- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Targeting the Expression of Long Noncoding RNAs in Murine Satellite Cells from Single Myofibers
Published: Vol 11, Iss 21, Nov 5, 2021 DOI: 10.21769/BioProtoc.4209 Views: 3804
Reviewed by: Gal HaimovichPhilipp WörsdörferAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2021
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






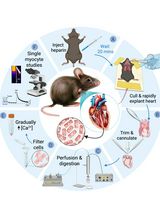
Abstract
LncRNAs have been recently implicated in the epigenetic control of muscle differentiation and their functional characterization has traditionally relied upon in vitro models of myogenic differentiation. However, the use of experimental paradigms to specifically target lncRNAs expression in muscle stem cells (MuSCs), also known as satellite cells, represents an important requisite to interrogate their function in more physiological contexts. Since isolation and culture of single myofibers preserves satellite cells within their physiological niche underneath the surrounding basal lamina, this procedure represents the optimal approach to follow satellite cell dynamics ex-vivo, such as activation from quiescence, expansion of committed progenitors, differentiation, and self-renewal. Here, we detail an optimized protocol to isolate viable single myofibers from the extensor digitorum longus (EDL) skeletal muscle of adult mice and to manipulate the expression of lncRNAs by antisense LNA GapmeRs-mediated knock-down (KD). Furthermore, we describe a method of EdU incorporation that, coupled to lncRNA KD and subsequent immunofluorescence analysis of proliferating, differentiating, and satellite cell-specific markers, permits the inference of lncRNAs function on muscle stem cells dynamics.
Graphic abstract:

Graphical representation of the single myofiber isolation method. Experimental workflow showing the main steps of the protocol procedure: EDL muscle harvesting from the mouse hindlimb; EDL digestion into single myofibers; transfection with antisense oligos and culture for 96h; immunofluorescence protocol and image outcome.
Background
Long noncoding RNAs (LncRNAs) represent a class of transcripts longer than 200 nucleotides, which are mostly devoid of protein-coding capacity (Rinn and Chang 2012; Fatica and Bozzoni, 2014). They are found both in the cytoplasm and in the nucleus, and modulate gene expression by influencing genome organization via epigenetic, transcriptional, and post-transcriptional mechanisms (Quan et al., 2015; Cai et al., 2016). As such, they are emerging as critical modulators of several biological processes, including stem-cell mediated regeneration and differentiation.
LncRNAs have been recently implicated in the epigenetic control of different steps of muscle differentiation (Lu et al., 2013; Ballarino et al., 2015 and 2018; Zhu et al., 2017; Desideri et al., 2020). However, examination of lncRNA function during myogenesis has traditionally relied on ablation studies performed on established myoblast cell lines (i.e., C2C12) or primary cells cultured in proliferating (high-serum) and differentiating (low-serum) conditions. These experimental settings allow discrimination of the role of lncRNAs only in the proliferation and differentiation steps, preventing their functional characterization on other crucial phases of in vivo myogenesis. Indeed, formation of new skeletal myofibers in vivo, or repair of damaged ones, relies on the activity of specialized adult muscle stem cells (MuSCs), located at the periphery of myofibers, between the sarcolemma and the basal lamina, that have been usually referred to as satellite cells (Mauro, 1961). In resting adult muscles, satellite cells are present in a post-mitotic quiescent state. However, upon injury, satellite cells activate and proliferate to expand a pool of committed myogenic progenitors that, upon expression of muscle-regulatory factors, enter the muscle differentiation program to form new muscle fibers (Figure 1). A small fraction of the expanded pool avoids terminal differentiation to self-renew and replenish the pool of quiescent stem cells (Biferali and Mozzetta, 2019).
Therefore, to infer the function of lncRNAs along the different stages of satellite cell-mediated myogenesis, it is important to rely on an experimental model that allows for monitoring of satellite cell activation from the quiescent state to their expansion and commitment to differentiation, and even to self-renewal (Mozzetta, 2016). To this end, we optimized previous protocols to isolate intact and viable single myofibers (Pasut et al., 2013; Mozzetta, 2016; Pegoli et al., 2020) from the extensor digitorum longus (EDL) muscle of wild-type mice for the purposes of subsequent manipulation of lncRNAs expression and phenotypical characterization by EdU incorporation and immunostaining (Cipriano et al., 2021).
This isolation procedure preserves satellite cells within their physiological niche underneath the surrounding basal lamina. Thus, once cultured ex-vivo in non-adherent conditions and in the presence of serum and growth factors, satellite cells associated with the myofibers start to proliferate, giving rise to a progeny visible as a round group of cells attached to the fiber (Figure 1). This cluster represents the progeny of a single satellite cell that, upon 72-96 h of culture, comprises either differentiating myoblasts or self-renewing cells. The protocol described here details how to interfere with lncRNAs expression by LNA GapmeRs-mediated knock-down and optimization of subsequent immunostaining assays for assessment of MuSCs-associated myofibers.

Figure 1. Satellite cells are present in a post-mitotic quiescent state (PAX7+; MYOD-; MYOG-; Ki67-; EdU-) on single myofibers. Upon injury or isolation, satellite cells become activated and start to express early muscle-regulatory factors (such as MYOD), undergoing the first replication within 48 h, in which one cell is a committed myogenic progenitor, while the other one can return to quiescence. The activated cell starts to proliferate, creating a cluster of round cells that then enter the muscle differentiation program by expressing Myogenin (MYOG) to form new muscle fibers.
Materials and Reagents
2 ml tube (Eppendorf, catalog number: 0030.120.094)
1.5 ml tube (Eppendorf, catalog number: 0030.120.086)
1,000 μl Tips (Axygen, catalog number: TF-1000-LRS)
200 μl Tips (Axygen, catalog number: TF-200-LRS)
50 ml tube (Corning, catalog number: 430829)
Syringe filter 0.22 μm (Millipore, catalog number: SLGS033SS)
50 ml syringe (Fisher BD, catalog number: 309653)
Petri dish, tissue culture treated (Falcon, catalog number:353003)
12 well plate, tissue culture treated (Falcon, catalog number: 353043)
60 mm plate, tissue culture treated (Falcon, catalog number: 353002)
Cover glasses (Thermo Scientific, Menzel-Glaser 12mm diameter #1)
Glass slides (Thermo Scientific, Menzel-Glaser AA00000102E01FST20)
C57BL/10 (WT) (JAX, catalog number: 000665)
Ice
Collagenase I (Sigma, catalog number: C0130)
Fibroblast growth factor (FGF) (Gibco, catalog number: PHG0026)
Chick embryo extract (CEE) (Life Science Production, catalog number: MD-004D-UK)
Anti-Pax7 mouse monoclonal (DSHB, catalog number: Pax7-s [1:10])
Anti-Ki67 rabbit polyclonal (Abcam, catalog number: ab15580 [1:100])
Alexa Fluor 488, rabbit (Invitrogen, catalog number: A21206 [1:250])
Alexa Fluor 594, mouse (Invitrogen, catalog number: A21203 [1:250])
Antisense oligos: LNA GapmeRs: non-targeting control (Qiagen, catalog number: 300610) or lncRNA sequence-specific custom GapmeRs
Lipofectamine 2000 (Invitrogen, catalog number: 11668019)
Optimem (Gibco, catalog number: 31985047)
Click-iT EdU Alexa Fluor 594 HCS Assay (Invitrogen, catalog number: C10354)
Ethanol (Sigma, catalog number: 32221)
Phosphate buffered saline (PBS) (Sigma, catalog number: D8537)
Triton-X 100 (Sigma, catalog number: T8787)
Glycerol (Applichem, catalog number: A2926)
Paraformaldehyde (PFA) (Sigma, catalog number: P6148)
Penicillin-Streptomycin (Sigma, catalog number: P0781)
4’,6-diamidino-2-phenylindole (DAPI) (Sigma, catalog number: 28718-90-3 [1:10,000])
Dulbecco’s Modified Eagle Medium (DMEM), high glucose, pyruvate (Gibco, catalog number: 41966-052)
Horse serum (Gibco, catalog number: 26050088)
Fetal Bovine Serum (FBS) (Corning, catalog number: 35-015-CV)
Digestion solution (see Recipes)
Wash solution (see Recipes)
Growth medium for fibers (see Recipes)
IF solutions (see Recipes)
Equipment
Micropipette (P200, P1000)
Surgical scissors (F.S.T, catalog number: 14060-10)
Jewelers’ forceps, Dumont No. 5, L 4 1/4 (Sigma, catalog number: F6521)
Microscope (Zeiss, model: Stemi DV4)
Axio Observer 3 inverted fluorescence microscope (ZEISS)
Shaker (Labnet, model: Rocker 25)
Vortex (Heidolph, model: Reax 2000)
Water Bath (Thermo Scientific, Precision Water Bath)
CO2 Incubator (Euroclone, model: S@fegrow 188)
Ice bucket
Software
Fiji image processing package (Open-source software (OSS) projects, https://imagej.net/Fiji)
ZEN 3.0 (Blue edition, ZEISS)
Procedure
Attention: Keep everything sterile if planning to culture the fibers.
Preparation
Fill an ice bucket with ice.
Prepare “digestion solution,” “wash solution,” and “growth medium for fibers” (see Recipes).
Pre-heat the “wash solution” and “growth medium for fibers” solution at 37°C.
Coat 2 ml tubes (one tube per sample) with FBS. Add 1 ml of FBS to the tube. Move it up and down until the sides of the tube are completely covered, and then discard the FBS.
Note: The tube needs to be coated to keep the myofibers in suspension; otherwise, they will stick to the tube’s wall. We use FBS for coating as this will also be in the growth medium.
Add 2 ml of the “digestion solution” to each coated tube. Keep it on ice.
Pre-heat the water bath at 37°C.
EDL isolation
Attention: This step must be performed as fast as possible before rigor mortis. For this reason, we suggest performing EDL isolation from one mouse at a time.
Note: Always handle the muscle from tendon to tendon and avoid touching the bulk of the muscle to preserve myofiber integrity.
Sacrifice the mouse using a method approved for your research, such as CO2 or cervical dislocation.
Wet the skin and the fur of the mouse with 70% ethanol.
Position the mouse under the hood. Pull up and cut the skin from the ankle to remove the skin and to expose the tibialis anterior (TA) and the extensor digitorum longus (EDL) (Video 1).
 Video 1. Procedure for EDL isolation. (This video was made at the Department of Biology and Biotechnologies of University Sapienza, according to guidelines approved by the Institutional Animal Care and Use Committee of the Department of Biology and Biotechnology of University Sapienza, the Italian Ministry of Health, and local authorities according to Italian law; Protocol N° 7FF2C.4–Authorization N° 746/2016-PR.)
Video 1. Procedure for EDL isolation. (This video was made at the Department of Biology and Biotechnologies of University Sapienza, according to guidelines approved by the Institutional Animal Care and Use Committee of the Department of Biology and Biotechnology of University Sapienza, the Italian Ministry of Health, and local authorities according to Italian law; Protocol N° 7FF2C.4–Authorization N° 746/2016-PR.)Carefully cut the tendon of the TA muscle from the ankle, grab the loose tendon with the tweezers, and gently pull up the muscle. Then cut the TA muscle under the knee to expose and have a good visualization of the EDL muscle (Video 1).
Cut the tendon of the EDL at the base, just over the ankle, and gently pull the muscle until it remains attached only by the tendon on the knee. By keeping the EDL from the ankle tendon, isolate the muscle cutting the tendon over the knee (Video 1). Place the muscle inside a coated tube with the “digestion solution” and leave the tube on ice.
Repeat Steps B4 and B5 with the other leg. Add the second muscle to the coated tube with the “digestion solution” and place it on ice.
Digestion
Incubate the tube from 45 min to 1 h in a pre-warmed bath at 37°C. Gently shake every 10 min.
Note: Invert the tubes a couple of times; do not shake vigorously to avoid breaking the myofibers.
Tip: It is possible to see the state of the digested muscle looking through the tube against a source of light. The muscle should be in a loose state.
At the end of the incubation, give a strong shake to release the myofibers from the digested muscle.
Tip: It is possible to see the released myofibers looking through the tube against light before and after the final shake.
Single myofiber isolation
Attention:
This step needs to be performed as fast as possible to avoid fiber contraction. When the myofibers are kept for too long on the “digestion plate,” they will start to shrink and contract, leading to myofiber damage.
To avoid contraction, pre-heat all the solutions to 37°C. The myofibers are sensitive to temperature and should be kept constantly at 37°C.
Tip: If it is possible, perform myofiber isolation from one mouse at a time. In any case, keep all the plates containing the fibers in the incubator while they are not being used.
Prepare two 60 mm plates per mouse with 4 ml of pre-heated “wash solution” for the cleaning step.
Transfer the digestion solution containing the fibers to one of the 60 mm plates with the “wash solution.”
Note: Myofibers should appear long, smooth, unbroken, and slightly translucent (Figure 2, red arrow).

Figure 2. Representative image of a newly isolated single myofiber (red arrow) in the washing plate. Black arrows indicate broken pieces that should be avoided when collecting the single myofiber.Coat the tip of a P200 micropipette by pipetting one time with the “wash solution.” Under the dissecting microscope (Zeiss; Stemi DV4), with the coated tip, take one by one every single myofiber in the direction of the fiber to avoid damage, collect multiple fibers in the tip, and gently release them into the second clean 60 mm plate containing 4 ml of pre-heated “wash solution” (cleaning step).
Tip: Slightly slope the plate (and consequently move the media) to better visualize the translucent myofibers.
Attention:
Avoid the broken or contracted pieces (Figure 2, black arrow) and take only the intact single myofibers.
Critical step! Be as gentle and as fast as possible. All the cleaning procedures over the hood should not take more than 10 min.
After picking all the visible intact myofibers, gently pipette the digested muscle to release the myofibers still attached to it. Under the microscope, it is possible to see the myofibers being released.
Tip: It is possible to perform this step using the tip of a P1000 micropipette cut with scissors or with a sterile glass Pasteur pipette previously broken at the neck.
Note: Be sure to coat the tip with the media before using it.
When more myofibers are released, pick out single myofibers with the coated tip of a P200 micropipette (as done in Step D3) and place them in the 60 mm plate used before in the cleaning step. In this case, also avoid the broken pieces, taking only the single myofibers.
When all the myofibers are in the cleaning plate, pick single myofibers in the direction of the fiber and plate them in the final dish (e.g., 24 well plate ~10-15 fiber/well or 12 well plate ~20-25 fiber/well) for culture with the pre-heated “Growth medium for fiber.”
Return the plates to the incubator at 37°C.
Attention: The myofibers are cultured in suspension, so the volume should not be too small to allow fibers to float in the medium (e.g., in a 12 well plate, it should not be less than 1 ml).
The fibers can be fixed and analyzed immediately or cultured for satellite cell amplification.
Culture and transfection
Attention: The myofibers are sensitive to changes in temperature.
Keep the myofiber in the incubator for at least 4 h before transfection. The first hour after plating is critical, so keep the myofibers in the incubator to stabilize them.
To perform the transfection, mix 1 μl/ml of lipofectamine with 49 μl of Optimem in one 1.5 ml tube per sample. In another 1.5 ml tube, add 1 μl/ml of antisense oligos (GapmeRs) from a stock at a concentration of 50 μM per sample and mix 49 μl of Optimem. After 5 min, mix the content of the lipofectamine tube with the content of the GapmeRs tube and wait for 20 min. Pipet 100 μl of the mix directly to the culture media in each well, drop by drop. Incubate overnight (13-14 h).
Notes:
The final volume should be 1 ml per well (for a 12 well plate).
We strongly recommended not performing more than one overnight (13-14 h) transfection to avoid myofiber disruption.
In the morning, change the media, paying attention to not remove fibers. To this end, gently remove as much medium as possible by tilting the plate on one edge and aspirating the medium from the opposite side. Always leave some media in the plate and add new pre-heated “growth media for fibers” on top of the plate, drop by drop. Let the myofibers sink for 10 min (preferably in the incubator), and then repeat the procedure once or twice more.
Note: Aspire the media from the surface very gently to avoid loss of the suspended myofibers.
Leave in culture until the desired time point. It is safe to keep them in culture for at least 96 h after plating (Figure 3).
Note: If left only for 96 h in culture, there is no need for media change to prevent fiber loss. For longer culture, we suggest changing the media every 2-3 days.

Figure 3. Experimental timeline used for the culture and transfection of single myofibers and EdU incorporation pulse.
EdU incorporation
Add 1 μl/ml of EdU directly to the media, as indicated in the manufacturer’s instructions.
The recommended pulse is 24 h (Figure 3).
Note: The pulse should start 24 h before the selected time point.
Myofiber fixation
Attention: The myofibers are in suspension, so, after each step, let the fibers settle to the bottom for 10 min and then gently remove the supernatant close to the surface.
Note: Myofiber fixation is performed in the well.
Gently remove 500 μl of media from each well, paying attention not to remove fibers.
Attention: The fibers should be left in ~500 μl.
Add 1 ml of PFA 4% directly in the media to obtain a final concentration of 3% PFA. Leave for 30 min at room temperature.
Remove as much PFA as possible and add 1 ml of PBS to wash the myofibers for 10 min. Repeat this washing step three times. Then leave the myofibers in at least 1 ml of PBS.
Note: It is possible to keep them at 4°C for a few weeks. To avoid PBS evaporation, carefully close the lid of the plate with parafilm and, if EdU incorporation was performed, keep the plate in the dark.
Edu detection and immunofluorescence
Note: For immunofluorescence analysis, transfer approximately 50 myofibers in 2 ml tubes per condition.
Coat a 2 ml tube/each sample with FBS (as described in Step A3).
Under the microscope, remove as much supernatant as possible, and then pick the single myofibers (in the direction of the myofiber) with a coated P200 micropipette tip and transfer them to the bottom of the 2 ml coated tube.
Attention: When pipetting the myofibers, always coat the micropipette tip with FBS to prevent myofiber loss or disruption (they tend to attach to the sides of the uncoated tube or tip).
Let the fibers settle down to the bottom of the tube for 10 min and then remove the supernatant. Leave the myofibers in no less than 150 μl of PBS.
Tip: It is a good trick to compare the volume with another 2 ml tube containing 150 μl of water.
Add 500 μl of Triton 0.5% to each tube and incubate for 10 min at room temperature.
Remove the supernatant and wash with 1 ml of PBS for 10 min.
For EdU detection, myofibers are stained using Click-iT EdU Alexa Fluor 594 HCS Assay (Invitrogen):
Dilute the 10× solutions (Click-iT EdU buffer additive Component E 10× and Click-iT reaction buffer Component C 10×) provided in the kit ten times to 1× by adding the proper volume of water (e.g., 50 μl of E 10× + 450 μl of water).
Then prepare the Click-iT reaction cocktail mix, mixing 425 μl of Component C solution 1×, 20 μl of CuSO, 1.25 μl of Alexa Fluor azide, and 50 μl of Component E solution 1× to obtain ~500 μl of the mix.
Note: Reagents should be added to the mix in this specified order.
Then add 200 μl of cocktail mix per sample of fibers to be stained. Incubate 30 min in the dark at room temperature, and then wash with 200 μl of reaction rinse buffer provided in the kit.
Notes:
Perform the EdU detection assay before standard immunofluorescence.
If you perform EdU detection, all the steps of the immunofluorescence should be performed in the dark.
Remove the supernatant and wash with 1 ml of PBS for 10 min.
Remove the supernatant and add 1 ml of block solution (10% FBS in PBS). Leave for 1 hour at room temperature, preferably on a shaker in slow agitation.
Let the tube settle for 10 min in a vertical position and then remove the supernatant, leaving the myofibers in ~150 μl. Add the primary antibodies diluted in 10% FBS in PBS (Ki67 at 1:100 and Pax7 at 1:10). The final volume should not be less than 300 μl. Incubate the tube overnight (13-14 h) at 4°C.
Tip: When preparing the mix with the antibodies, we recommend calculating the needed concentration of the antibody for the final volume but adding 150 μl less of FBS 10% in the mix (this volume is already present in the tube with the myofibers).
Remove the supernatant and wash twice with 1 ml of 0.1% FBS in PBS for 10 min.
Remove the supernatant, leaving the myofibers in ~150 μl. Add the secondary antibodies diluted in 10% FBS in PBS (Alexa Fluor 488 and 594, both at 1:250). The final volume should not be less than 300 μl. Incubate for 1 hour at room temperature in the dark.
Remove the supernatant and wash once with 1 ml of 0.1% FBS in PBS for 10 min.
Remove the supernatant and wash once with 1 ml of PBS for 10 min.
Add 500-800 μl of DAPI (1:10,000 in PBS) and incubate for 10 min at room temperature.
Remove the supernatant and perform two washes with 1 ml of 0.1% FBS in PBS.
Mount the fibers on the cover glass by removing all the supernatant and leaving the myofibers in as little volume as possible.
Add 5-10 μl of Glycerol:PBS. Cut a small piece of the tip of a P200 micropipette with scissors and coat it with FBS. Use it to gently take the myofibers from the bottom of the 2 ml tube and place them on the glass cover, drop by drop, spanning the center of the glass slide.
Note: This is a critical step, so be careful not to discard myofibers that might remain attached to the tip. Check the tip content before discarding it, by looking inside the tip against a source of light. If necessary, pipette more Glycerol:PBS.
Tips:
It is possible to see the myofibers on the glass slide at this stage.
Check the bottom of the 2 ml tube, looking through it against a light source to ensure that there are no myofibers left.
Delicately close the cover glass with a rectangular coverslip.
Let the mounted fibers dry in the dark and then seal the cover glass on the sides with nail polish.
Image with a fluorescence microscope (Figure 4).
Note: It is suggested to use a 40× or 63× objective to visualize the satellite cell clusters and 10× or 20× objective for the entire myofiber.
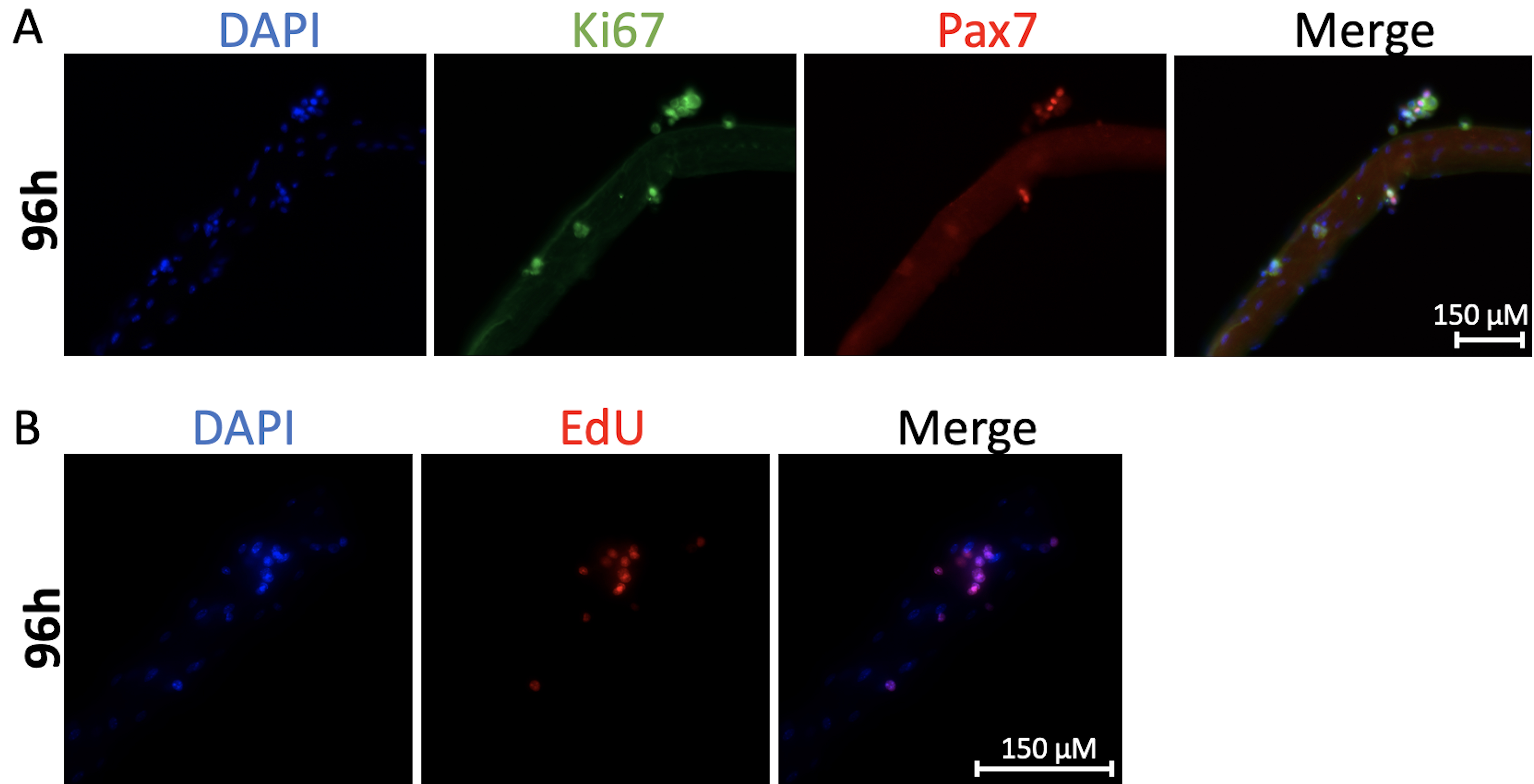
Figure 4. Example of immunofluorescence images obtained with our protocol. A. Representative images of immunofluorescence performed on a single myofiber kept in culture for 96 h. Ki67, proliferation marker (green) and Pax7, satellite cells specific marker (red). B. Representative images of EdU detection (red) in a single myofiber kept in culture for 96 h and with a 24 h EdU pulse. DAPI (blue) was used to visualize the nuclei.
Data analysis
The data obtained with the single myofiber immunofluorescence and EdU detection can be used to perform different analyses, depending on the biological question that is being addressed.
For example, we wanted to understand the role of LncRewind in the context of the activation and proliferation of satellite cells (Cipriano et al., 2021), so we performed co-staining with Pax7, a specific marker for satellite cells, and Ki67, a marker for proliferation, on the single myofibers isolated from five mice. To obtain statistically significant results, we suggest quantifying at least 50 fibers per condition, isolated from a minimum of three mice. Exclude short and broken myofibers from the analysis.
For the statistical analysis, apply unpaired Student’s t-tests when comparing two conditions and one-way Anova with Tukey’s multiple comparison test when comparing more than two conditions.
We used the data to execute different analyses:
We performed a manual count under the microscope of the number of Pax7+ cells and Pax7+Ki67+ double positive cells per cluster, comparing the results among the different conditions (Control vs. KD). For the statistical analysis, we compared 40 clusters per condition.
We also counted the mean number of clusters [composed by a number of nuclei (n) > 2], pairs (n = 2), or single Pax7+ (n = 1) cells per fiber. For the statistical analysis, we compared 80 fibers per condition.
We also compared the total number of Pax7+ cells per fiber between control and KD. In this case, we used the data from 50 fibers per condition for the statistical analysis.
Notes
There is great variability among different mice, even with the same sex and age.
The mean number of myofibers that we collect per experiment is approximately 100 per mouse, from two EDL muscles, but approximately 20% do not survive during culture.
We also noticed that performing this procedure with more than three mice at a time can be detrimental, as the cleaning step after isolation should be performed quickly to avoid fiber contraction.
We also performed myofiber isolation from gastrocnemius muscle, dividing it into four longitudinal pieces. However, in our hands, the results are cleaner and the myofibers are stronger just with the EDL muscle. We found that the EDL muscle was ideal to use in this procedure because its small size allows the digestion of the entire muscle without the need for manual trituration that could cause myofiber damage.
Recipes
Digestion solution (2 ml per mouse)
Collagenase I 10 mg
DMEM high-glucose + Pyruvate (Gibco) 2 ml
Pen/Strep 100× 20 μl
Attention:
The quantity and the state of the Collagenase I is critical for the success of this procedure
Always keep the collagenase I and the solution on ice. When using mice with fluorescent reporter genes, we recommend keeping this solution in the dark.
Measure the quantity of Collagenase I very carefully.
Place it in a 50 ml tube. In a sterile environment, add the media and the antibiotic and vortex carefully until the Collagenase I is completely dissolved.
Filter the solution through a 0.22 μm filter with a 50 ml syringe into a new 50 ml tube.
Tip: It is possible to lose some of the volume in the filtering step, so it is better to start with a larger volume.
Wash solution (50 ml)
DMEM high-glucose + Pyruvate (Gibco) 45 ml
Horse serum 10% 5 ml
Mix the ingredients
Before use, pre-heat the solution at 37°C
Note: Ensure that the solutions are sterile.
Tip: If the fibers are to be transfected, we recommend not adding any antibiotics in this solution, as they will interfere with the transfection efficiency.
Growth medium for fibers
DMEM high-glucose + Pyruvate (Gibco), 39.5 ml
FBS 20%, 10 ml
FGF 2.5 ng/ml
Chick embryo extract (CEE) 1%, 500 μl
Mix the ingredients and filter them through a 0.22 μm filter with a 50 ml syringe into a new 50 ml tube.
Before use, pre-heat the solution at 37°C.
Note: Ensure that the solutions are sterile.
Tip: If the fibers are to be transfected, we recommend not adding any antibiotics in this solution, as they will interfere with the transfection efficiency.
IF solutions
PFA 4% in PBSDissolve 4 g paraformaldehyde powder in 100 ml PBS.
Heat the solution to 65°C until the paraformaldehyde is completely dissolved.
Store 10 ml aliquots at -20°C.
Triton X-100 0.5% in PBS
Mix 50 μl of Triton with 9.95 ml of PBS
FBS 10% in PBS
Mix 5 ml of FBS with 45 ml of PBS
FBS 0.1% in PBS
Mix 50 μl of FBS with 50 ml of PBS
Glycerol:PBS 3:1
Mix 3 ml of Glycerol with 1 ml of PBS
Acknowledgments
This work was supported by the Italian Ministry of University and Research (SIR, Scientific Independence of Young Researcher n. RBSI14QMG0), Italian Association for Cancer Research (AIRC; MyFIRST grant n.18993), AFM-Telethon, and the CNCCS (Collection of National Chemical Compounds and Screening Center). Sapienza University (prot. RM120172B7D32C04 and RM11916B7A39DCE5) and POR FESR Lazio 2020-T0002E0001 to MB. Illustrations have been created under a paid subscription with biorender.com.
This protocol is adapted from Cipriano et al. (2021; DOI: 10.7554/eLife.54782).
Competing interests
The authors declare that no competing interests exist.
Ethics
Animal experimentation: For the experiments described in this study, C57BL/10 wild-type mice were used, and differences that were observed in both male and female mice were included in experiments. With respect to housing, nutrition, and care, animals were treated according to the guidelines of good laboratory practice (GLP). All experimental protocols were approved and conformed to the regulatory standards (Protocol N° 7FF2C.4–Authorization N° 746/2016-PR, Cipriano et al. eLife 2021; 10: e54782. DOI: https://doi.org/10.7554/eLife.54782 20 of 25 Research article Cell Biology). All animals were kept in animal cages with at least five animals, at a temperature of 22°C ± 3°C, and with humidity between 50% and 60%.
References
- Ballarino, M., Cazzella, V., D'Andrea, D., Grassi, L., Bisceglie, L., Cipriano, A., Santini, T., Pinnaro, C., Morlando, M., Tramontano, A. et al. (2015). Novel long noncoding RNAs (lncRNAs) in myogenesis: a miR-31 overlapping lncRNA transcript controls myoblast differentiation. Mol Cell Biol 35(4): 728-736.
- Ballarino, M., Cipriano, A., Tita, R., Santini, T., Desideri, F., Morlando, M., Colantoni, A., Carrieri, C., Nicoletti, C., Musaro, A., et al. (2018). Deficiency in the nuclear long noncoding RNA Charme causes myogenic defects and heart remodeling in mice. EMBO J 37(18): e99697.
- Biferali, B. and Mozzetta, C. (2019). Epigenetic Regulation of Muscle Stem Cells During Skeletal Muscle Regeneration and Disease. (Chapter 13) In: Epigenetics and Regeneration. Elsevier:309-332.
- Cai, L., Chang, H., Fang, Y. and Li, G. (2016). A Comprehensive Characterization of the Function of LincRNAs in Transcriptional Regulation Through Long-Range Chromatin Interactions. Sci Rep 6: 36572.
- Cipriano, A., Macino, M., Buonaiuto, G., Santini, T., Biferali, B., Peruzzi, G., Colantoni, A., Mozzetta, C., and Ballarino, M. (2021). Epigenetic regulation of Wnt7b expression by the cis-acting long noncoding RNA Lnc-Rewind in muscle stem cells. Elife 10: e54782.
- Desideri, F., Cipriano, A., Petrezselyova, S., Buonaiuto, G., Santini, T., Kasparek, P., Prochazka, J., Janson, G., Paiardini, A., Calicchio, A., et al. (2020). Intronic Determinants Coordinate Charme lncRNA Nuclear Activity through the Interaction with MATR3 and PTBP1. Cell Rep 33(12): 108548.
- Fatica, A. and Bozzoni, I. (2014). Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet 15(1): 7-21.
- Lu, L., Sun, K., Chen, X., Zhao, Y., Wang, L., Zhou, L., Sun, H. and Wang, H. (2013). Genome-wide survey by ChIP-seq reveals YY1 regulation of lincRNAs in skeletal myogenesis. Embo J 32(19): 2575-2588.
- Mauro, A. (1961). Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol 9: 493-495.
- Mozzetta, C. (2016). Isolation and Culture of Muscle Stem Cells. Methods Mol Biol 1480: 311-322.
- Pasut, A., Jones, A. E. and Rudnicki, M. A. (2013). Isolation and culture of individual myofibers and their satellite cells from adult skeletal muscle. J Vis Exp (73): e50074.
- Pegoli, G., Lucini, F., Mozzetta, C. and Lanzuolo, C. (2020). Single Myofiber Isolation and Culture from a Murine Model of Emery-Dreifuss Muscular Dystrophy in Early Post-Natal Development. J Vis Exp (161). doi: 10.3791/61516.
- Quan, M., Chen, J. and Zhang, D. (2015). Exploring the secrets of long noncoding RNAs. Int J Mol Sci 16(3): 5467-5496.
- Rinn, J. L. and Chang, H. Y. (2012). Genome regulation by long noncoding RNAs. Annu Rev Biochem 81: 145-166.
- Zhu, M., Liu, J., Xiao, J., Yang, L., Cai, M., Shen, H., Chen, X., Ma, Y., Hu, S., Wang, Z., et al. (2017). Lnc-mg is a long non-coding RNA that promotes myogenesis. Nat Commun 8: 14718.
Article Information
Copyright
![]() Macino et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Macino et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Macino, M., Biferali, B., Cipriano, A., Ballarino, M. and Mozzetta, C. (2021). Targeting the Expression of Long Noncoding RNAs in Murine Satellite Cells from Single Myofibers. Bio-protocol 11(21): e4209. DOI: 10.21769/BioProtoc.4209.
- Cipriano, A., Macino, M., Buonaiuto, G., Santini, T., Biferali, B., Peruzzi, G., Colantoni, A., Mozzetta, C., and Ballarino, M. (2021). Epigenetic regulation of Wnt7b expression by the cis-acting long noncoding RNA Lnc-Rewind in muscle stem cells. Elife 10: e54782.
Category
Developmental Biology > Cell growth and fate > Myofiber
Molecular Biology > RNA > Transfection
Cell Biology > Cell isolation and culture > Cell isolation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.