- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vivo Imaging of Calcium Activities from Pancreatic β-cells in Zebrafish Embryos Using Spinning-disc Confocal and Two-photon Light-sheet Microscopy
(*contributed equally to this work) Published: Vol 11, Iss 23, Dec 5, 2021 DOI: 10.21769/BioProtoc.4245 Views: 4649
Reviewed by: Vishal ParekhSonal Patel PatelAnonymous reviewer(s)

Original research article
The authors used this protocol in:

Jan 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Visualizing the function of pancreatic β-cells in vivo has been a long-sought goal for β-cell researchers. Unlike imaging of β-cells in mammalian species with conventional positron emission tomography and single-photon emission computed tomography, which only provides limited spatial-temporal resolution, transparent zebrafish embryos are a unique model that allows high-resolution fluorescent imaging of β-cells in their native physiological microenvironment in vivo. Here, we detail a protocol for real-time visualization of individual β-cell function in vivo in a non-invasive manner, through combination of a novel transgenic zebrafish reporter line Tg (ins:Rcamp1.07) with both a commercial spinning-disc confocal microscope and an in-house developed super-resolution microscope (2P3A-DSLM). The protocol described here allows for the longitudinal monitoring of dynamic calcium activities from heterogeneous β-cells in early developing zebrafish embryos and is readily adaptable for use in imaging other important processes in islet biology, as well as screening new compounds that can promote β-cell function or maturation using a living whole organism system.
Background
With conventional methods, such as immunostaining microscopy to observe the expression of maturation markers in fixed samples or ELISA to measure the amount of insulin content, β-cell functionality or maturity is usually evaluated as a whole. Whether all β-cells within each islet mature at a synchronized pace during early development remains a mystery. Deconvolution of the function of individual β-cells using advanced imaging microscopy will overcome this problem. The primary function of a mature β-cell is to quickly secrete insulin in response to increased blood glucose. Glucose-triggered Ca2+ influx in pancreatic β-cells is essential for insulin secretion, thus often serves as a functional marker to evaluate β-cell maturity (Pagliuca et al., 2014; Rezania et al., 2014). Ca2+ transients in primary β-cells can be imaged in isolated islets when β-cells are labeled with cell-permeable Ca2+ dyes or a genetically encoded fluorescent Ca2+ reporter. However, to non-invasively image Ca2+ transient in β-cells in vivo remains the unmet challenge due to the non-transparent nature of the pancreas tissue deeply buried in the human abdomen.
In this protocol, we used zebrafish as a model organism in which β-cell development is highly conserved with mammals since its maturation processes can be visualized in the transparent, externally located, and rapidly developing embryos (Huang et al., 2001). To overcome the negative impacts of phototoxicity caused by long-term laser exposures and the effect of autofluorescence from tissues on imaging qualities (e.g., signal-to-noise ratios), we generated a novel Tg (ins:Rcamp1.07) zebrafish reporter line, in which every β-cell is specifically labeled with Rcamp1.07, a red fluorescent Ca2+ indicator. With this reporter line, we use a commercially available spinning-disc confocal microscope, as well as our home-made high-resolution two-photon three-axis digital scanning light-sheet microscope (2P3A-DSLM) (Zong et al., 2015; Zhao et al., 2019), to visualize real-time glucose-stimulated Ca2+ responses in individual β-cells in a living organism. With the spinning-disc confocal microscope, dynamic calcium activities from pancreatic islets in living zebrafish embryos can be easily visualized, but individual β-cells are difficult to discern. In contrast, the 2P3A-DSLM setup enables both the reconstruction of a clear three-dimensional structure of pancreatic islets and the high resolution of dynamic calcium activities from individual β-cells. With the 2P3A-DSLM imaging strategy, we have longitudinally monitored the functional status of β-cells in early developing zebrafish embryos and observed the heterogeneity of β-cell functional development, as islets mature in vivo during embryonic development.
Future imaging of panoramic β-cell function and mass in living zebrafish may serve as a unique tool for drug screening and unraveling mechanisms of many important but obscure processes in islet biology, such as neogenesis, regeneration, cell identity, and functional changes during normal development and disease progression.
Materials and Reagents
Confocal imaging chamber/35mm culture dishes with cover glass bottom (Cellvis, catalog number: D35-20-1-N)
10 cm plastic Petri dish (Greiner, catalog number: 633185)
1 ml syringe (Becton Dickinson, catalog number: 309659)
3 ml plastic transfer pipette (Leiwo, catalog number: B089FD9Q17)
Glass capillary (1 mm × 90 mm) (Narishige, catalog number: G-1)
Fine sharp forceps (Fine Science Tools, catalog number: 11295-51)
Cell-Tak tissue adhesive (Corning, catalog number: 354240)
Tg (ins:Rcamp1.07) zebrafish embryos (24hpf-5dpf)
Tricaine (Sigma-Aldrich, catalog number: A5040)
PTU (Sigma-Aldrich, catalog number: P7629)
Ultra-pure agarose (Invitrogen, catalog number: 16500100)
D-glucose (Sigma-Aldrich, catalog number: G7021)
NaCl (Sigma-Aldrich, catalog number: S7653)
KCl (Sigma-Aldrich, catalog number: P9333)
CaCl2·2H2O (Sigma-Aldrich, catalog number: C5080)
MgSO4·7H2O (Sigma-Aldrich, catalog number: 63138)
1 M Tris-HCl pH 9.0 (BIO BASIC, catalog number: SD8146)
1× E3 medium (see Recipes)
1,000× PTU stock solution (0.2 M) (see Recipes)
20× Tricaine stock solution (4 g/L) (see Recipes)
40 mM D-glucose stock solution (see Recipes)
1 M D-glucose stock solution (see Recipes)
1% ultra-pure agarose (see Recipes)
Equipment
Olympus spinning-disc confocal microscope (Olympus, catalog number: IX83P2ZF)
Home-made two-photon light-sheet microscope (2P3A-DSLM; for more information about how this microscope setup is designed and developed, see details in the original article by Zong et al., 2015)
Olympus epifluorescent microscope (Olympus, model: BX50, catalog number: 18342)
Incubator with constant climate chamber (Memmert, catalog number: HPP110eco)
Microwave oven
Heat block (Bechmark Scientific, catalog number: BSH-1001)
pH meter (Mettler-Toledo, catalog number: 30266626)
Scale (Mettler-Toledo, catalog number: ML54)
Software
MetaMorph (Molecular Devices, https://www.moleculardevices.com/products/cellular-imaging-systems/acquisition-and-analysis-software/metamorph-microscopy#gref)
HCImage (Hamamatsu, https://hcimage.com)
Fiji/ImageJ (National Institute of Health, https://imagej.net/Fiji/Downloads)
GraphPad Prism 6.0 or above versions (GraphPad Software, https://www.graphpad.com)
Procedure
In vivo imaging of calcium activities from pancreatic β-cells in zebrafish embryos using a spinning-disc confocal microscope
Prepare zebrafish embryos
Set up male and female adults in the afternoon one day before. Usually, one male and three females in a mating tank are enough.
Collect embryos the next morning in Petri dishes filled with approximately 25 ml of E3 medium and discard any unfertilized or dead embryos using wide-bore plastic transfer pipettes.
At 24 h post fertilization (hpf), examine the fluorescence under an epifluorescent microscope and select Rcamp1.07-positive embryos of Tg (ins:Rcamp1.07) zebrafish. Transfer the positive embryos to a new Petri dish filled with approximately 25 ml of E3 medium containing 1× PTU. Change the E3 medium containing 1× PTU and discard any dead embryos daily.
Note: Supplementing PTU into E3 medium helps to reduce the pigmentation and thus improves imaging quality. Daily medium change is not necessary, though we recommend refreshing E3 medium daily, especially for the first 2 days of incubation, as unfertilized and abnormally developed embryos are commonly seen before 48 hpf. Closer inspection can be done under a stereotype microscope.
Dissect embryos out from their chorion membranes using two fine sharp forceps when imaging early-stage embryos (e.g., 12-36 hpf embryos). Specifically, use one forceps to hold the chorion and make a tear in the chorion with the other forceps. The chorion is then held in a region opposite to that of the tear, and the embryo is gently pushed through the opening by passing the chorion and embryo between the tips of the other forceps. Alternatively, many embryos older than 48 hpf may have spontaneously hatched. In this case, no dechorionation is required to dissect embryos out from the chorion membranes.
Note: It is critical that the tips of the forceps are sharp and that their ends can touch. Early-stage embryos are particularly fragile, so great care must be taken to minimize damage.
Coat confocal imaging chamber and mount zebrafish embryo
Coat each Cellvis confocal imaging chamber with 2-3 μl of Cell-Tak tissue adhesive in the center region of the glass coverslip bottom. Spread the Cell-Tak tissue adhesive using a pipette tip and let it dry until a very thin coating layer is formed.
Note: It takes about 5-10 min to dry the Cell-Tak tissue adhesive, and we recommend using the coated chamber immediately.
Select one embryo and orient it on the coated region of the imaging chamber, with the right side of the embryo body facing the bottom of the chamber. This reduces the actual distance between the objective lens and the islet of the embryo (Figure 1A).
Immediately fill the imaging chamber with 200 μl of E3 medium containing 1× tricaine, ensuring the embryo is fully covered (Figure 1B).
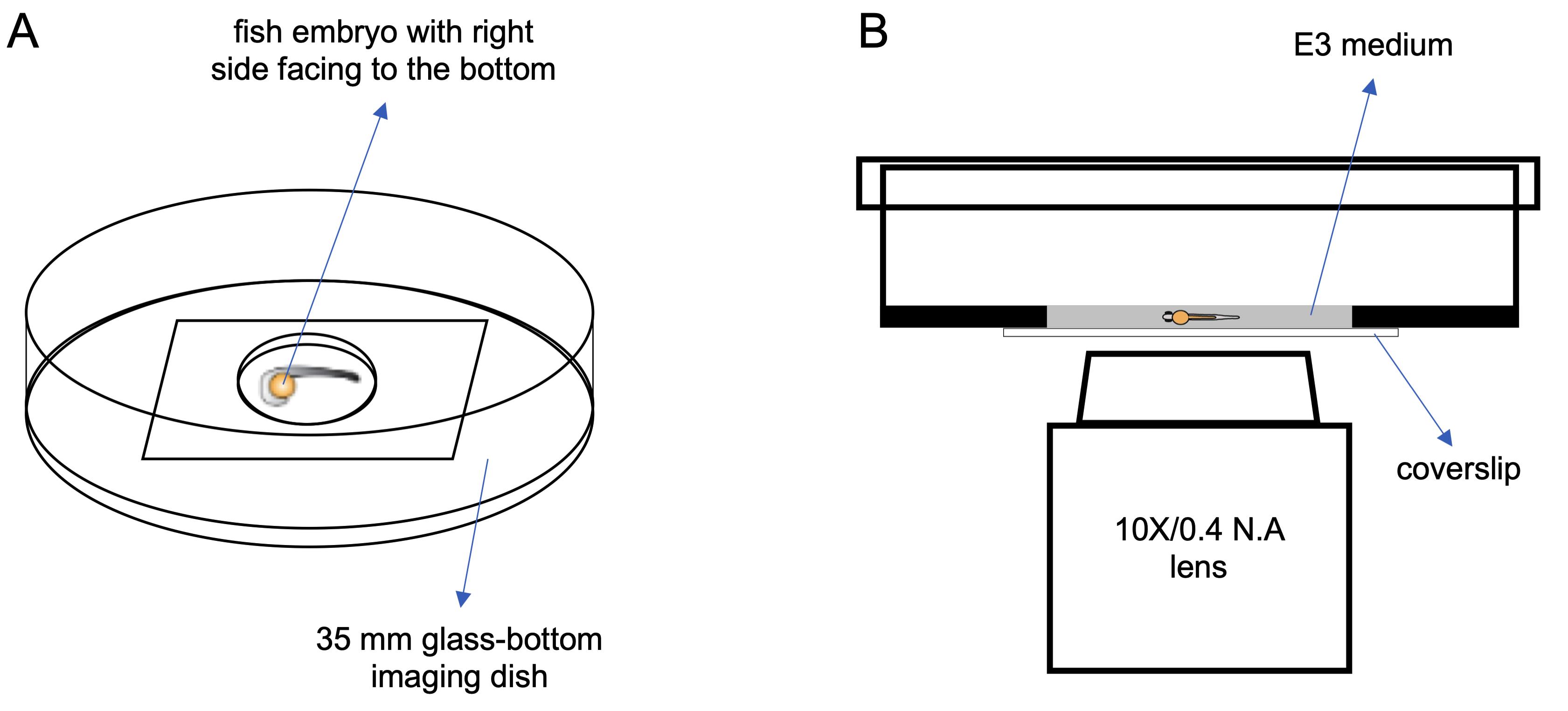
Figure 1. An illustration of zebrafish embryo mounting method and the spinning-disc confocal imaging setup. A. Top view shows the method of mounting zebrafish embryo. B. Side view shows the spinning-disc confocal setup for imaging calcium activities in living zebrafish embryo.
Image dynamic calcium activities from β-cells in zebrafish embryo using a spinning-disc confocal microscope
Find the zebrafish embryo and fine adjust to image Rcamp1.07 signals with the most islet β-cells on the optimal focal plane.
Use a 561 nm laser to illuminate Rcamp1.07 signals under a spinning-disc confocal microscope equipped with a 10×/0.4 N.A objective and use a 1.6× pre-amplifier to magnify images.
Use E3 medium containing 1× tricaine for 10-15 min for basal calcium activities. Gently add an equal volume (200 μl) of 40 mM D-glucose stock solution to the imaging chamber to reach a final concentration of 20 mM during stimulation. Image for 20-40 min for stimulated calcium activities (Figure 2).
For time-lapse imaging, capture images once per second (1Hz) with 100 ms exposure time using 561 nm laser under spinning-disc confocal microscope (Figure 2).
Collect all the time-lapse images using MetaMorph software. Process and analyze raw image data using Fiji software (for details, see Data analysis).
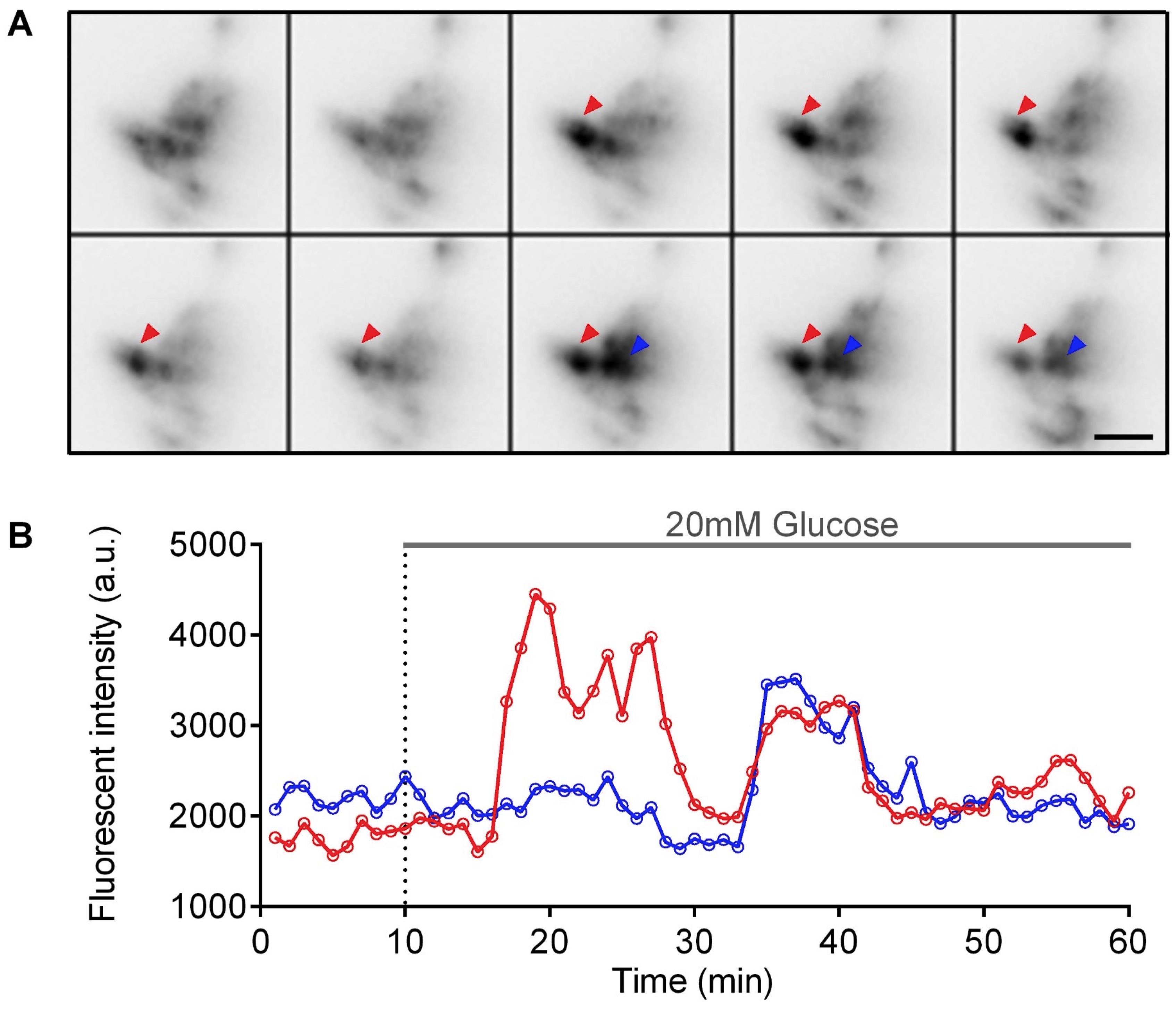
Figure 2. Visualization of glucose-stimulated calcium activities from β-cells in Tg (ins:Rcamp1.07) embryos under spinning-disc confocal microscope. A. Representative images of calcium activities from pancreatic islets in a living Tg (ins:Rcamp1.07) embryo at 72 hpf at basal condition (indicated by the first two images in the first row of the montage picture) and after 20 mM glucose stimulation (indicated by remaining images of the montage picture). Arrows (red and blue) indicate two active regions showing the glucose-stimulated calcium activities from the principal islet. Time-lapse images were captured using MetaMorph software, and montage picture was generated using Fiji software. B. Representative time-course traces of the calcium activities in the regions marked in (A). Graph was analyzed and generated using GraphPad Prism 6.0 software. Scale bar: 20 μm.
In vivo imaging of calcium activities from pancreatic β-cells in living zebrafish embryos using a two-photon light-sheet microscope
Transfer zebrafish embryo to the agarose
Heat up one aliquot (1 ml) of 1% agarose and keep it warm in a heat block set to 38-40°C.
Supplement tricaine to a final concentration of 1× into the agarose and mix gently.
Select one dechorionated Rcamp1.07-positive embryo of Tg (ins:Rcamp1.07) zebrafish and transfer it into the warm 1% ultra-pure agarose using a plastic transfer pipette (Videos 1-2).
Note: When transferring embryos for agarose mounting, it is unavoidable to bring additional E3 medium along with the embryos. The additional E3 medium, if in a large volume, will change the final concentration of agarose solution, and as such, it may result in uneven embryo mounting in agarose. Transfer as minimal volume of additional E3 medium as possible in this step.
 Video 1. Zebrafish embryo transfer. Video 2. Agarose mounting strategy.
Video 1. Zebrafish embryo transfer. Video 2. Agarose mounting strategy.
Mount the zebrafish embryo in an agarose cylinder
Fill the self-designed mounting device with 1% agarose solution using a syringe-attached glass capillary (Figure 3A) before taking up the zebrafish embryo.
Take up an embryo by pulling the syringe very gently and place the embryo with its head pointing to the opening end of the capillary.
Note: Avoid generating bubbles inside of the capillary.
Keep the capillary horizontal until the agarose becomes fully solid (Figure 3A).
Partially plug out a short agarose cylinder containing the zebrafish embryo, locate it vertically using the device holder equipped on the 4D-stage of light-sheet microscope (Figure 3B-3C ), and immerse the agarose-embedded embryo in the light-sheet cubic imaging chamber pre-filled with 49 ml of E3 medium containing 1× tricaine.
Note: The authors find that illuminating zebrafish embryos in agarose cylinders rather than in glass capillaries will greatly increase the signal-to-noise ratio and enhance image contrast.
Rotate the device holder to orient the embedded embryo with the right side of the embryo body facing towards the illuminating objective of the light-sheet microscope. This allows light penetration through the zebrafish embryo and more effective illumination of Rcamp1.07 signals (Videos 1-2).
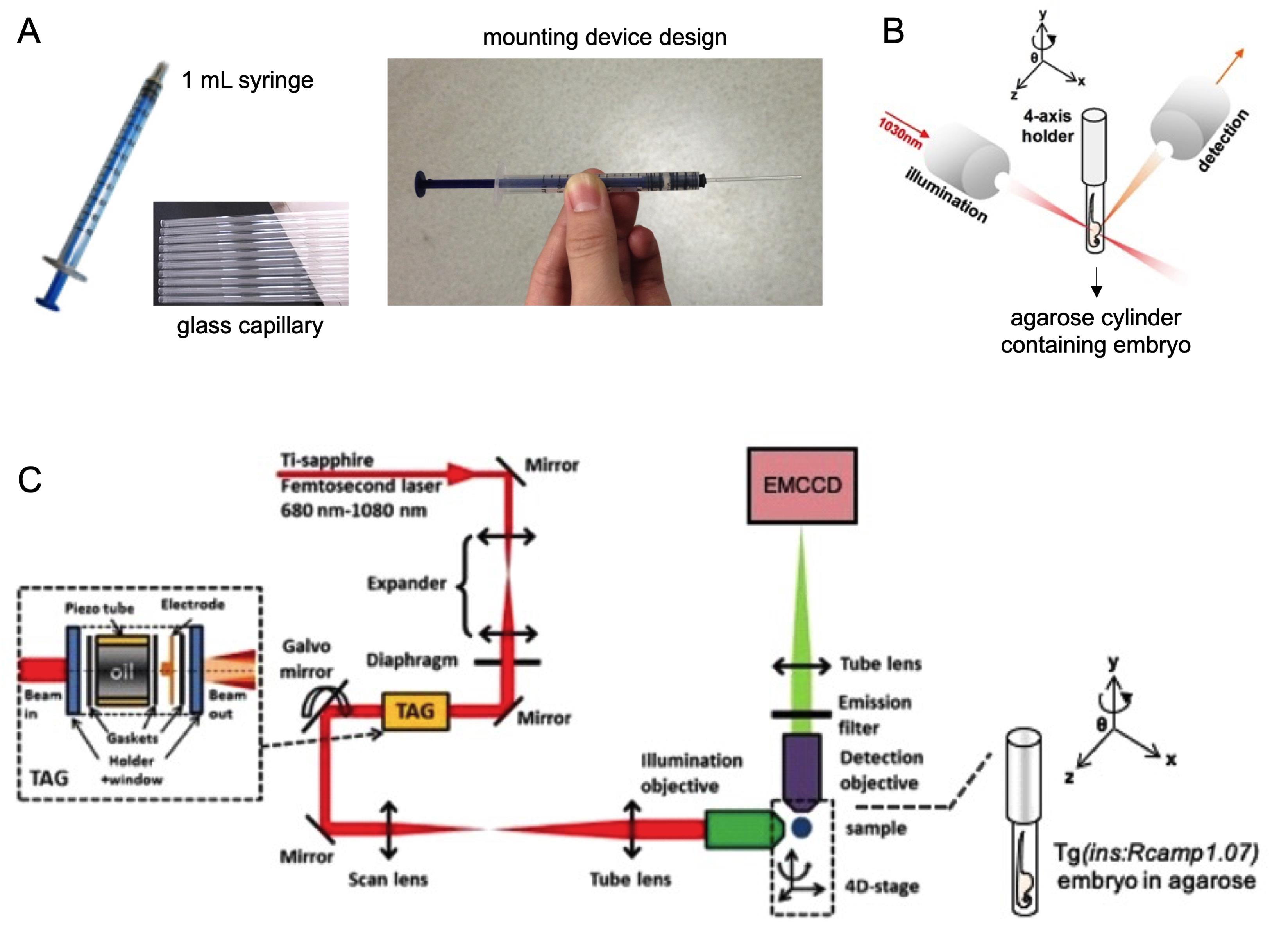
Figure 3. An illustration of zebrafish embryo mounting method and the 2P3A-DSLM two-photon light-sheet microscopy imaging setup. A. A self-designed mounting device comprised of a syringe-attached with glass capillary. B-C. To improve image quality, partially plug out a short agarose cylinder containing the zebrafish embryo and locate it vertically using the device holder equipped on the 4D-stage of the light-sheet microscope.
Image dynamic calcium activities from β-cells in zebrafish embryo using 2P3A-DSLM
Find the zebrafish embryo and fine adjust the 4D-stage to place the Rcamp1.07-positive islet β-cells on the best focal plane.
Use a 1030 nm laser to illuminate Rcamp1.07 signals under 2P3A-DSLM equipped with two 40×/0.8 N.A water lenses or a two-photon light-sheet setup with similar parameters.
Capture time-lapse images in E3 medium containing 1× tricaine to record basal calcium activities. Add 1 ml of 1 M D-glucose stock solution to the cubic imaging chamber to reach a final concentration of 20 mM during stimulation. Capture time-lapse images under the high glucose E3 medium to record stimulated calcium activities (Figure 4).
Note: Depending on specific experimental design and research interests, the stimulation time might be different. In most cases, recording time-lapse images for 15-60 min upon glucose stimulation is required. This allows the time required for glucose penetration and uptake by fish embryos and for capturing the calcium responses to glucose stimulation from β-cells.
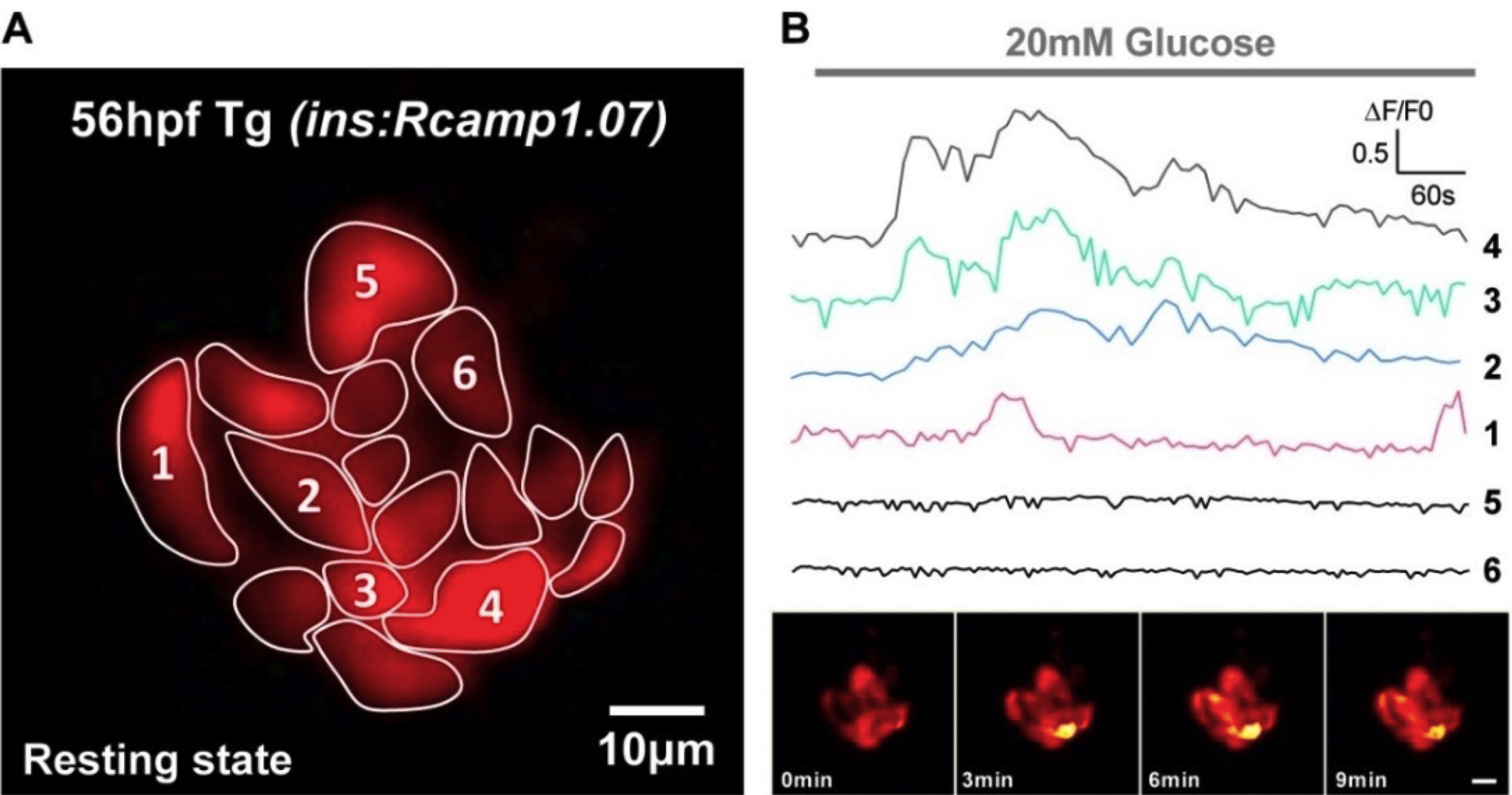
Figure 4. Visualization of glucose-stimulated calcium activities from β-cells in Tg (ins:Rcamp1.07) embryos under 2P3A-DSLM. A. The visibility of Rcamp1.07 fluorescence in β-cells under resting condition represents the basal level of intracellular calcium. B. Robust increase in fluorescent intensity of Rcamp1.07 under 20 mM glucose stimulation distinguishes the glucose-responsive β-cells (labeled with 1, 2, 3 and 4) from the glucose-non-responsive β-cells (labeled with 5 and 6). Scale bar: 10 μm.For 2D time-lapse imaging experiments, optically section the islet into 5-6 planes (z-step = 10 μm). Capture each plane with a 150-200 ms exposure time, using the 1030 nm laser under 2P3A-DSLM or a similar two-photon light-sheet setup and imaging procedure (Mahou et al., 2014). With this strategy, imaging the entire islet takes 1.5-2 s per imaging cycle (Figure 4).
For fast volumetric imaging experiments and reconstruction of calcium activities within the entire islet, optically section the islet into 25 planes (z-step = 2 μm). Capture each plane five times, each with 8 ms exposure, and average into one single image.
Note: This averaging process will greatly increase image contrast by increasing signal-to-noise ratios). With this strategy, imaging the entire islet takes approximately 1 s (25 × 5 × 8 ms = 1 s) per imaging cycle (see Videos 3, 5, and 6 in our original eLife research article [Zhao et al., 2019]).
Collect all the time-lapse images using HCImage software and process raw image data using Fiji software (details see Data analysis).
Data analysis
For image processing and analysis, use Fiji or ImageJ software.
For background subtraction, open Fiji/ImageJ → load images → Process → Subtract background → rolling ball radius 80 in the Rcamp1.07 channel (Figure 5A).
For removing noise caused by camera, open Fiji/ImageJ → load images → Process → Noise →Remove outliers → Radius: 2 pixels; Threshold: 80; Which outliers: Bright (Figure 5B).
For 3D rendering, open Fiji/ImageJ → load stack images → Image → Stack → 3D-projection → Projection method: Brightest Point; Axis of rotation: Y-Axis; Slice spacing inches (depends on z-step size) → Check the box of Interpolate (Figure 5C).
For image drift correction during long-term time-lapse imaging, open Fiji/ImageJ → load stack images → Plugins → Registration → StackReg → Transformation: Rigid Body (Figure 5D).
For statistical analysis, use GraphPad Prism 6.0 software or above versions. Perform unpaired Student’s two-tailed t-tests to compare data between two indicated groups and one-way ANOVA by Dunnett’s test for multiple comparisons with the control group.
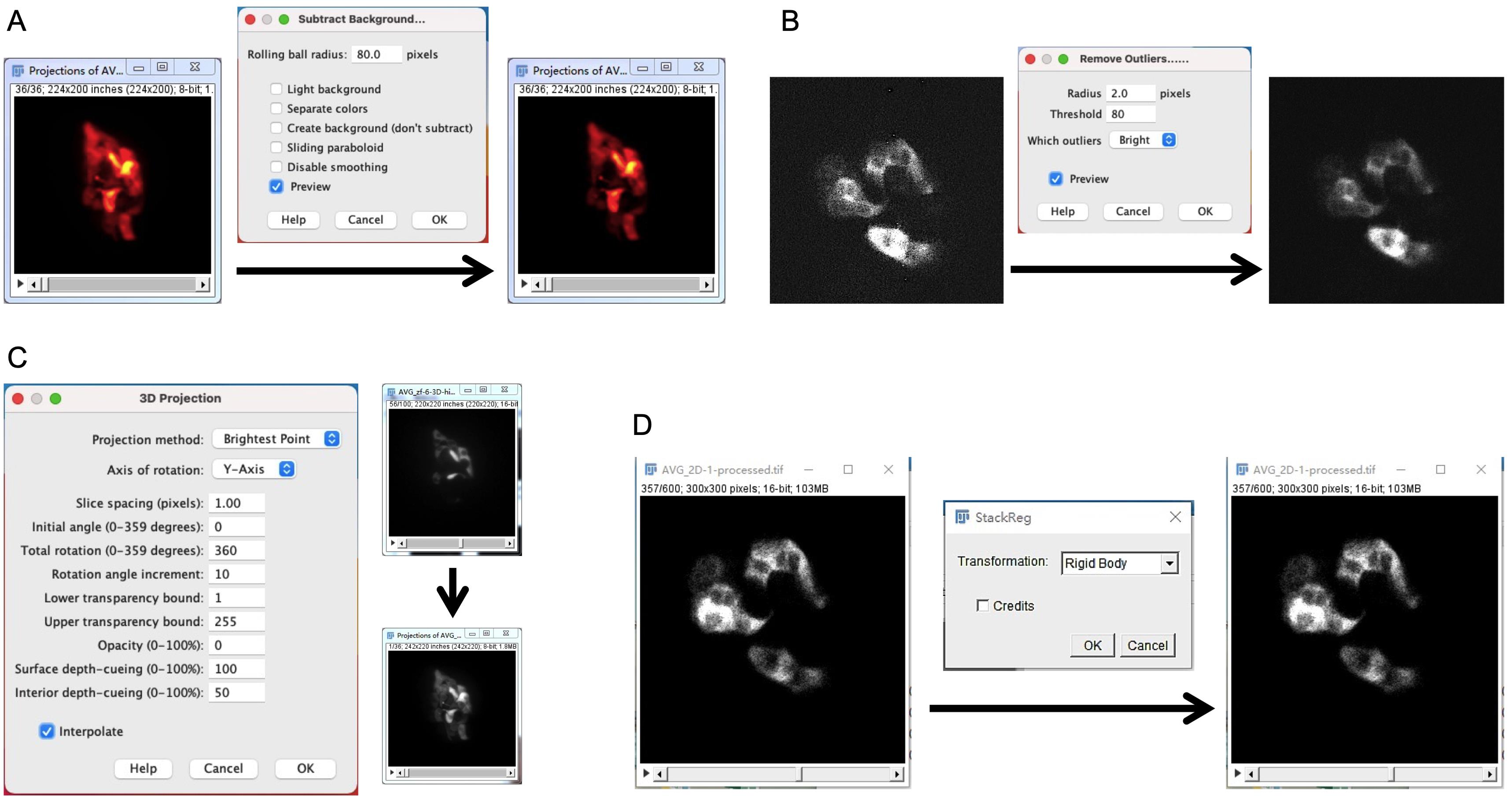
Figure 5. Illustration of image processing methods using Fiji or ImageJ software. A-D: Snapshots for each of the steps describe background subtraction (A), remove outliers (B), 3D rendering (C), and image drift correction (D).
Recipes
1× E3 medium
5 mM NaCl
0.17 mM KCl
0.33 mM CaCl2·2H2O
0.33 mM MgSO4·7H2O
Add 20 ml of 50× stock solution into a 1 L vessel and fill up to 1 L with MilliQ water.
1,000× PTU stock solution (0.2 M)
Add 30.44 g of PTU to 1 L of ethanol and store in air-tight bottle.
20× Tricaine stock solution (4 g/L)
Add 4 g to Tricaine in 979 ml of 1× E3 medium and add 21 ml of 1 M Tris-HCl (pH 9.0) to adjust the pH to 7.0.
40 mM D-glucose stock solution
Add 7.2064 g of D-glucose to 1 L of 1× E3 medium, sterilize using 0.22 μm filter, and store at 4°C.
1 M D-glucose stock solution
Add 18.016 g D-glucose to 100 ml of 1× E3 medium, sterilize using 0.22 μm filter, and store at 4°C.
1% ultra-pure agarose
Add 1 g of agarose to 100 ml of 1× E3 medium, heat in a microwave oven until the agarose is fully melted, and the solution is completely clear.
Aliquot 1 ml of warm agarose solution into a 1.5 ml microfuge tube, and store at -20°C.
Acknowledgments
This work was supported and funded by the grants from the National Science and Technology Major Project Program (2016YFA0500400), National Natural Science Foundation of China (91854112 and 91750203, 31327901, 31521062, 31570839, 31301186), and Beijing Natural Science Foundation (L172003). The protocol was first described in our original research paper, “In vivo imaging of β-cell function reveals glucose-mediated heterogeneity of β-cell functional development” published in eLife (Zhao et al., 2019; doi:10.7554/eLife.41540).
Competing interests
No potential conflict of interests was declared by the authors.
Ethics
Animal care, generation of transgenic zebrafish lines and all animal experimentation were approved by the IACUC of Peking University in China (reference number: IMM-ChenLY-2).
References
- Huang, H., Vogel, S. S., Liu, N., Melton, D. A. and Lin, S. (2001). Analysis of pancreatic development in living transgenic zebrafish embryos. Mol Cell Endocrinol 177(1-2): 117-124.
- Mahou, P., Vermot, J., Beaurepaire, E. and Supatto, W. (2014). Multicolor two-photon light-sheet microscopy. Nat Methods 11(6): 600-601.
- Pagliuca, F. W., Millman, J. R., Gurtler, M., Segel, M., Van Dervort, A., Ryu, J. H., Peterson, Q. P., Greiner, D. and Melton, D. A. (2014). Generation of functional human pancreatic beta cells in vitro. Cell 159(2): 428-439.
- Rezania, A., Bruin, J. E., Arora, P., Rubin, A., Batushansky, I., Asadi, A., O'Dwyer, S., Quiskamp, N., Mojibian, M., Albrecht, T., et al. (2014). Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 32(11): 1121-1133.
- Zhao, J., Zong, W., Zhao, Y., Gou, D., Liang, S., Shen, J., Wu, Y., Zheng, X., Wu, R., Wang, X., et al. (2019). In vivo imaging of β-cell function reveals glucose-mediated heterogeneity of β-cell functional development. eLife 8: e41540.
- Zong, W., Zhao, J., Chen, X., Lin, Y., Ren, H., Zhang, Y., Fan, M., Zhou, Z., Cheng, H., Sun, Y. and Chen, L. (2015). Large-field high-resolution two-photon digital scanned light-sheet microscopy. Cell Res 25(2): 254-257.
Article Information
Copyright
![]() Zhao et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Zhao et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Zhao, J., Liang, S., Zhao, Y., Zong, W., Tran, E., Chen, L. and Liu, Y. (2021). In vivo Imaging of Calcium Activities from Pancreatic β-cells in Zebrafish Embryos Using Spinning-disc Confocal and Two-photon Light-sheet Microscopy. Bio-protocol 11(23): e4245. DOI: 10.21769/BioProtoc.4245.
- Zhao, J., Zong, W., Zhao, Y., Gou, D., Liang, S., Shen, J., Wu, Y., Zheng, X., Wu, R., Wang, X., et al. (2019). In vivo imaging of β-cell function reveals glucose-mediated heterogeneity of β-cell functional development. eLife 8: e41540.
Category
Developmental Biology > Cell signaling > Intracellular calcium
Biophysics > Microscopy > Two-photon laser scanning microscopy
Biological Sciences > Biological techniques
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.