- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Urea Denaturation, Zinc Binding, and DNA Binding Assays of Mutant p53 DNA-binding Domains and Full-length Proteins
Published: Vol 11, Iss 20, Oct 20, 2021 DOI: 10.21769/BioProtoc.4188 Views: 4080
Reviewed by: Gal HaimovichAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
In the cell, the thermodynamic stability of a protein – and hence its biological activity – can change dramatically as a result of perturbations in its amino acid sequence and the concentration of stabilizing ligands. This interplay is particularly evident in zinc-binding transcription factors such as the p53 tumor suppressor, whose DNA-binding activity can critically depend on levels of intracellular zinc as well as point mutations that alter either metal binding or folding stability. Separate protocols exist for determining a protein’s metal affinity and its folding free energy. These properties, however, are intimately connected, and a technique is needed to integrate these measurements. Our protocols employ common non-fluorescent and fluorescent zinc chelators to control and report on free Zn2+ concentration, respectively, combined with biophysical assays of full-length human p53 and its DNA-binding domain. Fitting the data to equations that contain stability and metal-binding terms results in a more complete picture of how metal-dependent proteins can lose and gain DNA-binding function in a range of physiological conditions.
Graphic abstract:
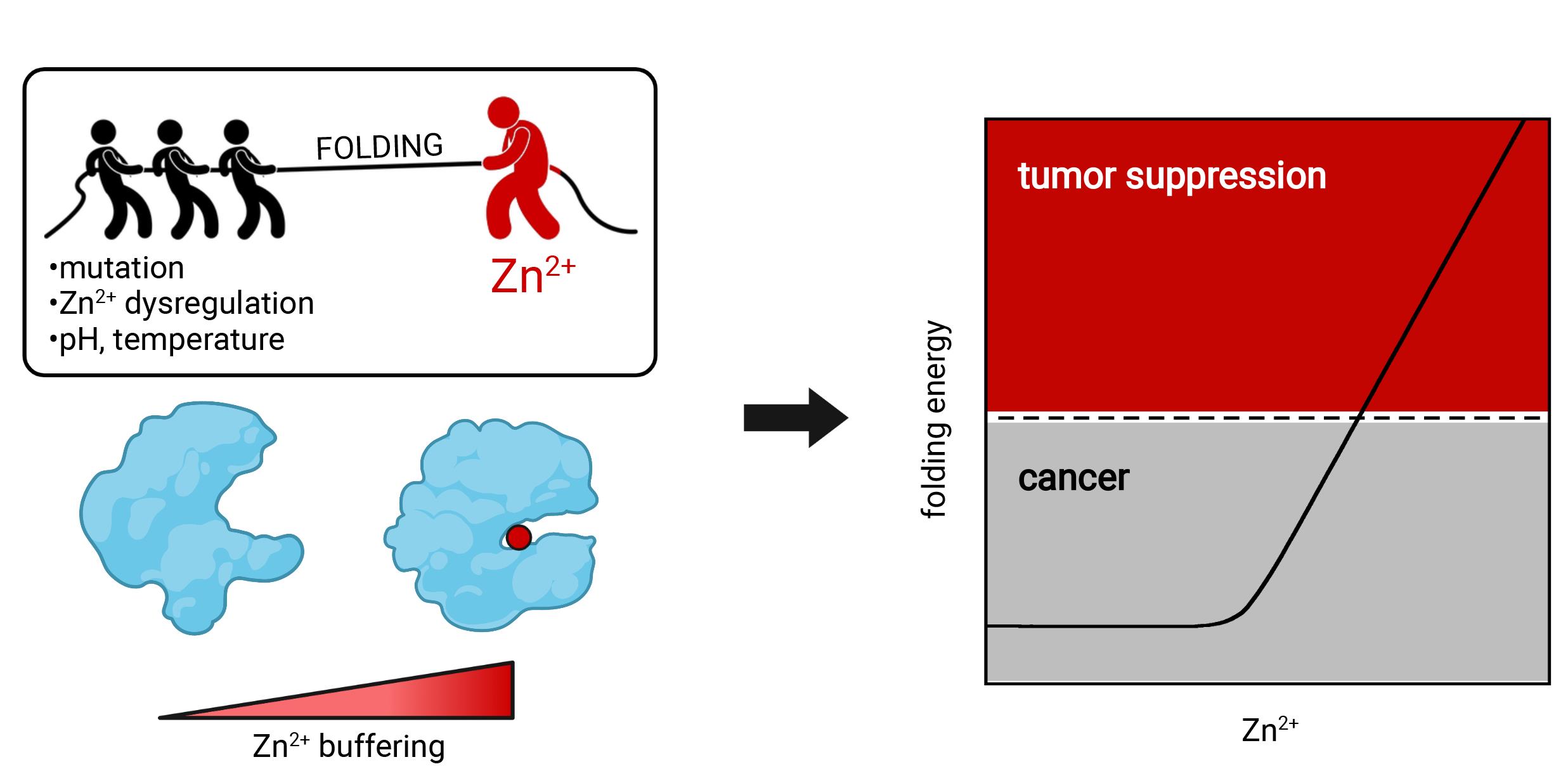
Figure 1. Raising intracellular zinc can restore tumor-suppressing function to p53 that has been unfolded by missense mutation or cellular conditions
Background
Zinc-binding proteins comprise about ten percent of the human proteome, with many additional proteins recognizing other d-block transition metals such as iron, copper, and manganese (Andreini et al., 2006; Barber-Zucker et al., 2017). Transition metals serve two general functions: to stabilize protein structure and to provide active centers for enzyme catalysis and electron transfer reactions. Transcription factors are well-represented members of the zinc-binding class of proteins that employ the metal for conformational stabilization. Whether or not a transcription factor is bound to zinc in the cell depends on two obvious factors and a third that is less well established. The first two are the dissociation constant of Zn2+ and the folded protein (KZn), and the concentration of available Zn2+ in the cell ([Zn2+]free). KZn and [Zn2+]free can be affected by pathological conditions such as mutation of zinc-coordinating residues and dysregulation of zinc homeostasis pathways, respectively. The third factor is the free energy of protein folding (ΔGfold), which begins to dominate as a protein becomes unstable and ΔGfold goes from a negative to a positive value (Figure 1). Because the high-affinity zinc-binding pocket typically exists only in the folded state, conditions that populate the unfolded state (such as elevated temperature or the presence of a missense mutation) sap metal-binding energy and can cause Zn2+ to dissociate.
p53 is a zinc-dependent transcription factor and tumor suppressor that is the most frequently mutated protein in human cancer. Most tumorigenic mutations are of the missense type and map to p53’s central DNA-binding domain (DBD) (Baugh et al., 2018). Mutations at nearly every codon position of DBD are represented in the p53 cancer patient database (Bouaoun et al., 2016). Our work (Blanden et al., 2020) and that of others (Bullock et al., 2000) have established that most mutations cause p53 to lose DNA-binding activity by at least one of the following three mechanisms: increasing KZn (zinc-binding class), increasing ΔGfold (stability class), or increasing the dissociation constant of folded DBD for its cognate DNA sequences (KDNA; DNA-contact class). Here, we provide protocols and equations for determining KZn, ΔGfold, and KDNA, as well as for quantifying the linkage between KZn and ΔGfold. Full-length p53 (FL-p53) expresses poorly in Escherichia coli, is unstable, and is prone to zinc loss and aggregation. We therefore include protocols for its purification, de-metallation, and re-metallation. The procedures herein should be applicable to other metal- and DNA-binding proteins.
Materials and Reagents
Black polystyrene 96-well plates, round bottom, non-treated (Corning Costar, catalog number: 3792)
Quartz cell for fluorimetry experiments, 5 mm × 5 mm (Starna Cells, Inc., catalog number: 3-5.45-Q-5)
Semi-micro polystyrene cuvettes (Laboratory Product Sales, catalog number: L321371)
PYREX Fernbach-style culture flask with baffles (Corning, catalog number: 4423-2XL)
HiTrap heparin HP affinity column (Cytiva, catalog number: 17040703)
HisTrap HP column (Cytiva, catalog number: 17524701)
HiLoad 16/600 Superdex 200 pg size exclusion column (Cytiva, catalog number: 28989335)
Superdex S200 Increase 10/300 GL (Cytiva, catalog number: 28990944)
Econo-Pac 10DG desalting prepacked gravity flow columns (Bio-Rad, catalog number: 7322010)
Dialysis tubing, 8,000 MWCO (BioDesign Inc., catalog number: D106)
Plasmids for bacterial expression of RBP-p53 and DBD are pending submission to Addgene. Contact the corresponding author for plasmid requests.
BL21(DE3) competent cells (New England Biolabs, catalog number: C2527)
5’-Cy3 labeled oligonucleotides with p53 binding elements sequence and its complementary unlabeled DNA (custom order from Eurofins Genomics). Nucleotide sequences are in Supplementary File 1A of Blanden et al. (2020).
Acetic Acid (Fisher Scientific, catalog number: A38C-212)
β-mercaptoethanol (βME) (Millipore Sigma, Calbiochem, catalog number: 444203)
Coomassie Brilliant Blue G-250 (GoldBio catalog number: C-460-50)
Ethylenediamine tetraacetic acid (EDTA) (MilliporeSigma, Aldrich, catalog number: ED2SS)
Ethyleneglycol-bis(-aminoethylether)-N,N,N’,N’-tetraacetic acid (EGTA) (Honeywell Chemicals, Fluka, Sigma-Aldrich, catalog number: 03779)
FluoZinTM-3, tetrapotassium salt (Thermo Fisher Scientific, catalog number: F24194)
Isopropyl-D-1-thiogalactopyranoside (IPTG) (GoldBio, catalog number: I12481C, store at -20°C)
Imidazole (Fisher Scientific, Acros Organics, catalog number: 301870010; Thermo Scientific, Alfa AesarTM, catalog number: A10221)
Guanidine hydrochloride (MP Biochemicals, catalog number: 105696)
N-(2-hydroxyethyl)ethylenediamine-N,N’,N’-triacetic acid, trisodium salt (HEDTA) (Sigma-Aldrich, Honeywell Chemicals, Fluka, catalog number: 34461)
HEPES (Millipore Calbiochem EMD OmniPure, catalog number: 5310)
Kanamycin monosulfate (VWR, Amresco Life Science, catalog number: 0408-100G)
Luria broth, Miller (BD, DifcoTM, catalog number: 244610)
Lysozyme, hen egg white (GoldBio, catalog number: L-040, store at -20°C)
4-(2-pyridylazo)resorcinol monosodium salt hydrate (PAR) (MilliporeSigma, Aldrich, catalog number: 178268)
Potassium acetate (VWR, Avantor, J.T. Baker, catalog number: 2912)
PreScission protease (Cytiva, catalog number: 27084301, store at -20°C)
SDS-PAGE Mini-Protean TGX precast protein gels (Bio-Rad, catalog number: 4561033)
Sodium chloride (Fisher BioReagents, catalog number: BP358)
Sodium dodecyl sulfate (SDS) (Fisher Scientific, catalog number: BP166)
Tris(hydroxymethyl) aminomethane, Tris base (Fisher BioReagents, catalog number: BP152)
Tris-(2-carboxyethyl)phosphine hydrochloride (TCEP) (GoldBio, catalog number: TCEP10, store at -20°C)
TweenTM 20 (Fisher BioReagents, catalog number: BP337)
Urea (Milliporesigma, Calbiochem EMD OmniPure, catalog number: 9510)
Zinc reference standard in 5% HNO3 (Fisher Scientific, SPEX CertiPrep, catalog number: PLZN2-3Y)
Buffers 1-6 (prepared from Tris base, imidazole, sodium chloride, β-mercaptoethanol, tris-(2-carboxyethyl)phosphine hydrochloride, and guanidine hydrochloride; see Recipes)
Equipment
LM10 Microfluidizer (Microfluidics)
J6-MI Centrifuge with JS-4.2 swinging bucket rotor (Beckman Coulter, catalog numbers: 360293 [centrifuge], 339080 [rotor])
Sorvall Legend XTR centrifuge with Piramoon Technologies FIBERlite F15-8 x 50C fixed angle rotor (Thermo Scientific, catalog numbers: 75217421 [centrifuge], 75003663 [rotor])
MaxQ refrigerated console shaker (Thermo Fisher Scientific catalog number: SHKE480HP)
BioLogic DuoFlowTM Medium-Pressure QuadTec Chromatography 10 System (Bio-Rad, catalog number: 7604137)
FluoroMax-4 Spectrofluorimeter (Horiba Scientific)
Microlab 540B dispenser (replaced by Microlab 600 dispenser) (Hamilton, catalog number: ML615-DIS)
SpectraMax i3x Multi-mode Microplate Reader (Molecular Devices)
Rhodamine Fluorescence Polarization Cartridge (Molecular Devices, catalog number: 0200-7009POS)
Fluorescein Fluorescence Polarization Cartridge (Molecular Devices, catalog number: 0200-7010POS)
EvolutionTM 201 UV-Visible Spectrophotometer (Thermo Fisher Scientific, catalog number: 840-210800)
Software
Any scientific graphic software capable of nonlinear least-squares curve fitting can be used, including recent versions of the following programs:
KaleidaGraph (Synergy Software; https://www.synergy.com/)
SigmaPlot (Systat Software; https://systatsoftware.com/)
GraphPad Prism (GraphPad Software; https://www.graphpad.com/)
Igor Pro (WaveMetrics; https://www.wavemetrics.com/)
Winmaxc32.exe (requires Microsoft Windows PC or emulation thereof). Winmaxc32 is part of the MaxChelator suite of downloads and programs available at somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator
Procedure
General tips for purifying, storing, and handling FL-p53 and DBD
FL-p53 aggregates at room temperature and concentrations >10 μM, as do some mutants of DBD. Anything that p53 contacts must be <10°C (preferably 4°C) until the experiment begins. All buffers and columns must be pre-chilled.
Experimental steps (preparing protein solutions, performing dilutions, etc.) should be carried out on ice or in a cold room. Protein incubation steps should take place in cold rooms unless the experimental goals dictate equilibration at higher temperatures. Fluorimeters, plate readers, and other instruments should be set to 10°C whenever possible.
Reducing agent (βME or TCEP) must always be present. Do not use DTT as it binds weakly to metals.
Once purification is started, we recommend that all steps should be completed within ~24 h (DBD) or ~48 h (FL-p53) and that the purified proteins be immediately flash frozen in aliquots and stored at -70°C. This is to minimize sample degradation, aggregation, and loss of activity that may occur over time.
In addition to Tris buffer, HEPES, MOPS, and Bis-tris propane are all acceptable for experiments (pH 7.0-7.5). Do not use phosphate buffer as it can form insoluble complexes with Zn2+.
Freeze p53 as rapidly as possible (see Protocol A).
Thaw p53 as rapidly as possible, without allowing temperature to rise above 10°C. Frozen tubes are thawed by immersing in room temperature water with gentle inversion until ice is mostly gone, then transferred to an ice bucket.
Prepare ZnCl2 stock solutions by diluting the zinc reference standard solution into 5 mM HCl to a final concentration of 200 mM ZnCl2. Measure the exact Zn2+ concentration using 4-(2-pyridylazo)resorcinol (PAR) as described (Hunt et al., 1985). Briefly, dissolve 9.48 mg of PAR in 20 ml of 20 mM Tris pH 7.0 (final concentration 0.2 mM PAR). Aliquot 1 ml of PAR to two tubes. Prepare a 100-fold dilution of the stock ZnCl2 solution into 5 mM HCl. Add 5 μl of HCl to the first PAR tube and 5 μl of the 1:100 ZnCl2 solution to the second PAR tube. Blank the spectrophotometer (500 nm) using the first PAR tube and read the A500 of the second PAR tube. Calculate [Zn2+] of the stock solution by dividing A500 by the PAR·Zn2+ extinction coefficient (66,000 M-1 cm-1) and multiplying by the dilution factor (20,000).
Full-length p53 purification
Note: Protocol A is specific for the construct in which FL-p53 is inserted into a cleavable expression tag (His-tagged, circular permutant of Thermoanaerobacter tengcongensis ribose binding protein [RBP]). The gene is identical to that described previously (Ha et al., 2021), except that the FL-p53 gene is inserted into RBP at the location indicated in that paper. This expression system greatly increases the yield of soluble, active p53. Refer to Table 1 for properties of p53 constructs.
Table 1. Properties of p53 constructs
Construct # Amino acids MW (g/mol) ε280 (M-1 cm-1) FL-p53-RBP (fusion protein) 734 78,760 38,120 FL-p53 (cleaved) 410 45,123 34,280 DBD 220 24,682 15,930 Transform plasmid containing the above gene into competent E. coli BL21(DE3) cells (any DE3 strain is acceptable) and spread on Luria broth (LB) plates containing 30 µg/ml kanamycin. Incubate at 37°C until small colonies are seen (8-12 h). Cells should be freshly transformed for each growth.
Pick ~5 colonies and inoculate into 0.75 L LB in a 2.8 L baffled Fernbach flask containing 30 µg/ml kanamycin. Shake at 250 RPM and 30°C. We find that p53 yields are greatest when cultures are not allowed to exceed OD600 ~0.2 until induction with IPTG. We therefore advise against inoculating a small overnight starter culture, as this will typically overgrow. We inoculate the 0.75 L culture with multiple colonies instead of one because this reduces time to IPTG induction. p53 is not toxic to E. coli, it does not appear to be undue pressure for mutation or plasmid loss, and we do not observe reduced p53 yield when inoculating with more than one colony.
When OD600 reaches 0.5-0.8 (6-8 h), transfer flask to an 18°C shaking incubator and induce with 50 mg/l isopropyl β-d-1-thiogalactopyranoside (IPTG). Continue shaking at 18°C for 12-16 h.
Harvest cells by centrifugation (4,500 × g, Beckman J6, 30 min, 4°C).
Discard supernatant and freeze cell pellets on dry ice. These can be stored for several months at -80°C or thawed for immediate purification.
Add Buffer 1 (see Recipe section for instructions to prepare all Buffers) to frozen cell pellets (40 ml buffer per 750 ml of original culture). Thaw cell pellets by swirling in a room temperature water bath until the ice is nearly gone, then transfer to ice bucket.
Lyse cells by adding 10 mg hen lysozyme per 750 ml of original starting culture, incubating 15-30 min on ice, then twice passing the cells through a microfluidizer (Microfluidics LM10) using the manufacturer’s instructions. Ensure that washing buffers are pre-chilled and microfluidizer lysis components are in an ice bath.
Centrifuge lysate (16,800 × g, Sorvall Legend XTR, 30 min, 4°C).
Load supernatant (2 ml/min, Bio-Rad DuoFlow medium pressure chromatography system) onto a nickel-NTA column (5 ml HisTrap HP, Cytiva) that was equilibrated in Buffer 1. Wash with Buffer 1 until A280 of the flow through is below 0.1 (typically 20-40 column volumes, or 100-200 ml).
Elute with at least 20 column volumes Buffer 2 (≥ 100 ml), collecting 5 ml fractions. Pool fractions for which A280 > 0.2.
To cleave FL-p53 from RBP, add ≥ 60 units of PreScission protease per 750 ml of original culture to the pooled fractions and transfer to a dialysis bag (8,000 molecular weight cutoff). Dialyze against 4 L of pre-chilled Buffer 3 for 4-6 h at 4°C.
Remove RBP by loading the dialyzed solution (2 ml/min, Bio-Rad DuoFlow chromatography system) onto a 5 ml HiTrap heparin HP column (Cytiva), previously equilibrated in Buffer 3. Wash with Buffer 3 until A280 of the flow through is below 0.02 (typically 10 column volumes, or 50 ml).
Elute FL-p53 with a linear gradient of 100% Buffer 3 to 100% Buffer 4, using the gradient mixing feature of the BioLogic DuoFlow. The gradient takes place over 100 ml, and the flow rate is 2 ml/min. FL-p53 should elute as the only peak, starting approximately one-third the way into the gradient and eluting in ~20 ml.
Perform a final clean-up step by injecting the above eluent onto a HiLoad Superdex S200 pg 16/600 size exclusion column (Cytiva). Conditions are 1 ml/min Buffer 5, Bio-Rad DuoFlow chromatography system.
Following these steps, FL-p53 is >90 % pure as judged by SDS-PAGE with Coomassie brilliant blue staining (Figure 2A). It elutes from an analytical Superdex S200 Increase column (Cytiva) with an apparent molecular weight of 226 kDa, consistent with that of the native tetramer, and a small population (8 %) of higher molecular weight aggregates that elute in the void volume (Figure 2B). Less than 0.1 % of the RBP expression tag co-purifies with FL-p53.
FL-p53 is stable for up to a week at 4°C. For long-term storage, flash freeze aliquots by pre-chilling 1.5 ml centrifuge tubes on dry ice or liquid N2, then adding 0.5 ml of protein solution. Store at -70°C.
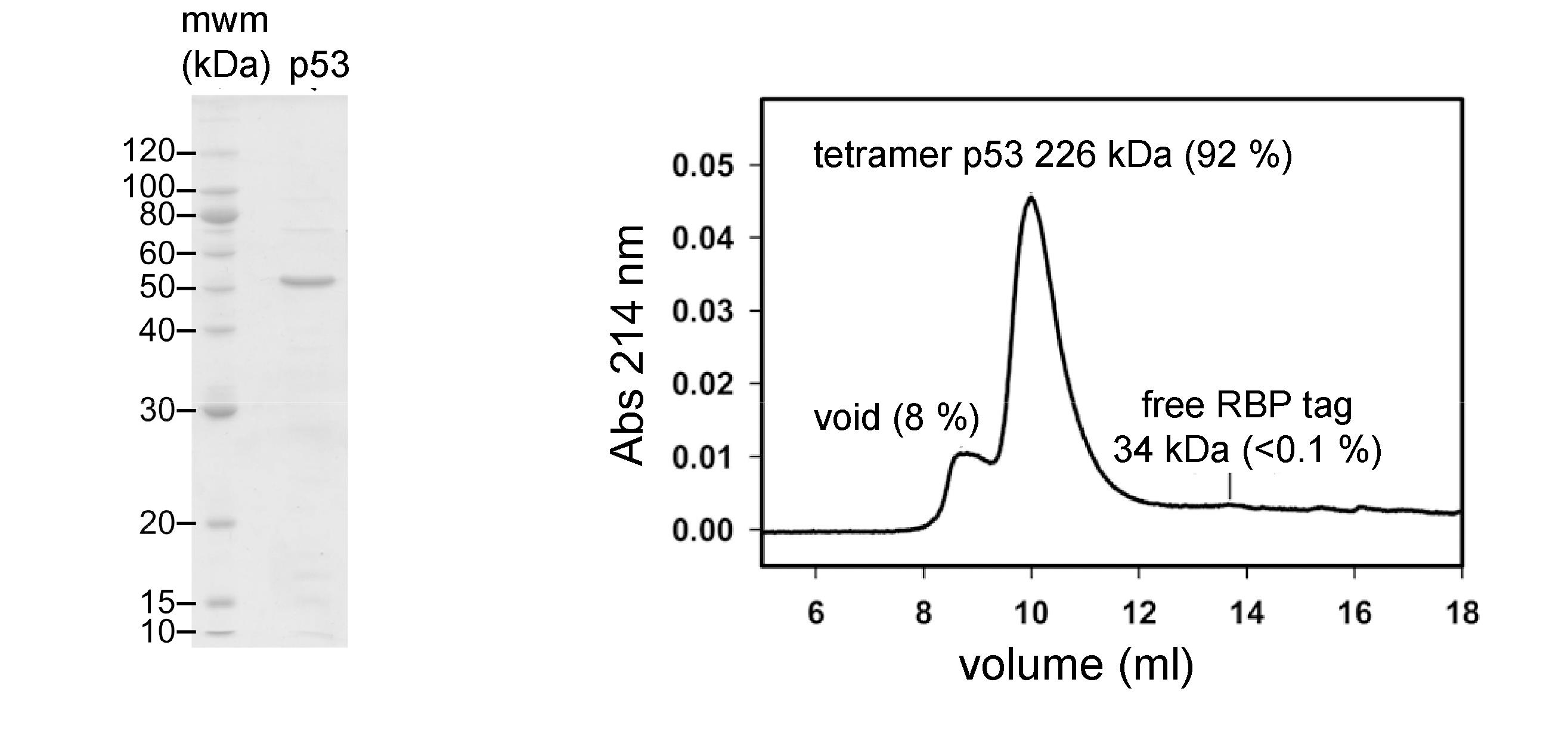
Figure 2. Validation of purified FL-p53. (A) FL-p53 is judged to be >90% pure by SDS-PAGE with Coomassie brilliant blue staining. (B) FL-p53 elutes as an apparent tetramer from a Superdex S200 Increase size exclusion column, with a minor population of larger aggregates and an almost undetectable RBP purification tag.
DBD purification
Note: WT and mutant DBD generally express well in E. coli, so they do not contain tags of any sort.
Express and harvest cells as in Steps A1-A5.
Add Buffer 3 to frozen cell pellets and thaw as in Step A6.
Lyse cells as in Steps A7 and A8.
Purify DBD as in Steps A12-A15 (HiTrap heparin column only). DBD does not contain a HisTag, so the HiTrap nickel-NTA column is not used. Heparin, being highly negatively charged like nucleic acids, is considered a pseudo-affinity column for purifying DNA and RNA binding proteins. Aided by the high expression levels of DBD, the heparin and Superdex columns are usually sufficient to obtain >95% pure DBD.
De-metallation procedure
Note: WT and most mutants of FL-p53 and DBD purify with ~1 equivalent of bound Zn2+ and are natively folded. Some zinc-binding class mutants purify with <1 equivalent of bound Zn2+, and the metal appears to be ligated to non-native sites. For the latter mutants, de-metallating and re-metallating can restore proper zinc-binding and DNA-binding activities. Extra care should be taken to ensure that apo proteins remain cold, as removing zinc increases their propensity to aggregate.
Thaw 2 ml of FL-p53 or DBD as described in the general tips section.
Add 20 µl of 0.5 M EDTA, 60 µl of 10% acetic acid (final pH ~4.3) and vortex gently.
Incubate 1 min and neutralize pH by adding 0.5 ml of 1.5 M Tris (pH 7.5) with brief vortexing.
Remove EDTA/Zn2+ by loading 2.5 ml above solution onto a 10DG desalting column (Bio-Rad) previously equilibrated with Buffer 5. After the solution is fully loaded, wash with 0.5 ml of Buffer 5 and elute with 3 ml of the same buffer (Buffer 5).
Zinc-free (apo) FL-p53 and DBD are less stable and more prone to aggregation compared to zinc-bound proteins. Apo proteins should be prepared fresh for each round of experiments and not refrozen.
Re-metallation procedure
To 1 ml of apo p53 in Buffer 5, add 10 µl of 200 mM EGTA (pH 7.5 in ddH2O) and briefly vortex.
Re-metallate by adding 8 µl of 200 mM ZnCl2 (in 10 mM HCl) and briefly vortexing. It is important that ZnCl2 is only introduced after EGTA has been added and vortexed, as unbuffered Zn2+ will cause the proteins to precipitate.
Incubate 1 h on ice and remove excess EGTA/Zn2+ as in Step C4.
Determining DBD stability parameters by urea or guanidine hydrochloride denaturation
Note: Sample preparation and data analysis steps in Protocol E are applicable to any protein. For p53, however, the Trp fluorescence application applies to DBD only (not FL-p53). FL-p53 contains additional Trp residues that do not report on folding of DBD. Hence, Protocol E is used to compare thermodynamic parameters of WT and mutant DBD. Fluorescence plate readers generally lack the sensitivity, spectral resolution, and thermal control that are required for this Protocol, so we recommend using a dedicated benchtop fluorimeter.
Place 6.4 ml of Buffer 5 in one tube (Tube A) and 6.4 ml of Buffer 6 in another tube (Tube B). Add 1.6 ml of 10 μM DBD to each tube and mix thoroughly. Note that Buffer 6 is identical to Buffer 5 except it contains 7.5 M urea or 6.25 M GdnHCl (see Recipes). Thus, the purpose of this step is to generate two solutions that have the same protein concentration (2 μM) and only differ by the absence (Tube A) or presence (Tube B) of denaturant.
Place 26 empty 1.5 ml microcentrifuge tubes on ice (these are referred to as dilution tubes).
Use a two-syringe automated diluter (Hamilton Microlab 540B) to deliver a linear combination of volumes from Tube A and Tube B to each dilution tube. Program the diluter such that the first delivery dispenses 500 µl Tube A + 0 µl Tube B to dilution tube 1, the second delivery dispenses 480 µl Tube A + 20 µl Tube B to dilution tube 2, the third delivery dispenses 460 µl Tube A + 40 µl Tube B to dilution tube 3, and so on. When completed, the tubes will contain linearly spaced concentrations of denaturant (starting from zero in dilution tube 1 and ending at 6 M urea or 5 M GdnHCl in dilution tube 26) and the same concentration of p53 (2 µM), the latter being critical for consistent fluorescence spectra. If no diluter is available, the samples may be prepared by hand using a repeat pipettor. Set the repeat pipettor to dispense 20 µl aliquots. Deliver 25 shots of Tube A to dilution tube 1, 24 shots of Tube A to dilution tube 2, and so on. Change pipet tips and repeat this process except delivering aliquots of Tube B and working backwards from dilution tube 26 to dilution tube 1.
Touch vortex dilution tubes and incubate at the desired temperature. The maximum temperature is ~25°C for WT DBD (and is typically lower for mutants), above which the proteins aggregate. Equilibration times decrease with increasing temperature (10-15 h if T <15°C, 6-10 h if T > 15°C).
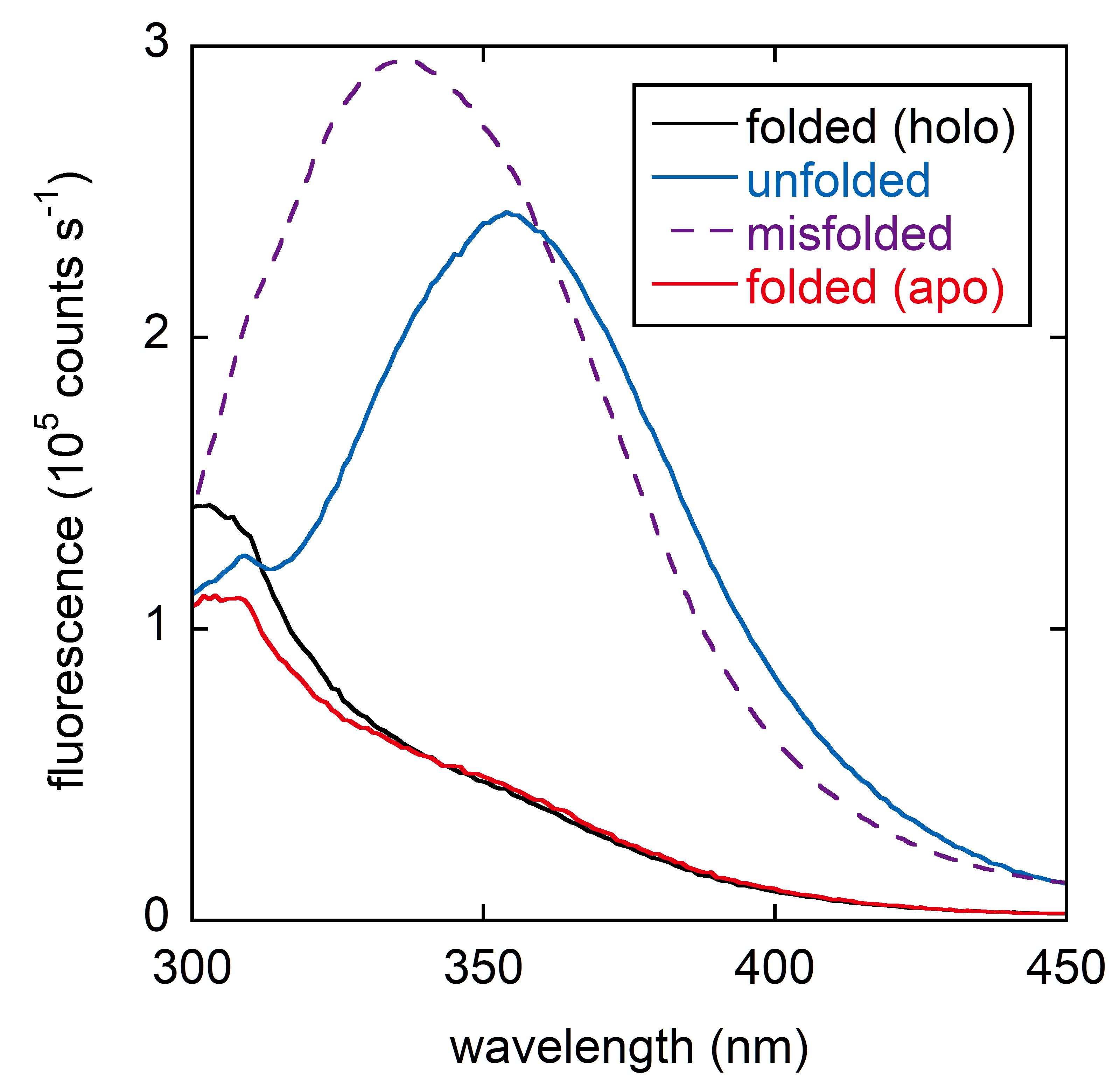
Figure 3. Characterizing DBD conformations by Trp/Tyr fluorescence. Trp fluorescence is almost fully quenched in folded DBD (both holo and apo) and unquenches when DBD is unfolded in 6 M urea, displaying a maximum at 355 nm. When p53 aggregates or otherwise misfolds, Trp fluorescence exhibits a characteristic intense peak centered at 338-340 nm. Removing Zn2+ causes a slight quenching of Tyr fluorescence at 300-310 nm. Sample conditions are 2 µM WT DBD, 20 mM Tris (pH 7.5), 0.15 M NaCl, and 10°C.Record Trp/Tyr emission spectra (excitation: 280 nm with 3 nm bandpass; emission: 300-450 nm with 4 nm bandpass, 1 nm interval, 0.15 s per nm, 5 mm × 5 mm quartz cuvette, Horiba FluoroMax 4 fluorimeter) (Figure 3). For maximum reproducibility, scan samples starting with the lowest denaturant concentration and ending with the highest denaturant concentration, leaving the cuvette in place and using a fresh transfer pipet to remove as much solution as possible between samples.
Plot the emission spectra and choose the wavelength (Fobs) that displays the greatest change between folded and unfolded states (355 nm for DBD; Figure 3). The signature of aggregated p53 is a strong peak centered at 338 nm. If this is observed, the data cannot be fit to the two-state equation (Eq. 1).
Plot Fobs (y-axis) vs. [denaturant] (x-axis) and use scientific graphic software with nonlinear least-squares curve-fitting capability (KaleidaGraph, SigmaPlot, R, GraphPad Prism, Igor Pro, etc.) to fit Fobs to Eq. 1. There are six adjustable parameters: the fluorescence values of the native and unfolded states (FN and FU), the slopes of the native and unfolded baselines (βN and βU), the cooperativity parameter (m), and the free energy change in the absence of denaturant (ΔGfold). R is the ideal gas constant.
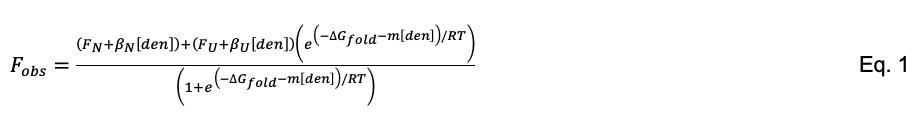
Eq. 1 is derived from the linear extrapolation model (Santoro and Bolen, 1988), which specifies that the free energy of protein folding is linear with respect to denaturant concentration, with a slope of m and a y-intercept of ΔGfold. ΔGfold and m are the parameters of primary interest in Eq. 1.
Measuring KZn by direct binding and intrinsic Trp/Tyr fluorescence
Note: For methods that employ a metal buffering system to measure KZn (Protocol F and Protocol G), the total zinc concentration is always set to be greater than the total protein concentration. The same KZn value is therefore obtained regardless of whether one starts with zinc-bound or zinc-free proteins or a mixture thereof. Protocol F and Protocol G can be applied to many metal-binding proteins (not just those that bind zinc) because chelators are available with a range of dissociation constants for various metals. However, Protocol F and Protocol G require that the protein exhibit distinct fluorescence spectra in their metal-bound and metal-free states (Protocol F), or their folded and unfolded states (Protocol G).
Choose the zinc buffering system using the “Evaluate Chelators” function of Winmaxc32 (Figure 4, top). Select the chelator such that its Kd for Zn2+ is within 10-fold of the KZn value of the p53 mutant. For example, EGTA (Kd = 7.8 nM) is a good choice for a protein with KZn in the low nanomolar range.
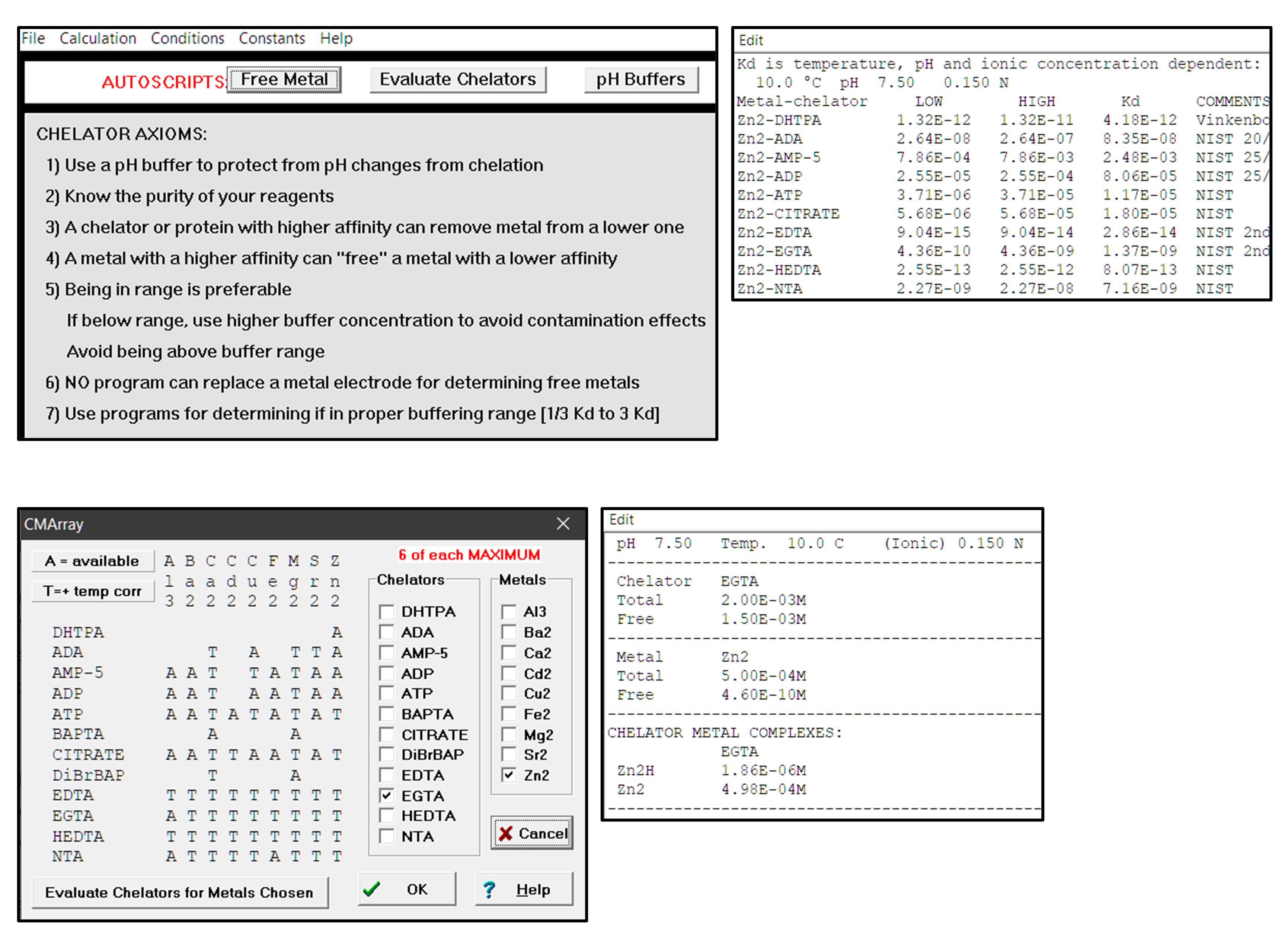
Figure 4. Choosing metal chelators and calculating free metal concentrations using Winmaxc32. Top: After entering temperature (10°C), pH (7.5), ionic strength (0.15 M), and desired metal (Zn2+), a list of common chelators and their metal binding affinities is displayed. Bottom: to calculate free metal concentration, select the chelator and enter total chelator and metal concentrations in the dilution tubes (2 mM EGTA and 50 μM Zn2+ in this example). Concentrations of total, free, and complexed species are then displayed, with free [Zn2+] (4.6 × 10-10 M) being the value that is entered into Eq. 2.Place 5.53 ml of Buffer 5 in one tube (Tube C) and 1.25 ml of Buffer 5 in another tube (Tube D).
To Tube C, add 70 µl of 200 mM chelator and vortex.
To Tube D, add 16 µl of 200 mM chelator and 12.8 µl of 200 mM ZnCl2. Vortex.
Add 1.4 ml of 10 μM p53 to Tube C and 0.32 ml of 10 μM p53 to Tube D. Vortex. Identical in purpose to Step E1, Steps F2-F5 generate two solutions that have the same protein concentration (2 μM) and chelator concentration (2 mM), differing only by the absence (Tube C) or presence (Tube D) of ZnCl2.
Place 0.5 ml aliquots of Tube C into 11 1.5 ml microcentrifuge tubes (dilution tubes) on ice.
Perform 1.5× serial dilutions by adding 1 ml of Tube D to the first dilution tube, mixing by pipetting up and down 3×. Change pipet tips, transfer 1 ml of first dilution tube to second dilution tube, mix, repeat. Leave the eleventh dilution tube as is. You now have 12 zinc concentrations ranging from [Zn2+]tot = 0 (the 11th dilution tube) to [Zn2+]tot = 1.6 mM (the remaining contents of Tube D).
Incubate at 4°C to allow for zinc binding and unbinding to come to equilibrium. For p53, this can require up to 24 h for WT proteins, with shorter times being recommended for mutants with faster zinc dissociation rates.
Record Trp/Tyr emission spectra as in Step E5 (10°C). For maximum reproducibility, scan samples starting with the lowest [Zn2+]tot (no-zinc control) and ending with the highest [Zn2+]tot, leaving the cuvette in place and using a fresh transfer pipet to remove as much solution as possible between samples.
Plot the emission spectra and choose the wavelength that displays the greatest change between zinc-free and zinc-bound states (306 nm for DBD (Figure 3), 350 nm for FL-p53).
Calculate [Zn2+]free for each sample using the “Free Metal” function of Winmaxc32 (Figure 4, bottom).
Plot Fobs (y-axis) vs. [Zn2+]free (x-axis) and fit Fobs to Eq. 2, where Fapo and Fholo are the fluorescence values of the zinc-free and zinc-bound proteins, respectively (Figure 5).

Eq. 2 is a straightforward derivation of the standard quadratic equation for one-site ligand binding, made simpler because the free ligand concentrations are known.
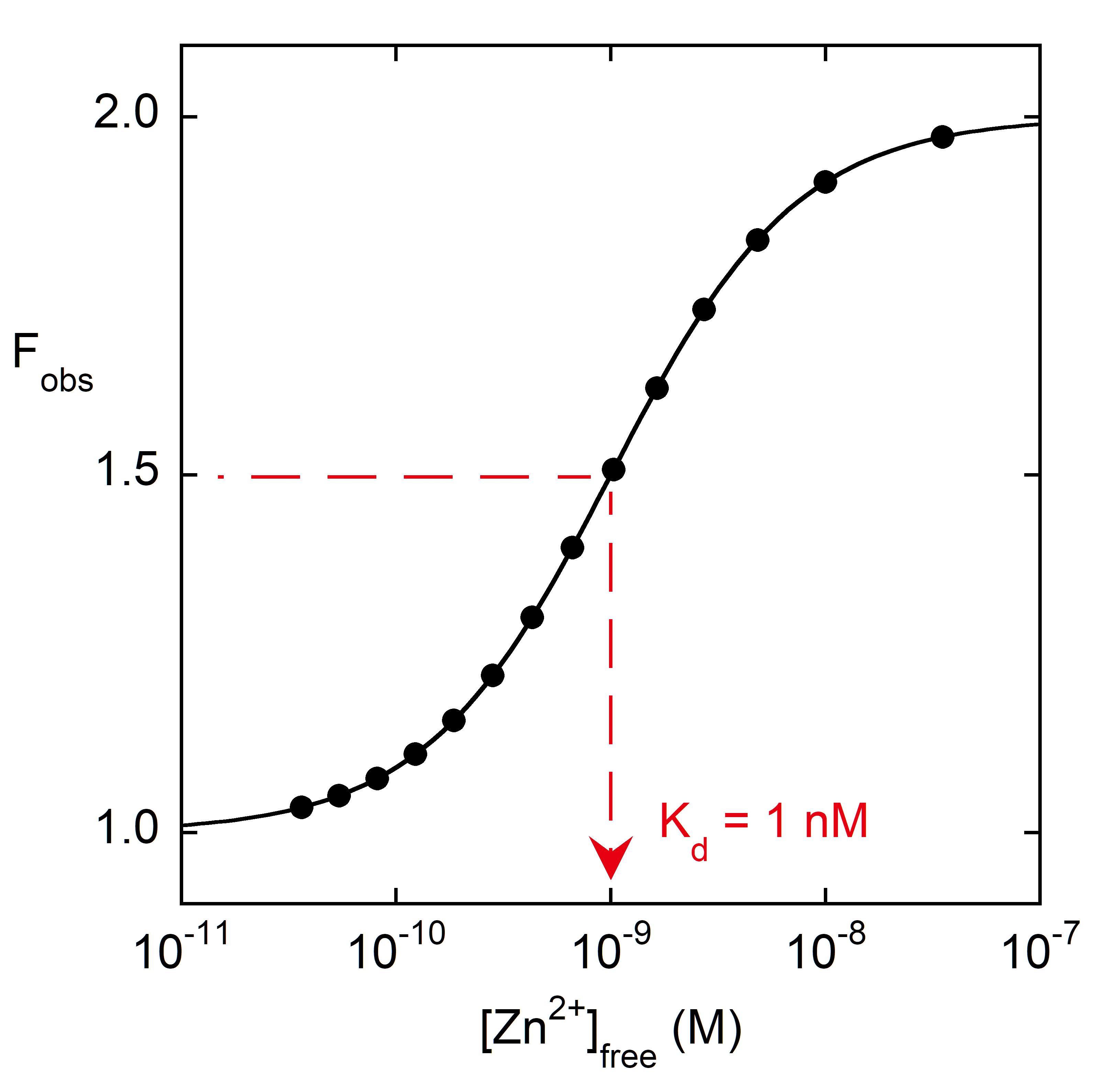
Figure 5. Fitting Tyr fluorescence to Eq. 2 to obtain KZn. In this simulated data set, Fapo and Fholo were set to 1.0 and 2.0, respectively, and KZn was chosen to be 1 nM. [Zn2+]free values were calculated from the dilutions described in the protocol, using Winmaxc32 (EGTA chelator). The black line is the fit of the simulated data to Eq. 2. The fitted parameters are Fapo, Fholo, and KZn. KZn can also be estimated from the half-way point in the sigmoidal line (red dashed arrow).
Measuring KZn by change in ΔGfold (‘hockey-stick’ plot)
Note: This method is useful because it provides three parameters: KZn, ΔGfold of the metal-free protein (ΔGapo), and the extent to which the protein is stabilized by increasing metal concentration. The latter behavior can reveal the presence of additional, non-native metal-binding sites.
Choose the zinc buffering systems using the “Evaluate Chelators” function of Winmaxc32. A chelator can buffer [Zn2+]free within ~100-fold of its zinc Kd, so one typically chooses 2-3 chelators with Kd above and below KZn, depending on the range of [Zn2+]free that one wishes to cover. For example, if your protein has a KZn value of 10-10 M, EGTA (Kd = 7.8 nM) is appropriate to buffer [Zn2+]free at levels comparable to and greater than KZn, and hydroxyethyl ethylene diamine triacetic acid (HEDTA; Kd = 7.3 pM) is suitable to buffer [Zn2+]free at levels comparable to and less than KZn.
Use the “Free Metal” function of Winmaxc32 to calculate the concentrations of ZnCl2 and chelator needed to achieve the desired [Zn2+]free, keeping in mind the requirements [chelator] > [Zn2+]total and [Zn2+]total > [p53]total.
Prepare Tube A and Tube B as in Step E1, with the additional step of adding ZnCl2 and chelator to each tube according to the calculations in Step G2.
Perform Steps E2-E7 to obtain ΔGfold at each [Zn2+]free (Figure 6).
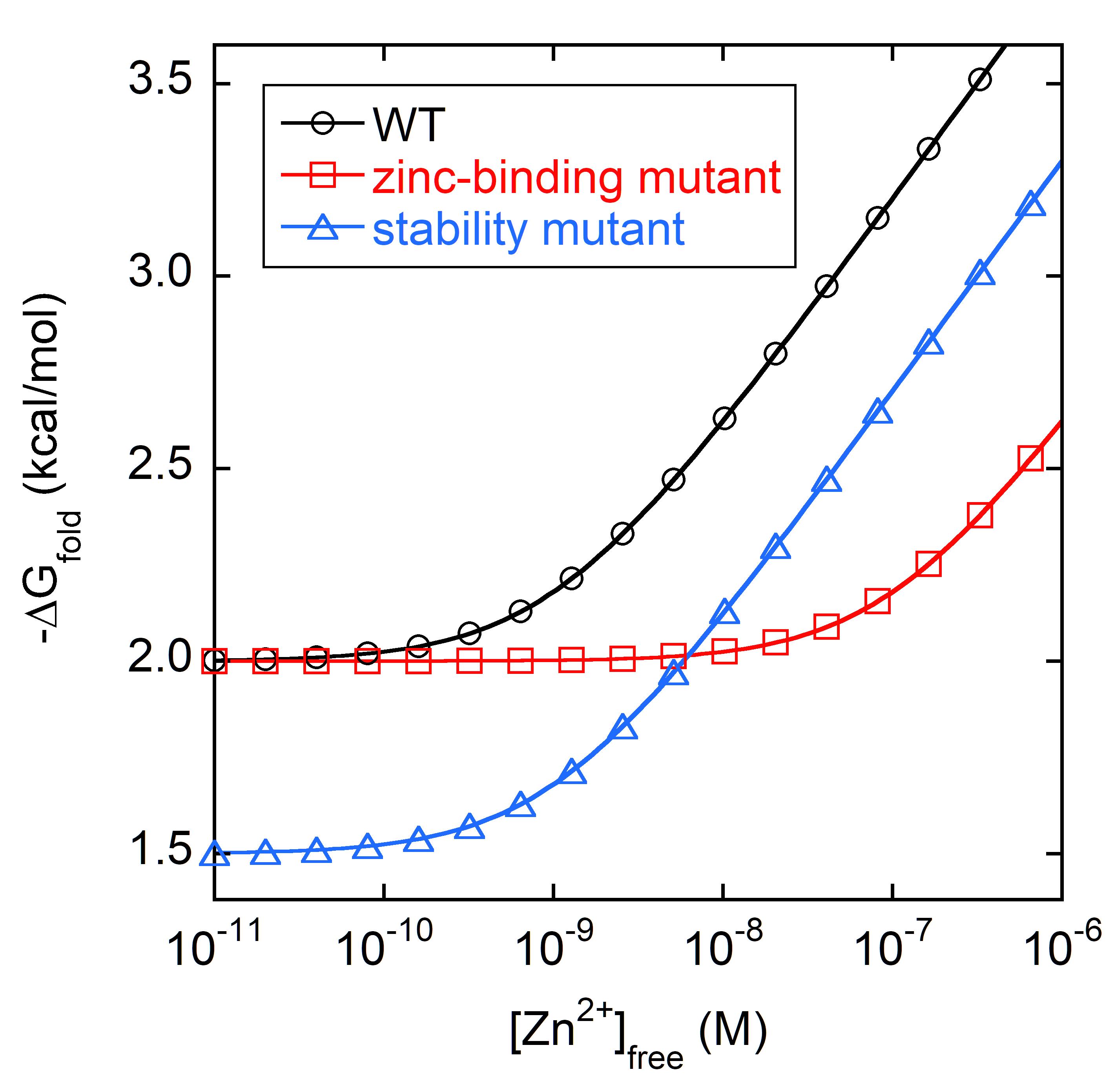
Figure 6. Hockey-stick plot for determining KZn and ΔGapo and classifying mutant types. The y-intercepts correspond to ΔGapo (-2 kcal/mol for a hypothetical WT protein; black) and the points at which the lines deflect upward correspond to KZn (1 nM for WT). A simulated zinc-binding mutant (red) has the same ΔGapo as WT, but its KZn is weakened by 100-fold. A simulated stability mutant (blue) has the same KZn as WT but is 0.5 kcal/mol less stable in the absence of zinc. Lines are best fits of the data to Eq. 3.Fit ΔGfold to Eq. 3 to obtain ΔGapo and KZn.
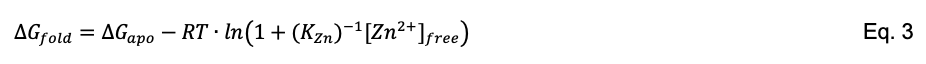
Eq. 3 is derived from the substrate stabilization model (Pace and McGrath, 1980). It describes the coupled equilibria of protein folding (Eq. 4, where U and N are unfolded and native states) and ligand binding (Eq. 5, where the ligand (Zn2+) binds to N but not to U):

The free energy changes for the equilibria of Eq. 4 and Eq. 5 are ΔGapo and ΔGbind, respectively, where ΔGbind = -RT·ln(1+(KZn)-1[Zn2+]free). The stability of the protein at any given ligand concentration is the sum of ΔGapo and ΔGbind, as given by Eq. 3.
The upward deflection of -ΔGfold signifies the onset of metal binding and stabilization (Figure 6). For one-site binding, the slope is theoretically constant regardless of protein or ligand. It will only change if additional binding events occur; for example, at high [Zn2+]free, the metal binds to sites in unfolded p53, causing -ΔGfold to eventually level off.
Measuring KZn using fluorescent zinc indicators
Note: This method is useful when no intrinsic signal change exists between the zinc-bound and zinc-free states of the protein. Instead, one detects the protein-metal interaction indirectly using a fluorescent zinc indicator and observing the decrease in fluorescence as increasing amounts of protein compete for Zn2+ binding. This technique is limited by the availability of fluorescent zinc indicators with a zinc Kd value close to that of the protein and by the solubility of the protein in cases where large concentrations are needed to compete for zinc binding. Here, we use FluoZin-3 (FZ3; Kd = 15 nM) as an example.
Prepare Tube E (250 μl 10 μM p53 in Buffer 5 containing 0.005 % TWEEN-20).
Prepare Tube F by adding 7.2 μl 10 μM fluozin-3 (FZ3) and 7.2 μl 10 μM ZnCl2 to 1.2 ml Buffer 5 containing 0.005 % TWEEN-20.
Transfer 100 μl aliquots of Tube F into 11 wells of a fully black, 96-well microtiter plate.
Perform 2-fold serial dilutions by pipetting 100 μl of Tube E into the first well, mixing by pipetting up and down three times, changing tips, and repeating from the first well to the second well and so on. Transfer 100 μl of Tube E to the empty twelfth well.
Allow binding to come to equilibrium (2 h at 4°C).
Record fluorescence using a plate reader (Molecular Devices SpectraMax i3x) with excitation at 490 nm (9 nm bandpass) and emission at 520 nm (15 nm bandpass).
Plot the observed fluorescence (Fobs; y-axis) against the logarithm of total p53 ([p53]tot; x-axis). The data will take on the shape of a sigmoid (Figure 7), and the goal is to obtain the midpoint of inhibition (IC50). IC50 is not a parameter derived from physical law, so the form of the sigmoidal function is not particularly important. We find Eq. 6 to be convenient for this purpose:

where the fitted parameters are the fluorescence baseline (F0), the amplitude of the fluorescence change (A), a steepness parameter (B), and log(IC50) (x0). The x term is log[p53]tot.
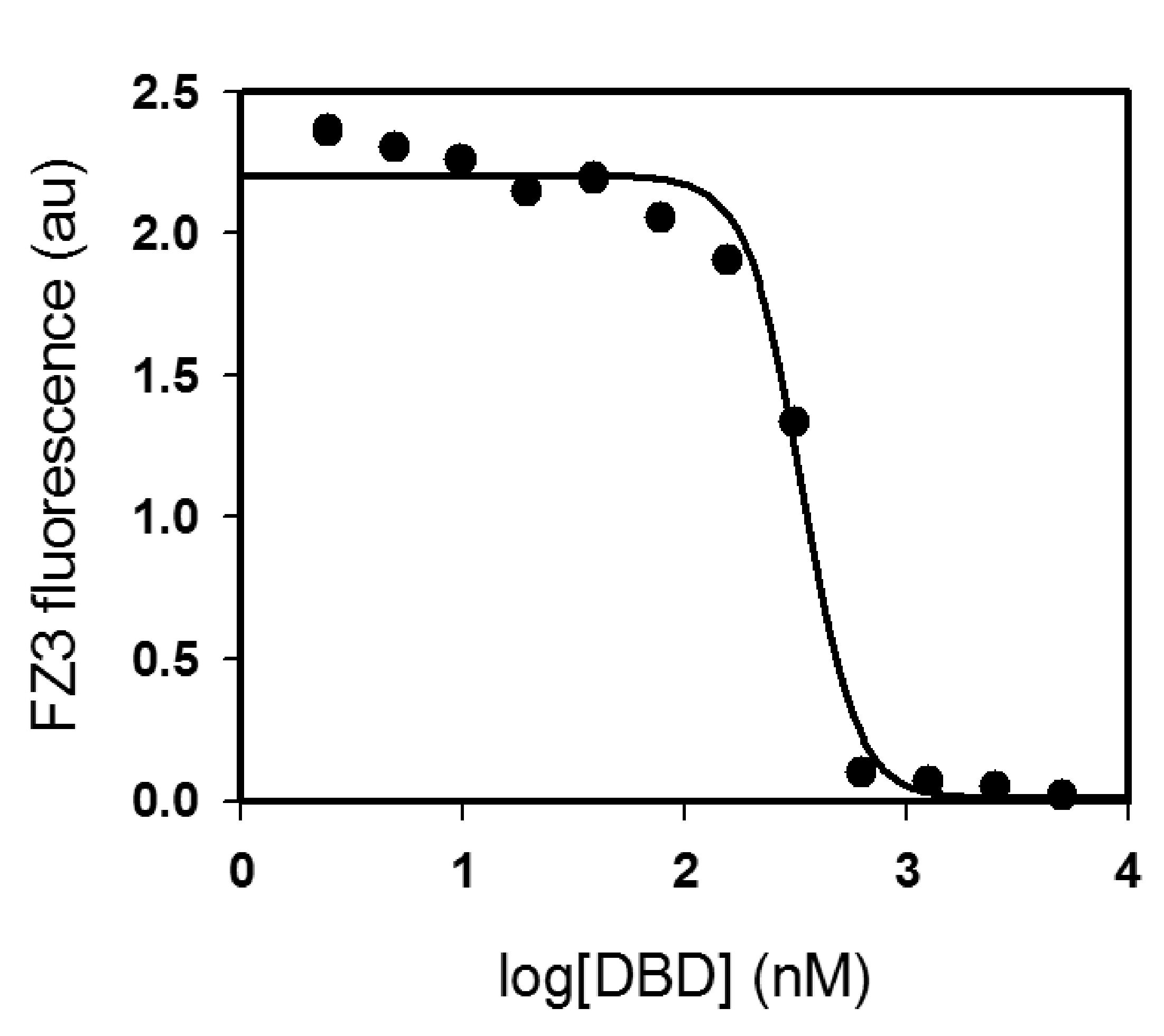
Figure 7. Fitting FZ3/p53 competition binding data to Eq. 6 to obtain IC50. Sample conditions are 30 nM FZ3, 50 mM Tris (pH 7.2), 0.1 M NaCl, 1 mM TCEP, 7 M urea, 2 h incubation on ice. Curve-fitting to Eq. 6 (line) yields IC50 = (237 ± 129) nM (n = 4). With a manufacturer reported zinc Kd of 15 nM for FZ3, this IC50 value corresponds to KZn = (44 ± 26) nM when solved with Eq. 7.Obtain KZn by entering the fitted IC50 value into the Munson-Rodbard solution (Munson and Rodbard, 1988) (Eq. 7) to the Cheng-Prusoff inhibition equation (Cheng and Prusoff, 1973), where Kd is the dissociation constant of the indicator for zinc and y0 is the ratio of bound to free FZ3 in the absence of protein.
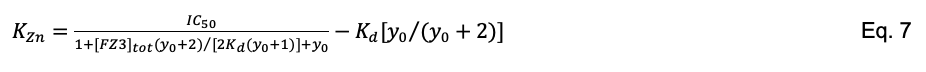
Note that when [FZ3]tot = [Zn2+]tot = 2·Kd, as we specify in this Protocol, y0 = 1 and Eq. 7 simplifies considerably.
Measuring KDNA using fluorescence anisotropy
From a commercial vendor, obtain DNA oligonucleotides (typically 20-25 nt) corresponding to each strand of the desired p53 response element. Order one of the strands labeled at either its 5’ or 3’ end with a green (fluorescein/Alexa 488/Cy2) or red (rhodamine/Alexa 568/Cy3) dye, depending on the configuration of the plate reader.
Anneal oligonucleotides by preparing a solution of 100 μM labeled oligonucleotide, 125 μM unlabeled oligonucleotide (in 30 mM HEPES (pH 7.5), 0.1 M potassium acetate), heating at 95°C for 2 min, and cooling to room temperature over 45 min.
Prepare Tube G by adding 1.25 μl 10 μM labeled oligonucleotide to 250 μl 10 μM p53 in Buffer 5 containing 0.005% TWEEN-20. For weakly binding p53 response elements and nonspecific DNA sequences, it may be necessary to increase p53 concentration.
Prepare Tube H by adding 6 μl 10 μM labeled oligonucleotide to 1.2 ml Buffer 5 containing 0.005% TWEEN-20.
Dispense 100 μl aliquots of Tube H into 11 wells of a fully black, 96-well microtiter plate.
Perform 2-fold serial dilutions by pipetting 100 μl of Tube G into the first well, mixing by pipetting up and down three times, changing tips, and repeating from the first well to the second well and so on. Transfer 100 μl of Tube G to the twelfth well.
Allow binding to come to equilibrium by incubating for 1 h in the dark.
Record fluorescence anisotropy using the SpectraMax i3x plate reader, equipped with the appropriate green or red anisotropy filter cartridge.
Obtain KDNA by fitting the observed anisotropy to the Hill binding equation (Eq. 8), where the adjustable parameters are the fluorescence anisotropy baseline (F0), the amplitude of the anisotropy change (A), and the Hill coefficient (n).

The sigmoidal shape of the data is very similar to those of Figure 5 and Figure 7, and KDNA is obtained as a fit parameter or by estimating the half-way point of the sigmoidal transition. Eq. 8 assumes that total p53 concentration is approximately equal to free p53 concentration. This requirement is not always met in the conditions of this experiment. However, the Protocol is designed to compare KDNA values for different p53 mutants and DNA sequences by determining total p53 concentrations that produce half-saturation rather than to measure KDNA values with high accuracy.
Data analysis
In urea and guanidine hydrochloride denaturation experiments (Protocol E and Protocol G), the error of the m-value is intrinsically large. This causes large uncertainties in ΔGfold, as ΔGfold is an extrapolated value based on m. To mitigate this uncertainty, it is often useful to fit replicate data sets as well as those of different DBD mutants using a shared m-value. One should only consider this option if the individual data sets fit adequately to the two-state model (Eq. 1) and if the variation in m-values is <50%. Graphing programs capable of shared-parameter curve fitting include SigmaPlot, R, and Igor Pro.
In Protocol H, the fluorescence baselines of FZ3 can exhibit slopes. These slopes are most likely caused by nonspecific reactions (potentially including binding to wells of the microtiter plate). It is straightforward to add compensatory parameters to Eq. 6, but sloping baselines rarely make an impact on the fitted IC50 value, which is the only relevant parameter in Eq. 6. Consequently, we find it more reliable to force-fit the data to Eq. 6 rather than risk over-parameterization.
Criteria for data inclusion/exclusion and a description of statistical tests can be found in Blanden et al. (2020) .
Recipes
Buffers used in Protocols: Prepare using double-distilled water and metal-free reagents. Adjust pH to indicated value after all components have been added.
Buffer 1
10 mM Tris
10 mM imidazole
300 mM NaCl
5 mM βME
pH 8.0
Buffer 2
10 mM Tris
200 mM imidazole
150 mM NaCl
5 mM βME
pH 8.0
Buffer 3
10 mM Tris
75 mM NaCl
10 mM βME
pH 7.5
Buffer 4
10 mM Tris
1,000 mM NaCl
10 mM βME
pH 7.5
Buffer 5
20 mM Tris
150 mM NaCl
2 mM TCEP
pH 7.5
0.005 % TWEEN-20 for Protocol H and Protocol I
Buffer 6
20 mM Tris
150 mM NaCl
2 mM TCEP
pH 7.5
7.5 M urea or 6.25 M GdnHCl, ultrapure
Acknowledgments
This work was supported by NIH grant R01 CA200800 to D.R.C. The protocols were derived from Blanden et al. (2020).
Competing interests
The authors declare no competing interests.
References
- Andreini, C., Banci, L., Bertini, I. and Rosato, A. (2006). Counting the zinc-proteins encoded in the human genome. J Proteome Res 5(1): 196-201.
- Barber-Zucker, S., Shaanan, B. and Zarivach, R. (2017). Transition metal binding selectivity in proteins and its correlation with the phylogenomic classification of the cation diffusion facilitator protein family. Sci Rep 7(1): 16381.
- Baugh, E. H., Ke, H., Levine, A. J., Bonneau, R. A. and Chan, C. S. (2018). Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ 25(1): 154-160.
- Blanden, A. R., Yu, X., Blayney, A. J., Demas, C., Ha, J. H., Liu, Y., Withers, T., Carpizo, D. R. and Loh, S. N. (2020). Zinc shapes the folding landscape of p53 and establishes a pathway for reactivating structurally diverse cancer mutants. Elife 9: e61487.
- Bouaoun, L., Sonkin, D., Ardin, M., Hollstein, M., Byrnes, G., Zavadil, J. and Olivier, M. (2016). TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum Mutat 37(9): 865-876.
- Bullock, A. N., Henckel, J. and Fersht, A. R. (2000). Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene 19(10): 1245-1256.
- Cheng, Y. and Prusoff, W. H. (1973). Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22(23): 3099-3108.
- Ha, J. H., Tu, H. C., Wilkens, S. and Loh, S. N. (2021). Loss of bound zinc facilitates amyloid fibril formation of leukocyte cell-derived chemotaxin 2 (LECT2). J Biol Chem 296: 100446.
- Hunt, J. B., Neece, S. H. and Ginsburg, A. (1985). The use of 4-(2-pyridylazo)resorcinol in studies of zinc release from Escherichia coli aspartate transcarbamoylase. Anal Biochem 146(1): 150-157.
- Munson, P. J. and Rodbard, D. (1988). An exact correction to the "Cheng-Prusoff" correction. J Recept Res 8(1-4): 533-546.
- Pace, C. N. and McGrath, T. (1980). Substrate stabilization of lysozyme to thermal and guanidine hydrochloride denaturation. J Biol Chem 255(9): 3862-3865.
- Santoro, M. M. and Bolen, D. W. (1988). Unfolding free energy changes determined by the linear extrapolation method. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochem 27(21): 8063-8068.
Article Information
Copyright
![]() Ha et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Ha et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Ha, J. H., Yu, X., Carpizo, D. R. and Loh, S. N. (2021). Urea Denaturation, Zinc Binding, and DNA Binding Assays of Mutant p53 DNA-binding Domains and Full-length Proteins. Bio-protocol 11(20): e4188. DOI: 10.21769/BioProtoc.4188.
- Blanden, A. R., Yu, X., Blayney, A. J., Demas, C., Ha, J. H., Liu, Y., Withers, T., Carpizo, D. R. and Loh, S. N. (2020). Zinc shapes the folding landscape of p53 and establishes a pathway for reactivating structurally diverse cancer mutants. Elife 9: e61487.
Category
Cancer Biology > Cancer biochemistry > Protein
Biochemistry > Protein > Isolation and purification
Biochemistry > Protein > Activity
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.