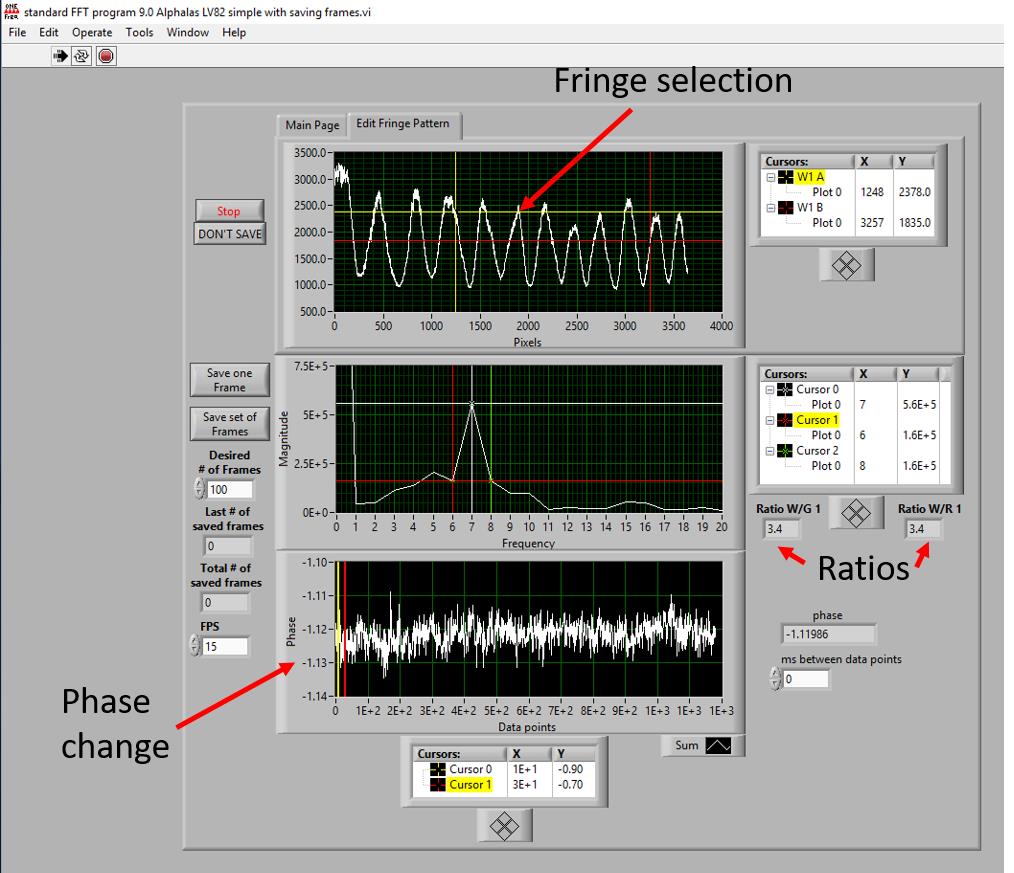
Figure 10. Snapshot of the software required for controlling the signal measurements. The signal measurement is controlled through the recording software. Three black windows are shown here. From the upper black window, we select the peaks which are representations of fringe patterns. The image in the bottom black window shows a direct phase change (indicated by a red arrow in the left side of the image), which will change during protein-protein interactions. This phase change signal is also displayed in numbers, which can be seen in the right part of the image below “Ratios” and is indicated as “phase’’.The values for the ratios should ideally be >2.5 and <3.8, and both values in the ratio should be nearly identical.
The bottom window is used for determining the phase change. Some fluctuations in the phase change signal can be observed. The smallest possible change should be in the range of 0.01. The phase change is reported in the unit of Rad. Figure 11 shows a flow diagram of the step-by-step protocol for signal recording.
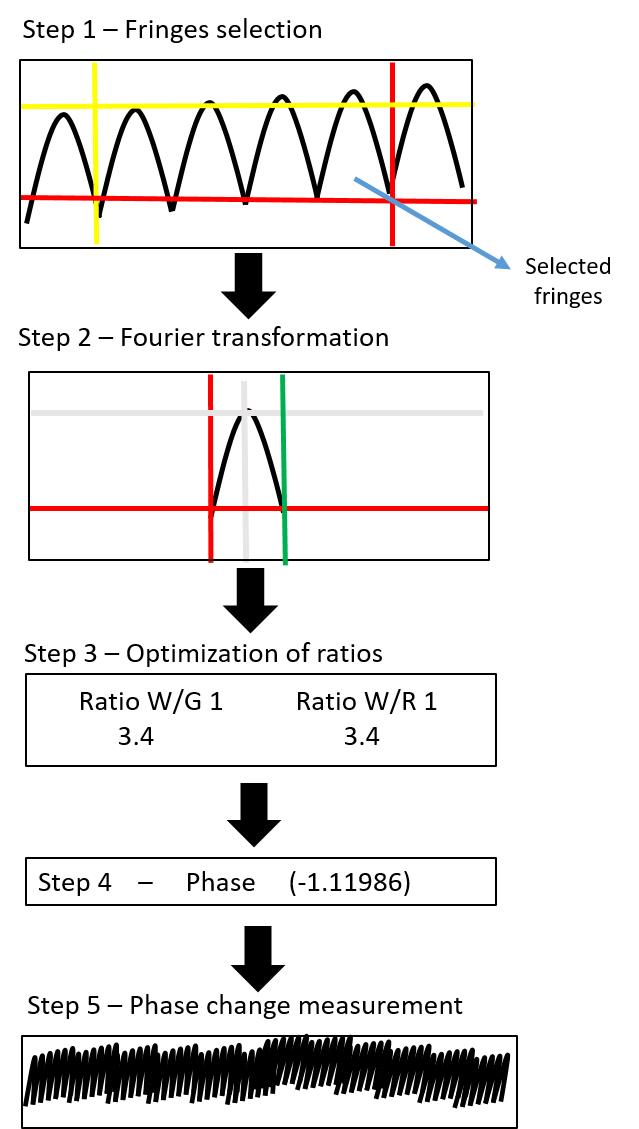
Figure 11. Flow diagram showing single steps of signal recording. This figure is the step-by-step representation of Figure 10, showing the signal recording steps in a schematic manner. In step 1, the fringes between red and yellow lines are selected. Step 2 is the Fourier transformation. Step 3 represents the ratio adjustment that is directly controlled from the signal and the instrument setup. Step 4 is the phase selection. The last step is the final phase change measurement.
Sample preparations (Figure 12)
Recoverin and GRK1 expression and purification were performed according to Abbas (Abbas et al., 2019).
Use lyophilized wild-type recoverin, N-terminus of GRK1 (first 25 amino acids) fused to GST, and only GST as a negative control. Measure the protein concentration of your samples (e.g., Bradford, 1976). See Abbas and Koch (2020) for general tips in protein quantification using the Bradford method.
Prepare samples with increasing concentration of recoverin (for example from 0 µM to 30 µM) and add a specific amount of the GST fusion protein (3 to 4 µM). Keep the total volume of each sample from 100 to 150 µl. For the background control, use only GST and prepare the sample in the same way. Figure 12 shows the sample preparation strategy.
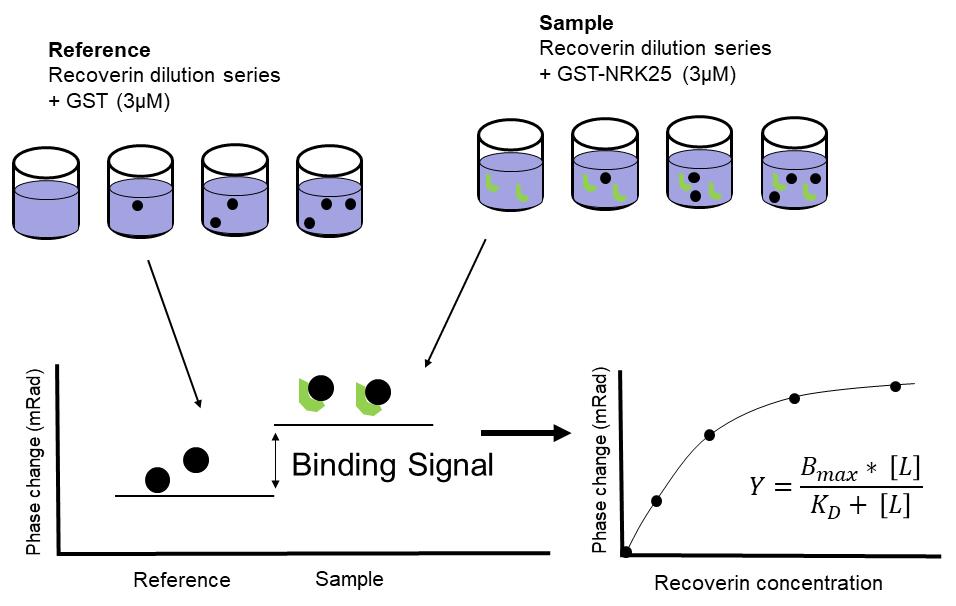
Figure 12. Scheme showing the experimental design of a protein-protein interaction study using BSI. Proteins for the background (reference) control and protein samples for positive binding signals are prepared in advance. In the present example, the reference control measurement is performed with recoverin and GST (the fusion part of the GRK1 fusion construct). The positive binding signal (sample signal) will originate from the interaction of recoverin with GST-NRK25 (shown in green). After subtracting the background control signal (reference) from the sample signal, the phase change of the resulting sample value (in mRad) is plotted as a function of increasing recoverin concentration.
Record signal
Start with cleaning the working bench and ensuring there is a sonicator placed near the BSI while performing your experiment.
Perform the measurements in a dark room (Figure 13), as the presence of light can distort the laser signal and influence its stability. Use a headlight while loading the sample onto the flow cell. Keep four 10 µl pipette sets with you and a box full of 10 µl pipette tips. Set all the pipettes to 10 µl.
Start with the negative control or reference sample measurements to save some time. Take the pipette with a filled tip and position it into the opening of the flow cell; due to pump pressure, it will start sucking the sample directly from the pipette tip without releasing it. As the sample will pass from the flow cell, use the switch control b to quickly turn off the outward flow from the flow cell. By doing so, the sample will stay inside the flow cell unless the flow cell is opened again using the switch control 11b (Figures 2 and 3). Keep the control switch 11b in the left hand while loading the sample.
As the microfluidic device is directly under the mirror, which is placed to reflect the laser beam towards the CCD camera, avoid changing the position of the mirror by bringing it into contact with the pipette.
After loading the sample, record the signal for 30 s. In between, pick the second pipette and fill it with a positive control recoverin sample and the GST-NRK25 fusion protein. Press the control switch 11b, which will open the flow cell. The sample that was already inside will be sucked away immediately. Now load the new sample in the same way and record the signal.
Every sample should be measured five times in a single experiment (technical replicates), along with five measurements of the positive control sample and five measurements of the negative control sample. A minimum of three biological replicates should be measured.
As you are using the same flow cell for the measurement of both samples (here recoverin and control), it is important to clean the flow cells in between. The following order can be followed for your convenience.
Load reference sample and measure.
Clean the flow cell with the same buffer that was used to prepare the sample.
Load recoverin sample and measure.
Wash once with 10 µl of 70% or 100% ethanol.
Wash five times with double-distilled water.
Equilibrate the channel two times with your sample buffer.
Start again with the reference sample (repeat from a to g).

Figure 13. An overview of the BSI system while performing an experiment. The laser beam is pointing to the mirror that reflects the beam to the chip in the chip holder (see Figure 8 for a different view of the set up). Switch B controls opening and closing of channels in the chip. Fringe patterns are on display in the CCD camera.
Data analysis
Open the data analysis program and click on “open file”. The program file is shown in Figure 14. Load the file for GRK1 and recoverin. The measurement was designed to record signals from mixtures of increasing concentrations of recoverin and a fixed concentration of GST-N-GRK25. Data analysis can be performed from any point. The data analysis program has three black windows (1, 2 and 3) that can show the recorded signals. Figure 14 shows the loaded file in window 2 and 3. The display needs to be zoomed in to see the recorded signals. To do so, we have two cursors in yellow and red color as shown in windows 1 and 2. An easy way to zoom in the signal is to set the x- and y-axis values as shown below window 1. Once you see the signals recorded from the reference and sample measurements (four times, window 2 in Figure 15), bring the yellow cursor to the left side of the signal and the red cursor to the right side of the signal. This will show the values for the number of points, the arithmetic mean, and the standard deviation (left side of window 3 in Figure 15). The minimum number of points that qualify for a good signal should be 800. Click on the button “create new table” (upper row), set the number of rows and columns, and click on the button “plot average” (right side next to window 3 in Figure 15). This action will bring the values to the table. By following these steps, values for all recorded signals will be extracted.
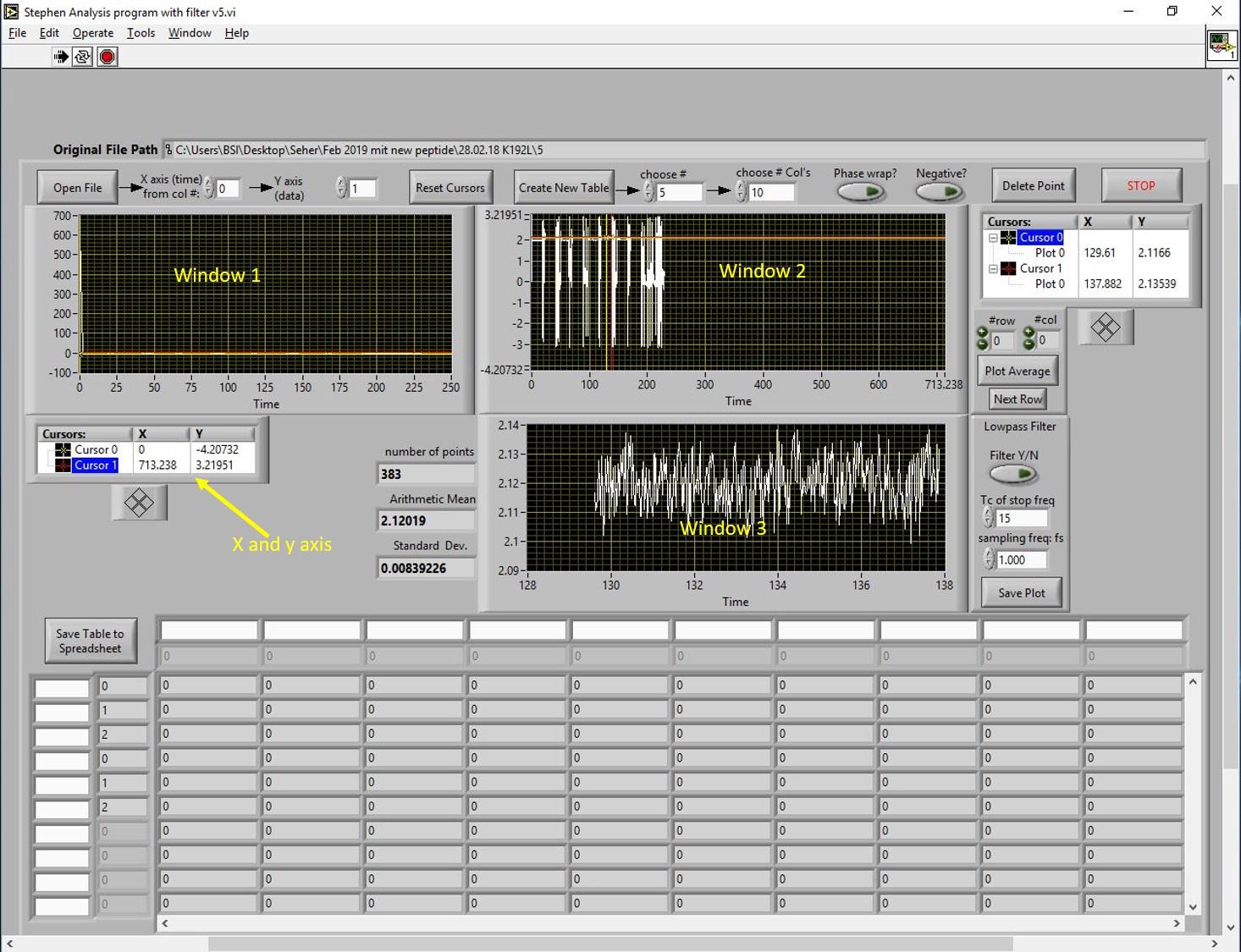
Figure 14. Data analysis. Data is analysed using data analysis software, which is shown above. Load the data and it appears in window 2 and 3.
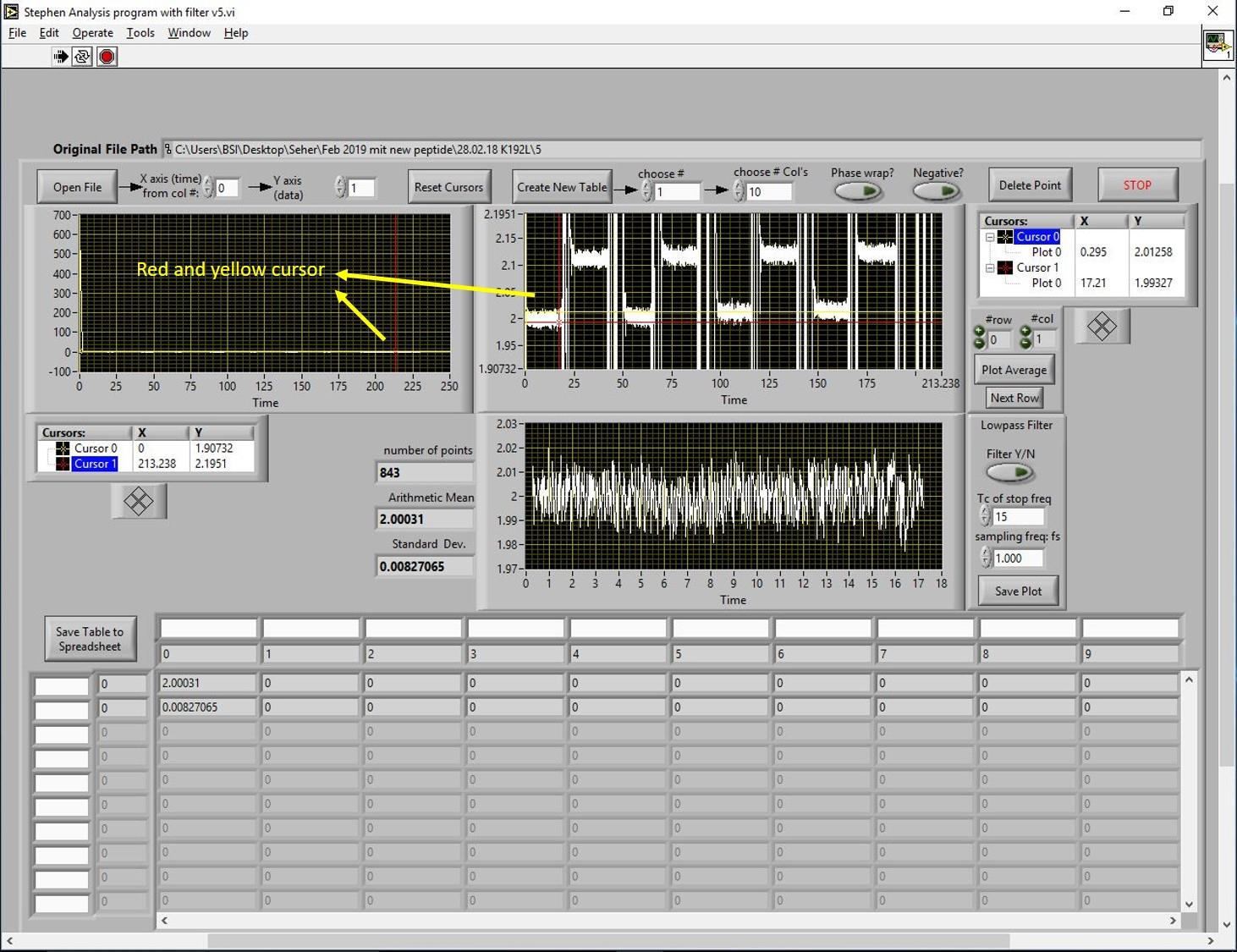
Figure 15. Data extraction. Data is extracted and plotted in the table at the bottom of software window. In the next step, values of the reference signal are subtracted from the sample signal.
See below for example:
Reference: 2.00031
Sample: 2.01134
Sample – Reference = 0.01103 Rad
Convert this value to mRad by
0.01103 * 1000 = 11.03 mRad
Calculate this value for all 9 recorded concentrations and use Sigma Plot 13 to plot the mRad signals as a function of increasing concentration of recoverin. In case a minor mRad signal was detected at zero concentration of recoverin, subtract this value from all data points before plotting the data. In the present experiment, we yielded an equilibrium dissociation constant KD = 5.1 ± 1.9 µM (Figure 16). Similar experiments performed previously by others and us (Sulmann et al., 2017; Abbas et al., 2019) reported KD values of 4.72 μM ± 0.72 μM and of 4.2 ± 1.9 μM, respectively. Both previous investigations and the present one in this contribution employed equal constructs of recoverin and GRK1 (fusion protein of GST and the first N-terminal 25 amino acids of GRK1, see background), but samples resulted from different biological preparations. KD values in the lower micromolar range are in agreement with values of the half-maximal inhibition (IC50) of GRK1 by recoverin (Gorodovikova et al., 1994; Klenchin et al., 1995), which is a physiological process during photoresponse recovery (see background). Sulmann et al. (2017) further tested the binding process of recoverin and the GST-GRK1 fusion protein in presence and absence of calcium showing lack of binding, when calcium was absent (Figure 1 in Sulmann et al., 2017).
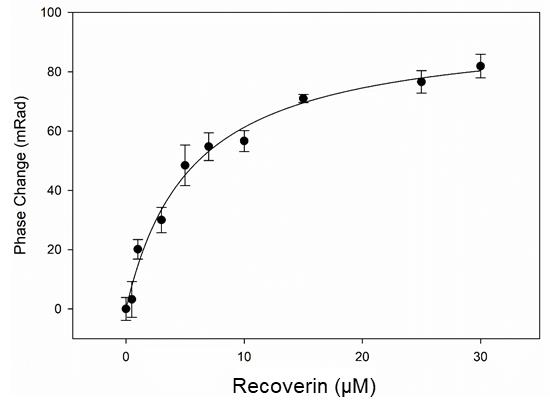
Figure 16. Interaction of recoverin and GRK1. The interaction data is plotted as a function of increasing concentration of recoverin in µM. The apparent dissociation constant (KD = 5.1 ± 1.9 µM) is extracted.
Notes
Never touch the microfluidic chip device without gloves.
Store the microfluidic chip device in 100% HPLC grade ethanol after finishing the experiment.
Always perform the experiment in a dark room. No direct light should interfere with the HeNe laser.
Turn ON the HeNe laser at least 3 h before starting the experiment. This will help to create a stable signal.
Use headlight to load the sample in microfluidic chip.
Recipes
BSI buffer
20 mM HEPES-KOH (pH 7.5)
100 mM NaCl
1 mM MgCl2
400 µM CaCl2
Acknowledgments
We thank the German Academic Exchange Program (DAAD) and the Deutsche Forschungsgemeinschaft (DFG, grant number GRK1885) for financial support. This protocol was modified from previous work (Sulmann et al., 2017; Abbas et al., 2019).
Competing interests
The authors declare not competing interest.
Ethics
Experiments were performed with recombinant proteins according to safety regulations in category S1.
References
- Abbas, S. and Koch, K. W. (2020). Quantitative determination of Ca2+-binding to Ca2+-sensor proteins by isothermal titration calorimetry. Bio-protocol 10(07): e3580.
- Abbas, S., Marino, V., Dell'Orco, D. and Koch, K. W. (2019). Molecular Recognition of Rhodopsin Kinase GRK1 and Recoverin Is Tuned by Switching Intra- and Intermolecular Electrostatic Interactions. Biochemistry 58(43): 4374-4385.
- Ames, J. B., Levay, K., Wingard, J. N., Lusin, J. D. and Slepak, V. Z. (2006). Structural basis for calcium-induced inhibition of rhodopsin kinase by recoverin. J Biol Chem 281(48): 37237-37245.
- Ames, J. B., Tanaka, T., Stryer, L. and Ikura, M. (1994). Secondary structure of myristoylated recoverin determined by three-dimensional heteronuclear NMR: implications for the calcium-myristoyl switch. Biochemistry 33(35): 10743-10753.
- Anderson, D. M., Ablonczy, Z., Koutalos, Y., Spraggins, J., Crouch, R. K., Caprioli, R. M. and Schey, K. L. (2014). High resolution MALDI imaging mass spectrometry of retinal tissue lipids. J Am Soc Mass Spectrom 25(8): 1394-1403.
- Archakov, A. I., Govorun, V. M., Dubanov, A. V., Ivanov, Y. D., Veselovsky, A. V., Lewi, P. and Janssen, P. (2003). Protein-protein interactions as a target for drugs in proteomics. Proteomics 3(4): 380-391.
- Bornhop, D. J., Kammer, M. N., Kussrow, A., Flowers, R. A., 2nd and Meiler, J. (2016). Origin and prediction of free-solution interaction studies performed label-free. Proc Natl Acad Sci U S A 113(12): E1595-1604.
- Bornhop, D. J., Latham, J. C., Kussrow, A., Markov, D. A., Jones, R. D. and Sorensen, H. S. (2007). Free-solution, label-free molecular interactions studied by back-scattering interferometry. Science 317(5845): 1732-1736.
- Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248-254.
- Dell'Orco, D. and Koch, K. W. (2016). Fingerprints of Calcium-Binding Protein Conformational Dynamics Monitored by Surface Plasmon Resonance. ACS Chem Biol 11(9): 2390-2397.
- Gorodovikova, E. N., Gimelbrant, A. A., Senin, II and Philippov, P. P. (1994). Recoverin mediates the calcium effect upon rhodopsin phosphorylation and cGMP hydrolysis in bovine retina rod cells. FEBS Lett 349(2): 187-190.
- Higgins, M. K., Oprian, D. D. and Schertler, G. F. (2006). Recoverin binds exclusively to an amphipathic peptide at the N terminus of rhodopsin kinase, inhibiting rhodopsin phosphorylation without affecting catalytic activity of the kinase. J Biol Chem 281(28): 19426-19432.
- Kammer, M. N., Kussrow, A. K., Olmsted, I. R. and Bornhop, D. J. (2018). A Highly Compensated Interferometer for Biochemical Analysis. ACS Sens 3(8): 1546-1552.
- Klenchin, V. A., Calvert, P. D. and Bownds, M. D. (1995). Inhibition of rhodopsin kinase by recoverin. Further evidence for a negative feedback system in phototransduction. J Biol Chem 270(27): 16147-16152.
- Koch, K. W. and Dell'Orco, D. (2015). Protein and Signaling Networks in Vertebrate Photoreceptor Cells. Front Mol Neurosci 8: 67.
- Komolov, K. E., Senin, II, Kovaleva, N. A., Christoph, M. P., Churumova, V. A., Grigoriev, II, Akhtar, M., Philippov, P. P. and Koch, K. W. (2009). Mechanism of rhodopsin kinase regulation by recoverin. J Neurochem 110(1): 72-79.
- Kumar, S., Mazumder, M., Gupta, N., Chattopadhyay, S. and Gourinath, S. (2016). Crystal structure of Arabidopsis thaliana calmodulin7 and insight into its mode of DNA binding. FEBS Lett 590(17): 3029-3039.
- Mizuno, H., Kihara, Y., Kussrow, A., Chen, A., Ray, M., Rivera, R., Bornhop, D. J. and Chun, J. (2019). Lysophospholipid G protein-coupled receptor binding parameters as determined by backscattering interferometry. J Lipid Res 60(1): 212-217.
- Sulmann, S., Kussrow, A., Bornhop, D. J. and Koch, K. W. (2017). Label-free quantification of calcium-sensor targeting to photoreceptor guanylate cyclase and rhodopsin kinase by backscattering interferometry. Sci Rep 7: 45515.
- Zernii, E. Y., Komolov, K. E., Permyakov, S. E., Kolpakova, T., Dell'orco, D., Poetzsch, A., Knyazeva, E. L., Grigoriev, II, Permyakov, E. A., Senin, II, et al. (2011). Involvement of the recoverin C-terminal segment in recognition of the target enzyme rhodopsin kinase. Biochem J 435(2): 441-450.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Abbas, S. and Koch, K. W. (2021). Label-free Quantification of Direct Protein-protein Interactions with Backscattering Interferometry. Bio-protocol 11(24): e4256. DOI: 10.21769/BioProtoc.4256.
Category
Biophysics > Scattering spectroscopy
Neuroscience > Cellular mechanisms > Intracellular signalling
Biochemistry > Protein > Interaction > Protein-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.