- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Analysis of Protein Stability by Synthesis Shutoff

Published: Vol 11, Iss 22, Nov 20, 2021 DOI: 10.21769/BioProtoc.4225 Views: 4280
Reviewed by: Khyati Hitesh ShahAashima KhoslaAnonymous reviewer(s)

Original research article
The authors used this protocol in:

Jan 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
In this protocol, we describe the analysis of protein stability over time, using synthesis shutoff. As an example, we express HA-tagged yeast mitofusin Fzo1 in Saccharomyces cerevisiae and inhibit translation via cycloheximide (CHX). Proteasomal inhibition with MG132 is performed, as an optional step, before the addition of CHX. Proteins are extracted via trichloroacetic acid (TCA) precipitation and subsequently separated via SDS-PAGE. Immunoblotting and antibody-decoration are performed to detect Fzo1 using HA-specific antibodies. We have adapted the method of blocking protein translation with cycloheximide to analyze the stability of high molecular weight proteins, including post-translational modifications and their impact on protein turnover.
Keywords: ShutoffBackground
The stability of any protein of interest can be examined, after blocking translation, by following its fate over time. Unstable proteins undergo either proteasomal or vacuolar degradation. Thus, the physiological relevance of protein turnover can be deciphered by blocking the proteasome, the vacuolar proteases, or by using stable mutant variants thereof. To bypass the need for antibodies binding specifically to it, tagged versions of the protein of interest can be constructed and expressed.
Here, we provide a detailed protocol to assess protein stability and its dependence on the proteasome. A low-cost and reliable western blot-based assay is presented, benefiting from the turnover properties of the protein Fzo1. The yeast mitofusin Fzo1 is a known target of post-translational ubiquitylation (Schuster et al., 2018), which is degraded under normal physiological conditions (Anton et al., 2013; Anton et al., 2011; Anton et al., 2019; Schuster et al., 2020; Simoes et al., 2018). Residues in Fzo1 and enzymes required for its turnover have been identified, whereby the use of respective mutations preventing this degradation can be used as a turnover control. In addition, increased instability, which depends on the proteasome, can happen during stress conditions or upon genetic alterations. Thus, as described below and exemplified in the representative figure, Fzo1 represents a good case-study example for protein stability analysis. Our protocol provides a detailed step-by-step description to analyze protein stability with a non-toxic preparation procedure and lower amounts of cycloheximide when compared to the commonly used protocol fromBuchanan et al. (2016). Cycloheximide (CHX) is widely known to block translational elongation by interfering with deacetylated tRNA and binding the ribosome on the 60S subunit (Klinge et al., 2011; Schneider-Poetsch et al., 2010). Furthermore, our protein extraction protocol using trichloroacetic acid (TCA) can be used for a western blot-based analysis of both small and high molecular weight proteins.
Materials and Reagents
Filter papers (Macherey-Nagel, catalog number: 742113)
Nitrocellulose membrane (Amersham, catalog number: 10600001)
Centrifugation tubes (15 ml, 1.5 ml) (VWR, catalogue number: 352096 [15 ml], Sarstedt, catalog number: 72.690.001 [1.5 ml])
X-ray films (Fujifilm, catalog number: 47410 19289)
Yeast Saccharomyces cerevisiae BY4741 isogenic to S288c (Mata his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) (Brachmann et al., 1998)
D(+)-Glucose monohydrate (Roth, catalog number: 6780.2)
Cycloheximide InSolution 100 mg/ml in DMSO (Sigma, catalog number: C4859)
MG132 InSolution 10 mM in DMSO (Calbiochem, catalog number: 474791)
NaOH (Merck, catalog number: 1.06498.5000)
β-Mercaptoethanol (Sigma-Aldrich, catalog number: M6250-100ML)
Trichloroacetic acid (TCA) (Roth, catalog number: 3744.1)
SDS (Roth, catalog number: CN30.3)
Tris (Roth, catalog number: 5429.5)
NaCl (Roth, catalog number: 3957.2)
37% HCl (VWR, catalog number: 20252.335)
Glycine (PanReac AppliChem, catalog number: 141340.0914)
DTT (MP, catalog number: 100597)
Bromophenol Blue sodium salt (USB, catalog number: US12370)
Glycerol (Serva, catalog number: 23176.01)
Color Prestained Protein Standard, Broad Range 11-245 kDa (New England Biolabs, catalog number: P7719)
Rotiphorese Gel 30 Acrylamide/Bis-acrylamide stock solution (Roth, catalog number: 3029.1)
Ammonium Peroxodisulfate (APS) (Merck, catalog number: 1.01201.0500)
Tetramethylethylenediamine (TEMED) (Sigma, catalog number: T9281-25ML)
Methanol (Roth, catalog number: 8388.5)
2-Propanol (VWR, catalog number: 20842.330)
Ponceau S (Serva, catalog number: 33429.02)
Acetic acid (VWR, catalog number: 20104.334)
Nonfat dried milk powder (Roth, catalog number: T145.3)
HA primary antibody (clone 3F10) (Roche, catalog number: 11867423001)
Sodium Azide (NaN3) (Merck, catalog number: 1.06688.0100)
Rat secondary antibody (Invitrogen, catalog number: 31470)
GE Healthcare AmershamTM ECLTM Prime (Amersham, catalog number: RPN2232)
TBS Buffer, pH 7.4 (see Recipes)
HU sample buffer (see Recipes)
Ponceau S solution (see Recipes)
Tris/Glycine buffer (see Recipes)
Transfer buffer (see Recipes)
Minimal yeast media which allows auxotrophic selection of yeast cells (see Recipes)
1 M DTT (see Recipes)
Rödel mix (see Recipes)
Equipment
Shaking incubator for yeast cultures (Infors-HT, Multitron Standard)
Bench-top centrifuge (VWR, model: Mega star 1.6)
Bench-top cooling centrifuge (Eppendorf, model: 5145R)
Vortex (Scientific Industries, Inc., model: Vortex-Genie 2)
Photometer (Hitachi, model: Double Beam Spectrophotometer U-2900)
Thermomixer (Eppendorf, model: Thermomixer comfort 5355)
Glass syringe (Hamilton, e.g., catalog number: 80565)
Note: Instead of a Hamilton syringe, capillary pipette tips (VWR, catalog number: 53509-015) can be used.
SDS-PAGE equipment (HoeferTM, model: SE 600 Series)
Western blot equipment (Peqlab, PerfectBlueTM, model: “semi-dry”-Blotter, SedecTM)
Tumbling table (Biometra, model: WT12)
Autoradiography cassette (Amersham, model: RPN11642)
Developer machine (AGFA, model: CURIX 60)
Procedure
Culture conditions
Preculture (Day 1)
Inoculate a small number of cells [starting optical density (OD)600 0.05 = 750,000 cells/ml] in 5 ml of selective minimal media supplemented with glucose (2%, w/v) and grow overnight to the exponential growth phase at 30°C, shaking at 160 rpm.
Note: 30°C is the optimal growth condition for wild-type yeast strains. For the analysis of essential proteins, and therefore genetically manipulated temperature-sensitive (ts) yeast strains, the growth temperature must be adapted accordingly. Furthermore, some proteins might be degraded only under certain conditions, for example, upon heat shock, which should therefore be tested for each case.
Main culture (Day 2)
Inoculate 50 ml of selective media supplemented with glucose (2%, w/v) with a starting OD600 = 0.3 in the morning, by diluting the yeast cells from the overnight culture. Let the cells divide at least 4-6 h to the exponential growth phase, up to a maximum OD600 = 2.
Protein synthesis shutoff by CHX treatment
Harvest cells an OD600 = 12 in total: measure the OD of the main culture and calculate the necessary volume (V) in milliliters by using V = 12 /measured OD600 of the main culture. Use 15 ml conical tubes for centrifugation (5 min, 4,100 × g, room temperature), keep 4 ml of growth media inside the tube, and discard the rest of the supernatant.
Example:
Measured OD600 of main culture = 1.3
Needed (V): 12 OD600 in ml
Calculate: V = 12 OD600/1.3 OD600 = 9.23 ml
9.23 ml will be transferred to a 15ml conical tube and cells harvested by centrifugation
Resuspend the harvested cells in 4 ml of media.
This step is optional, depending on the need for blocking the proteasome before translation inhibition. To inhibit the proteasome, 0.1 mM MG132 is added to the media, and cells are incubated for 1 h at 30°C, shaking at 160 rpm.
Note: If proteasomal inhibition is performed by MG132 treatment, cells have to be previously grown for at least 3 h in media containing proline and 0.003% SDS (Liu et al., 2007), to ensure that the MG132 is not exported from the cells immediately via several ABC (ATP binding cassette) transporters (Liu et al., 2007). Alternatively, cells lacking the ABC transporter Pdr5 and/or Snq2 can be used to perform proteasomal inhibition without using special culturing conditions. Importantly: Cells deleted for PDR5/SNQ2 are treated with only 0.05 mM MG132 due to increased drug permeability (Collins et al., 2010).
Add CHX to the cells to a final concentration of 100 µM and invert tubes gently up to five times.
Immediately transfer 1 ml of cell suspension containing 3 OD600 to a new reaction tube and harvest cells by centrifugation (5min, 16,100 × g, 4°C). Aspirate the supernatant and freeze the cell pellet at -20°C (time point (t) = 0).
Shake cells again for 1 h and afterwards repeat Step B5 (t = 1).
After an additional incubation of 2 h at 30°C, shaking at 160 rpm, 1 ml of media containing 3 OD600 will be harvested again (5 min, 16,100 × g, 4°C) (t = 3).
Note: The time points of the chase should be adjusted to the protein used in the study, e.g., a very unstable protein should be analyzed in a time frame of several minutes to 1 h, while stable proteins should be analyzed over several hours.
Sample preparation
Note: All following steps are performed on ice.
Cell pellets are resuspended in 1ml of sterile distilled water and 150 µl of Rödels mix, to break up the yeast cell wall and denature proteins by vortexing and incubating for 15 min on ice.
To precipitate the proteins, add 150 µl of 55% TCA to each sample. Invert the tubes and incubate for 10 min on ice.
Centrifuge for 10 min, 16,100 × g at 4°C.
Carefully remove the supernatant.
Centrifuge again 5 min, 16,100 × g at 4°C.
Remove the whole supernatant and resuspend the pellet in 50 µl of HU sample buffer (see Recipes) by shaking tubes for 20 min, at 45°C and with 1,400 rpm on a thermomixer.
SDS-PAGE and Immunoblotting
Proteins are separated using SDS-PAGE, as previously described (Schuster et al., 2018).
Gel equipment is assembled according to the manufacturer’s instructions.
Gel mixtures for stacking and separation gels are prepared as described in Table 1 but without APS and TEMED.
Note: The gel used to analyze the protein via SDS-PAGE depends on the size of the protein of interest. The smaller the size of the protein, the higher the percentage of the acrylamide and vice versa (Sambrook and Russell, 2006).
Table 1. Composition of one 12% SDS gel
12% Separation gel (ml) Stacking gel (ml) Acrylamide1 4 0.5 1.875 M Tris pH 8.8 2 - 0.6M Tris pH 6.8 - 0.6 MilliQ water 3.8 1.85 10% SDS 0.05 0.02 20% APS 0.05 0.015 TEMED 0.01 0.005 1 30% Acrylamide with 0.8% Bisacrylamide (37.5:1)
Add APS and TEMED to the separation gel mix, mix rapidly and thoroughly, and pour 9.5 ml of the mixture between two glass plates. Add 1 ml of 2-propanol on top of the gel mix to remove bubbles and smoothen the surface. Let the gel polymerize for at least 15 min.
Remove 2-propanol and wash 3× carefully with distilled water. Carefully remove excess water with a filter paper.
Put in the combs to the space between both glass plates before pouring the upper gel. Add APS and TEMED to the stacking gel mix. Mix well and pour the stacking gel mix on top of the separation gel until the space between both glass plates is full.
After polymerization of the upper stacking gel mixture (for at least 15 minutes), remove the combs and fill the wells with Tris/Glycine buffer.
Load a maximum of 15 µl per sample and include a pre-stained protein standard marker (e.g., NEB, P7719) in each gel.
Fill the lower chamber of the electrophoresis tank (anode) with Tris/Glycine buffer and insert gels into the system according to the manufacturer’s instructions.
Fill the upper cathode chamber with freshly prepared Tris/Glycine buffer. Follow the manufacturer’s instructions for information on volumes of buffers required.
Run gels at 200 V and 400 mA until the protein loading front (visible due to the bromophenol blue sodium salt in the HU-buffer, used as a tracking dye to monitor the progress of molecules moving through the polyacrylamide gel) reaches the end of the gel. To estimate the size of proteins, refer to the pre-stained protein standard on the gel. Ensure that the protein of interest stays inside the gel and does not run out.
Remove the gels from the running systems, disassemble the glass plate sandwich, and remove the stacking gel with a plastic wedge.
Transfer the acrylamide gels onto nitrocellulose membranes via semi-dry western blotting, as previously described in Schuster et al. (2018) .
Incubate membranes in Ponceau S (PoS) solution to stain all proteins transferred to the membrane. This staining is used to ascertain sufficient protein transfer and serves as a loading control. After imaging the membrane, destain it in 20 ml of TBS for at least 15 minutes.
Perform antibody detection. In the case exemplified in this protocol, HA-specific antibodies were used. Dilute the antibodies 1:1,000 in 5% milk in TBS. Incubate the membranes in the primary antibody overnight, at 4°C. The next day, remove the primary antibody, wash the membranes for 15 min with TBS, followed by 2 h incubation at room temperature with the secondary α-rat antibody diluted 1:5,000 in 5% milk in TBS. After removing the secondary antibody, wash the membrane with TBS three times for 10 min. The secondary antibody is coupled to the enzyme horseradish peroxidase (HRP), used to create an emission of light in a hydrogen peroxide-catalyzed oxidation of luminol. Western blots are developed by using light sensitive X-ray films and an enhanced chemiluminescence solution (Cytiva AmershamTM ECLTM Prime), containing hydrogen peroxide and luminol, as previously described in the protocol from Schuster et al. (2018) .
Note: Degradation of an unstable protein (e.g., Ubc6) during CHX treatment or its stabilization upon MG132 treatment can be used as a positive control for translational and/or proteasomal inhibition, respectively.
In Figure 1, a representative image is shown.
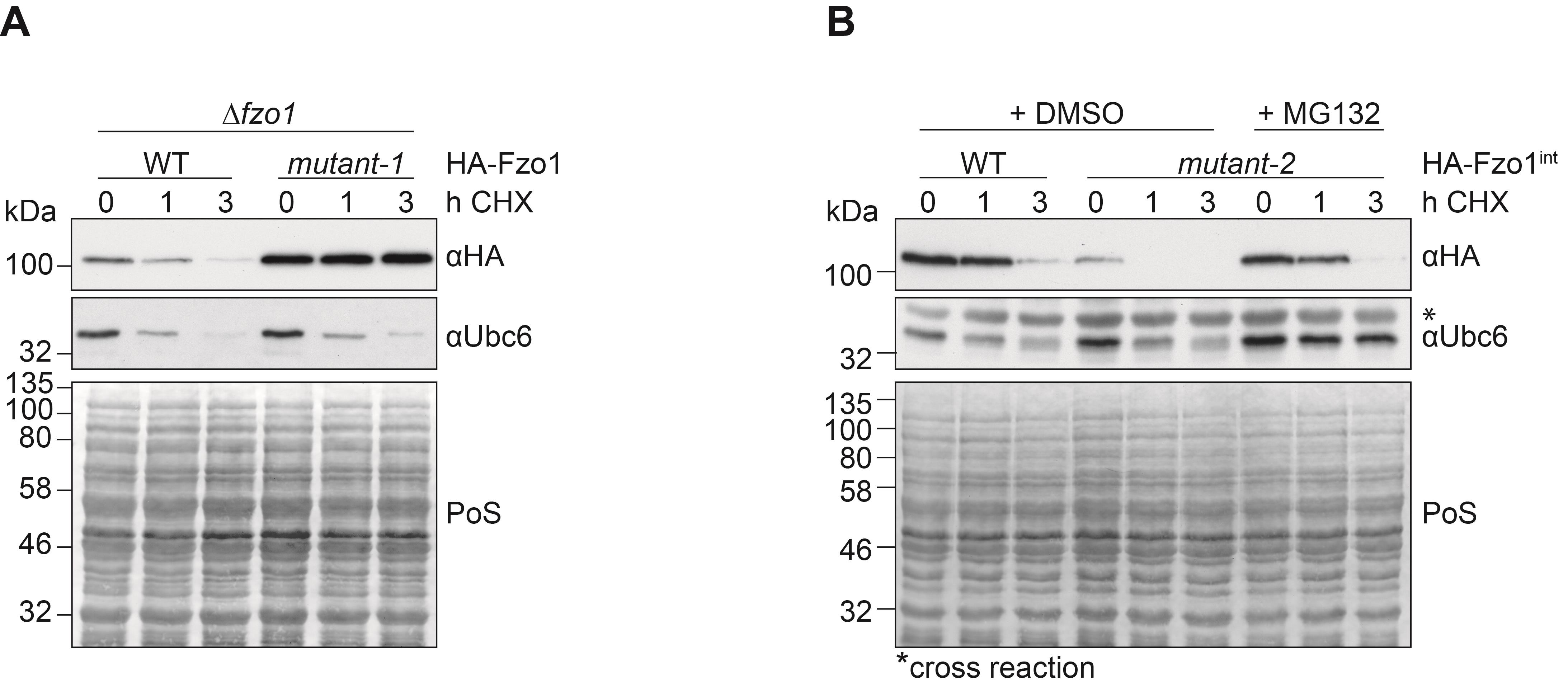
Figure 1. Stability of Fzo1 after translational shut down, with or without proteasomal inhibition. (A) Cells deleted for FZO1 and expressing HA-tagged Fzo1 were compared to cells expressing a mutant version of Fzo1 called mutant-1, which stabilizes the protein. (B) Cells lacking PDR5 and stably expressing genomically tagged versions of either HA-tagged wt Fzo1 or an unstable mutant version, called mutant-2, were analyzed. Cells were treated with 100 µM CHX for 1 or 3 h. When indicated, cells were incubated for 1 h with 0.05 mM MG132 prior to CHX treatment. (A+B) Protein levels were analyzed in total cellular extracts by SDS-PAGE and immunoblotting. Fzo1 was detected by using HA-specific antibodies. The unstable ER-protein Ubc6, observed in this analysis by using Ubc6-specific antibodies, was used as a control for proteasomal inhibition with MG132 and for translational inhibition with CHX (representative image). Ponceau S (PoS) staining was used as a loading control.
Recipes
TBS Buffer, pH 7.4
50 mM Tris
150 mM NaCl
Adjust pH to 7.4 using 37% HCl
HU sample buffer
8 M Urea
5% SDS
200 mM Tris pH 6.8
0.2 g/L Bromophenol blue sodium salt
100 mM DTT added freshly before use
Ponceau S solution
0.1% Ponceau S
5% Acetic acid
Tris/Glycine buffer
25 mM Tris
200mM Glycine
3.5mM SDS
Transfer buffer
200 mM Glycine
25 mM Tris
20% (v/v) Methanol
0.02% SDS
Adjust pH to 8.5 using 37% HCl
Minimal yeast media, which allows auxotrophic selection of yeast cells (Kaiser et al., 1994)
1.5 g Drop out mix (containing all amino acids except Adenine, Uracil, Leucine, Lysine, Histidine, and Tryptophane)
1.7 g Yeast Nitrogen Base
5 g Ammonium sulfate
Components listed above are dissolved in 1 l of distilled water, autoclaved and supplemented with 2% glucose and a CSM (complete supplement mixture) amino acid mix (containing Ade, Ura, Lys, His, Trp, Leu).
1 M DTT
1.54 g DTT in 10 ml of in sterile distilled water
Rödel mix
2 M NaOH
7.5% β-mercaptoethanol
Acknowledgments
Funding: Deutsche Forschungsgemeinschaft (DFG; CRC 1218 TP A03; EXC 2030 – 390661388; to ME-H); Center for Molecular Medicine Cologne (CMMC; CAP14, to ME-H and A02 to ME-H and M. Odenthal); Boehringer Ingelheim foundation (BIS; Exploration grant; to ME-H). This protocol was used in Simões et al. (2018; Doi: 10.7554/eLife.30015).
Competing interests
The authors declare they have no conflict of interest or competing interests.
References
- Anton, F., Dittmar, G., Langer, T. and Escobar-Henriques, M. (2013). Two deubiquitylases act on mitofusin and regulate mitochondrial fusion along independent pathways. Mol Cell 49(3): 487-498.
- Anton, F., Fres, J. M., Schauss, A., Pinson, B., Praefcke, G. J., Langer, T. and Escobar-Henriques, M. (2011). Ugo1 and Mdm30 act sequentially during Fzo1-mediated mitochondrial outer membrane fusion. J Cell Sci (7): 1126-1135.
- Anton, V., Buntenbroich, I., Schuster, R., Babatz, F., Simoes, T., Altin, S., Calabrese, G., Riemer, J., Schauss, A. and Escobar-Henriques, M. (2019). Plasticity in salt bridge allows fusion-competent ubiquitylation of mitofusins and Cdc48 recognition. Life Sci Alliance 2(6): e201900491.
- Buchanan, B. W., Lloyd, M. E., Engle, S. M. and Rubenstein, E. M. (2016). Cycloheximide Chase Analysis of Protein Degradation in Saccharomyces cerevisiae. J Vis Exp (110): 53975.
- Collins, G. A., Gomez, T. A., Deshaies, R. J. and Tansey, W. P. (2010). Combined chemical and genetic approach to inhibit proteolysis by the proteasome. Yeast 27(11): 965-974.
- Kaiser, C., Michaelis, S. and Mitchell, A. (1994). Methods in yeast genetics: a Cold Spring Harbor laboratory course manual. Plainview: Cold Spring Harbor Laboratory.
- Klinge, S., Voigts-Hoffmann, F., Leibundgut, M., Arpagaus, S. and Ban, N. (2011). Crystal structure of the eukaryotic 60S ribosomal subunit in complex with initiation factor. Science 334(6058): 941-948.
- Liu, C., Apodaca, J., Davis, L. E. and Rao, H. (2007). Proteasome inhibition in wild-type yeast Saccharomyces cerevisiae cells. Biotechniques 42(2): 158, 160, 162.
- Sambrook, J. and Russell, D. W. (2006). SDS-Polyacrylamide Gel Electrophoresis of Proteins. CSH Protoc 2006(4): pdb.prot4540.
- Schneider-Poetsch, T., Ju, J., Eyler, D. E., Dang, Y., Bhat, S., Merrick, W. C., Green, R., Shen, B. and Liu, J. O. (2010). Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol 6(3): 209-217.
- Schuster, R., Anton, V., Simoes, T., Altin, S., den Brave, F., Hermanns, T., Hospenthal, M., Komander, D., Dittmar, G., Dohmen, R. J. and Escobar-Henriques, M. (2020). Dual role of a GTPase conformational switch for membrane fusion by mitofusin ubiquitylation. Life Sci Alliance 3(1): e201900476.
- Schuster, R., Simões, T., den Brave, F. and Escobar-Henriques, M. (2018). Separation and Visualization of Low Abundant Ubiquitylated Forms. Bio-protocol 8(22): e3081.
- Simões, T., Schuster, R., den Brave, F. and Escobar-Henriques, M. (2018). Cdc48 regulates a deubiquitylase cascade critical for mitochondrial fusion. Elife 7: e30015.
Article Information
Copyright
![]() Buntenbroich et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Buntenbroich et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Buntenbroich, I., Simões, T. and Escobar-Henriques, M. (2021). Analysis of Protein Stability by Synthesis Shutoff. Bio-protocol 11(22): e4225. DOI: 10.21769/BioProtoc.4225.
- Simões, T., Schuster, R., den Brave, F. and Escobar-Henriques, M. (2018). Cdc48 regulates a deubiquitylase cascade critical for mitochondrial fusion. Elife 7: e30015.
Category
Microbiology > in vivo model > Fungi
Neuroscience > Cellular mechanisms > Mitochondria
Biochemistry > Protein > Degradation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.