- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Protocol for RNA-seq Expression Analysis in Yeast
Published: Vol 11, Iss 18, Sep 20, 2021 DOI: 10.21769/BioProtoc.4161 Views: 5883
Reviewed by: Shyam SolankiJihyun KimAnonymous reviewer(s)

Original research article
The authors used this protocol in:

Dec 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Genome-wide sequencing of RNA (RNA-seq) has become an inexpensive tool to gain key insights into cellular and disease mechanisms. Sample preparation and sequencing are streamlined and allow the acquisition of hundreds of gene expression profiles in a few days; however, in particular, data processing, curation, and analysis involve numerous steps that can be overwhelming to non-experts. Here, the sample preparation, sequencing, and data processing workflow for RNA-seq expression analysis in yeast is described. While this protocol covers only a small portion of the RNA-seq landscape, the principal workflow common to such experiments is described, allowing the reader to adapt the protocol where necessary.
Graphic abstract:
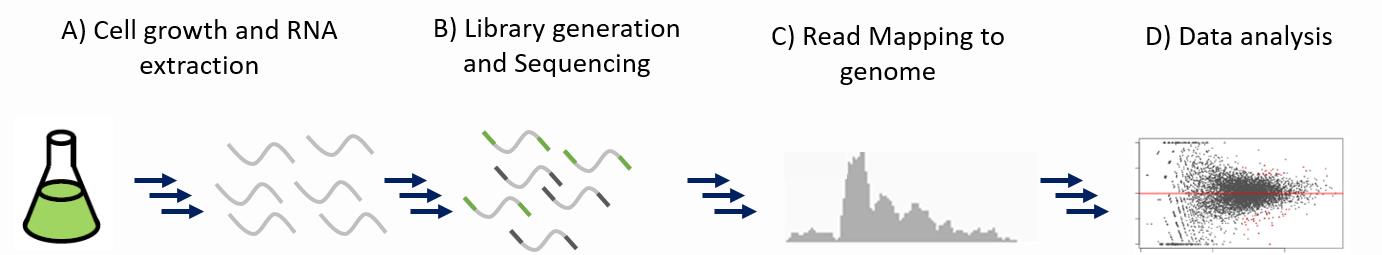
Basic workflow of RNA-seq expression analysis.
Background
Sequencing of RNA (RNA-seq) has – with the emergence of next-generation sequencing – become a powerful tool to measure the presence and quantity of RNA in a given cell population or even within a single cell. Since its initial uses (Bainbridge et al., 2006; Cheung et al., 2006; Emrich et al., 2007; Weber et al., 2007; Nagalakshmi et al., 2008), RNA-seq has seen a large variety of applications: from gene expression analysis by quantitating the relative amounts of RNA sequence reads to the discovery of novel transcripts or splice variants, ribosome profiling, or the detection of single nucleotide polymorphisms. Even though a manifold of specific RNA-seq uses exists, the basic workflow remains the same: RNA molecules are extracted, amplified, sequenced, aligned to the genome of the host organism, and, subsequently, the data is analyzed. Sample requirements are relatively low; typically, 1 μg down to 10 ng input RNA is sufficient for downstream amplification and library generation. For single-cell RNA-seq, as little as 10 pg is required since the low amount of input material is amplified prior to library generation (Haque et al., 2017). RNA-seq can be applied to any population of extracted RNA, independent of the source organism. Due to the wide applicability of RNA-seq, sample preparation kits that vary in complexity are commercially available (from RNA extraction to whole RNA-seq library generation, including the computational analysis).
An integral part of RNA-seq is sequencing of the extracted RNA population. Most commonly, sequencing is performed by the detection of fluorescently labeled nucleic acids bound to the surface of flowcells, e.g., using platforms such as Illumina and PAC Biosystem. To this end, the RNA fragments are converted into a cDNA library and amplified, and flowcell adapters are introduced. During each sequencing cycle, DNA polymerases attach fluorescently labeled nucleotides to the flowcell-bound library molecules, which are then detected by the sequencer, typically generating read lengths of 150–300 bp to several Kbp (for Illumina and PAC Biosystem, respectively). More recently emerging is sequencing by passage of nucleic acids through protein nanopores embedded in membranes (e.g., by Nanoporetech) (Logsdon et al., 2020), allowing for the sequencing of much longer fragments (up to Mbp). At the time of writing, the most commonly available sequencers (e.g., Illumina or PAC Biosystem) cost around $100 k for the instrument alone, whereas table-top sequencers using the nanopore technology are considerably cheaper (~$10 k), promising wider applicability in the near future. Due to the considerable cost of the most commonly available sequencing systems, resources are often shared among labs or institutes and managed by trained professionals that ensure the acquisition and integrity of high-quality sequencing data.
While it extends beyond the scope of this manuscript to describe all the applications of RNA-seq, this protocol aims to provide a workflow for RNA-seq expression analysis that can be used as a reference backbone, which the reader can adapt to their specific needs (e.g., RNA extraction from a different source or the addition of splice-aware alignment steps for genomes of higher eukaryotes). RNA-seq expression analysis is a powerful and commonly used tool to identify genes that are up- or downregulated in a stressed sample (e.g., in the presence of genomic mutations, UV light, drugs, chemical or nutrient stress) as compared with a relaxed sample (e.g., wild-type cell population). A gene is “upregulated” or “downregulated,” respectively, when more or less of its RNA is measured (i.e., expressed in the cell) under the stressed conditions as compared with the wild type.
Here, the workflow for RNA-seq expression analysis in S. cerevisiae is described, from cell growth to RNA extraction, library generation, data processing, and analysis. This protocol focuses on using commonly available lab resources wherever possible and utilizes open source and free-of-cost software packages provided by the bioinformatics community. This workflow has proven to be robust and useful for the analysis of gene expression profiles in libraries of histone point mutants in yeast (Braberg et al., 2020).
Materials and Reagents
1.5 ml Eppendorf tubes (e.g., Eppendorf, catalog number: 0030120086)
Petri dishes, plastic, 10-cm diameter (e.g., Falcon, catalog number: 353003)
Sterile pipette tips
Toothpicks (autoclaved)
Dry ice
Agar (e.g., Becton Dickinson, catalog number: 214030)
Bacto Peptone (e.g., Becton Dickinson, catalog number: 211677)
CHCl3 (Acid phenol, e.g., ThermoFisher, catalog number: AM9720)
DEPC-ddH2O (Diethyl pyrocarbonate-treated water, e.g., Invitrogen, catalog number: 750024)
EDTA (Ethylenediaminetetraacetic acid disodium salt dihydrate, e.g., Sigma-Aldrich, catalog number: E6635)
EtOH (Ethanol, e.g., Sigma-Aldrich, catalog number: 459836)
Formamide (e.g., Sigma-Aldrich, catalog number: 11814320001)
Glucose (e.g., Molekula, catalog number: 13002238)
HCl (Hydrochloric acid, e.g., Sigma-Aldrich, catalog number: 320331)
NaAc (Anhydrous sodium acetate, e.g., Sigma-Aldrich, catalog number: S2889)
NaOH (Sodium hydroxide pellets, e.g., Sigma-Aldrich, catalog number: 1064980500)
SDS (Dodecyl sulfate sodium salt, e.g., Merck, catalog number: 13760)
Tris (2-Amino-2-(hydroxymethyl)-1,3-propanediol, e.g., Sigma-Aldrich, catalog number: T1503)
Yeast extract (e.g., Serva, catalog number: 24540)
Yeast Extract Peptone Dextrose (YEPD) media (see Recipes)
1 M Tris-HCl solution, pH 7.5 (see Recipes)
0.5 M EDTA solution, pH 8.0 (see Recipes)
20% SDS solution (see Recipes)
Tris-EDTA-SDS (TES) solution (see Recipes)
3 M NaAc solution, pH 5.2 (see Recipes)
Equipment
Autoclave
Centrifuge and table-top centrifuge
Vortex
Flasks, autoclavable
Incubator
pH meter
Pipettes (1-ml, 200-μl, 20-μl, 2-μl)
Stir bar and stir plate, magnetic
Thermocycler
Software
bbmap (BBMap – Bushnell, B.; Version 38.90; sourceforge.net/projects/bbmap)
Biocmanager (https://cran.r-project.org/web/packages/BiocManager/vignettes/BiocManager.html; Version 3.12; https://bioconductor.org/install/)
bioconda (Grüning et al., 2018; http://bioconda.github.io/user/install.html)
bowtie2 (Langmead and Salzberg, 2012; Version 2.4.2; http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)
bwa (Burrows-Wheeler Aligner, Li and Durbin, 2009; Version 0.7.17; http://bio-bwa.sourceforge.net/)
DESeq2 (Love et al., 2014; Version 1.30.1, http://bioconductor.org/packages/release/bioc/html/DESeq2.html)
fastqc (Andrews, 2010; FastQC: a quality control tool for high throughput sequence data; Version 0.11.9; https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
htseq-count (Anders et al., 2014; Version 0.11.1; https://htseq.readthedocs.io/en/release_0.11.1/install.html)
Integrated Genomics Viewer (Robinson et al., 2011; Version 2.9.2; https://software.broadinstitute.org/software/igv/download)
R (R Core Team, 2017; Version 4.0.4; https://cran.r-project.org/bin/windows/base)
samtools (Li et al., 2009; Version 1.11; http://www.htslib.org/download/)
tophat (Langmead et al., 2009; Version 2.1.1; https://ccb.jhu.edu/software/tophat/index.shtml)
trimmomatic (Bolger et al., 2014; Version 0.40; http://www.usadellab.org/cms/?page=trimmomatic)
Procedure
Sample preparation and RNA extraction
It is of utmost importance when handling RNA that all materials and reagents are RNase-free. Furthermore, it must be noted that the computation of relative expression values described in Procedure D requires at least three biological replicates. Accordingly, for example, if the expression levels of a mutant strain are to be compared with a wild-type strain, six RNA samples need to be prepared (three for each strain), which can then be used to create six RNA-seq libraries.
Here, an efficient and reliable method to extract RNA despite the robust yeast cell wall is described (Collart et al., 2001):
With a sterile toothpick or pipette tip, pick single colonies of S. cerevisiae and inoculate 2 ml YEPD liquid media for growth at 30°C overnight.
Inoculate 10 ml liquid YEPD with 50 μl overnight culture.
Harvest the cells in the mid-log phase (OD600 ~1.0) by centrifugation, and transfer to a 1.5-ml Eppendorf tube. Resuspend in 300 μl DEPC-ddH2O, fast-spin in a table-top centrifuge (up to 9,500 × g), remove the supernatant, flash-freeze on dry ice, and store at -80°C.
Resuspend the cell pellet in 400 µl TES solution. Add 400 µl acid phenol (CHCl3), cap the tube, and vortex vigorously for 10 s (avoid leakage and handle carefully!). Incubate for 60 min at 65°C, vortexing every 15 min (Collart and Oliviero, 2001).
Place on ice for 5 min. Spin in a microfuge at 18,000 × g for 10 min at 4°C. Transfer the aqueous top layer to a clean tube (avoiding the white protein phase). Add 400 µl CHCl3 and vortex vigorously for 10 s. Spin in a microfuge at 18,000 × g for 10 min at 4°C. Transfer the aqueous top layer to a clean tube (pipette carefully and avoid the CHCl3 layer).
Add a 1/10 volume of 3 M NaAc, pH 5.2, and 2.5 volumes EtOH (-20°C). Precipitate at -80°C for at least 60 min. Spin in a microfuge at 18,000 × g for 10 min at 4°C. Carefully remove the supernatant and wash the pellet by vortexing in 70% EtOH (-20°C). Spin in a microfuge at 18,000 × g for 10 min at 4°C.
Resuspend the pellet in 100% formamide (at 4°C). Try an equal volume of liquid to pellet first, and move up from there. Most RNA should dissolve instantly. To aid solubilization, allow to sit at room temperature for 15 min, pipetting every 5 min. If the sample needs to be very concentrated, store at 4°C overnight.
Determine the concentration by diluting 1/100 in H2O and measuring at OD260/280 (OD260 1 ≈ 40 µg/ml for RNA). Remember to add formamide at a 1/100 dilution to the blank.
RNA-seq library generation and sequencing
Before sample preparation and submission to a sequencing facility, it is strongly recommended to discuss the aims of the project with the trained personnel. For successful library generation, the input RNA concentration is critical, commonly ranging from 1 μg down to 10 ng per sample. While it is possible to generate RNA-seq libraries from scratch (i.e., producing adaptors, buffers, polymerase, etc., using your own materials), it is strongly recommended to use commercially available kits that require minimum common lab resources and are, most importantly, more reliable in the hands of researchers unfamiliar with RNA-seq library formation.
During RNA-seq library generation, platform-specific adapters are attached to the extracted RNA molecules; therefore, the library kit must be chosen according to the sequencing platform to be used. Here, the QuantSeq 3’ mRNA-seq Library Prep Kit FWD for Illumina (Lexogen) was used for the generation of single-end (i.e., fragments will be sequenced from one end only), 50-bp reads, sufficient for RNA-seq expression analysis in yeast. For more complex eukaryotic genomes containing larger amounts of introns, and when longer reads are required, consider paired-end library kits and sequencing (i.e., fragments will be sequenced from both ends), after consultation with the sequencing facility staff.
Generate cDNA libraries containing sequencer- and sample-specific adapters by carefully following the steps described in the manufacturer’s manual.
Check the quality of the generated libraries and measure the cDNA concentration.
Sequence the cDNA library. Here, an Illumina HiSeq 4000 sequencer was used.
Check the quality of the raw read data, typically supplied in fastq format (Figure 1). The most common checks involve the number of reads per sample (should be the same order of magnitude for all sequenced samples, which means the files should be similar in size), the GC content (should match the overall GC content of the host organism), and the overall base quality. Several quality control tools exist; here, fastqc was used (see also Batut, 2021). It is strongly recommended that quality control is performed after each processing step to ensure the overall integrity of the data.

Figure 1. An example read sequenced on an Illumina platform in FASTQ format. Line 1 contains the basic read information, line 2 contains the actual sequence, and line 4 contains the quality score for each base in Phred33 or Phred64 code.
Processing of raw sequence data
C1. Preparation of data processing
The following steps describe the setup for the computational workflow described in Step C2, as well as the data analysis described in Procedure D. This workflow uses open source programs available on Linux operating systems (and its derivatives). While it is possible to process sequence files on Mac- or Windows-operated instruments, the reader is strongly recommended to use Linux-based utilities due to their wide applicability, timely updates, and community-based troubleshooting.
For most of the processing steps described in Step C2 and D, multiple tools exist; in particular, for the acquisition of genome assembly and gene annotations (15), creation of index files (16), adapter trimming (17), read alignment (18), and read filtering based on quality (19). While it extends beyond the scope of this manuscript to describe all the tools in detail, alternatives to the programs used in this protocol are suggested.
Several of the tools used here are available through so-called package managers, such as Bioconda or Biocmanager, allowing for easy installation of software and dependencies of most recent versions; hence, it is recommended to follow the installation order described here.
Install the Conda package manager via gitHub and R, bowtie2, and samtools using the specific channel, Bioconda.
curl -O https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
sh Miniconda3-latest-Linux-x86_64.sh
conda install -c bioconda R
conda install -c bioconda bowtie2
conda install -c bioconda samtools
Install Biocmanager and the DESeq2-package from within R.
R
if (!requireNamespace(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
BiocManager::install()
BiocManager::install(c(“DESeq2”))
C2. Processing of raw sequence read files
After passing quality checks, the sequence reads now undergo pre-processing and eventually, alignment to the genome of the host organism. First, genome assembly, gene annotation, and the genome index need to be prepared (C2.1 and C2.2). The sequence reads contain adapter contamination, random primer sequences, and low-quality tail reads, which need to be removed (C2.3) before the alignment of filtered reads to the genome of the host organism (C2.4). Finally, reads are filtered based on their quality score (C2.5) and indexed for downstream analysis (C2.6).
Download the S. cerevisiae genome assembly and gene annotation. Here, UCSC versions, sacCer3.fa and sacCer3.ensGene.gtf, were used, respectively (downloaded from http://hgdownload.cse.ucsc.edu/goldenPath/sacCer3/bigZips/). Besides the UCSC genome browser, other platforms allow the download of genome assemblies and gene annotations (e.g., NIH/NCBI or Ensembl). Since every platform maintains a slightly different naming convention, it is important that genome assembly and gene annotation are acquired from the same platform to avoid program errors during processing.
Create the index files based on the genome assembly (“sacCer3.fa”) from Step C2.1. Here, the index output files are stored with the base filename “sacCer3.” This index will be used by the alignment program in Step C2.4; hence, the indexing step must be adapted to the aligner of choice, and an example for bwa is given below. Caution: bwa is not a splice-aware alignment tool. If splice events need to be considered during analysis, aligners like tophat need to be used for index creation as well as alignment (Step C2.4).
bwa index sacCer3.fa
Remove the random primer sequence, adapter contamination, and low-quality tails. Here, bbmap was used for the library kit described in Step B1, according to the manufacturer’s recommended settings (see command below, from www.lexogen.com/quantseq-data-analysis). bbmap is a fast, splice-aware global alignment tool for RNA and DNA sequencing reads. The script “bbduk.sh” is used together with the polyA-tail sequence (polyA.fa.gz) and Illumina-specific adapter sequence information (truseq.fa.gz), located in the installation folder bbmap/resources/).
Adapter sequences and low-quality reads can also be removed using different tools such as “trimmomatic” (see the software section). Independent of the software used, parameters need to be adjusted according to the library kit and sequencing platform used. As a result of trimming, the size of the output file should be slightly smaller than that of the input file.
bbmap/bbduk.sh in=sample1.fastq out=sample1_trimmed.fastq ref=bbmap/resources/polyA.fa.gz,bbmap/resources/truseq.fa.gz k=13 ktrim=r forcetrimleft=11 useshortkmers=t mink=5 qtrim=t trimq=10 minlength=20; done
Create alignments of the pre-processed sequence reads from Step C1.1 using an alignment tool, such as tophat, bbmap, bowtie2, or bwa. Here, bwa was used. Depending on the sequence file size (i.e., the number of reads), genome size, and CPU used, this step can take several minutes to several hours. On an Intel® Zeon® CPU E5-2699 v3 @ 2.3GHz, alignment took about 3 s per 100 k reads.
The following command will align the input file reads (sample1_trimmed.fastq) to the genome index from Step C2.2 (sacCer3) and write the results to “sample1_trimmed_aligned.sam.”
bwa mem sacCer3.fa sample1_trimmed.fastq > sample1_trimmed_aligned.sam
Filter the data based on their quality by MAPQ filtering using samtools. Here, all reads with an average base read quality score less than 50 (i.e., the probability of correct mapping is > 99.999%, Figure 1) were removed from the mapped read files generated in Step C2.4. As a result of the quality filtering, the output file size will be smaller than the input file size. If no, or very few, reads remain, try filtering with a less stringent quality score (e.g., 20). If this recovers the number of reads, downstream analysis may still be possible, albeit less reliable.
samtools view -bq 50 sample1_trimmed_aligned.sam > sample1_trimmed_aligned_mapq50.bam
Sort the filtered, aligned reads from Step C2.5 and create the index files using samtools. This will create an index file with the same name as the input file, including the additional ending of “.bai.”
Aside from the quality check of the output file using tools such as fastqc, the mapped reads can be visualized using the Integrated Genomic Browser (IGV). In addition to ensuring that the overall mapping of reads is correct, tools like IGV allow confirmation of the presence of intended genomic mutations or gene deletions (Figure 2).
samtools sort sample1_trimmed_aligned_mapq50.bam -o sample1_trimmed_aligned_mapq50_sorted.bam samtools index sample1_trimmed_aligned_mapq50_sorted.bam
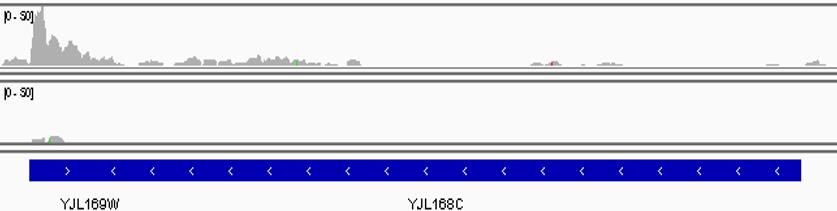
Figure 2. Snapshot of the IGV browser visualization. Here, reads (grey bars) mapping to the genomic region of Set2 (blue, YJL168C) in yeast are compared between wild type (upper lane) and the ∆Set2 mutant (lower lane).
Data analysis, calculation of expression values, and visualization of results
The sorted and indexed files prepared in C) contain all the reads that were successfully aligned to the host’s genome (here, S. cerevisiae), numbering typically from several hundreds of thousands to millions of reads per replicate. This vast amount of information is a major hurdle for analysis by the researcher. Differential gene expression (DGE) analysis aims to determine which, if any, genes show a higher or lower amount of aligned reads across the tested conditions. To this end, reads belonging to a feature (i.e., a gene) are summed for each replicate, and differential expression values are calculated across conditions considering the variance within a condition among replicates; hence, it is critical for DGE that several replicates of the same condition are considered (typically, n = 3). Gene expression values are usually reported as log2-fold changes, in conjunction with adjusted P-values describing the significance of the change (cut-offs vary, but typically P-values < 0.05 are considered reliable).
The number of aligned reads can differ strongly between replicates due to technical reasons (e.g., fluctuations in the amount of input RNA, variations in temperature of the thermocycler during library amplification, or differences in the binding capacity of the flowcell lanes); hence, reads must be normalized across replicates and conditions. Several normalization methods for the calculation of DGE values exist, such as Reads Per Kilobase of transcript per Million mapped reads (FPKM), Fragments Per Kilobase of transcript per Million mapped reads (RPKM), Transcripts per Million reads (TPM), or counts per feature (i.e., gene) (Dillies et al., 2013).
Here, the count-based normalization by DESeq2 was used, based on the assumption that most genes are not differentially expressed across conditions; therefore, the counts per feature are extracted from each file generated in Step C2.6 using htseq (Step D1), combined, and indexed (Steps D2 and D3). Finally, the counts are normalized, and DGE values are calculated using DESeq2 (Steps D4 and D5). The results are visually represented using MA plots, where log-fold changes are plotted against the mean expression values (Step D5).
Extract the counts for each sample using htseq-count. Here, the aligned, filtered, and sorted reads (e.g., sample1_trimmed_aligned_mapq50_sorted.bam) from Step C2.6 and the gene annotation file (sacCer3.ensGene.gtf) from Step C2.1 were used. This command generates a .txt file containing the number of reads assigned to each gene annotated in the gtf-file.
htseq-count -f bam sample1_trimmed_aligned_mapq50_sorted.bam sacCer3.ensGene.gtf > sample1_trimmed_aligned_mapq50_sorted_counts.txt
Count-based expression values are calculated using R and Dseq2; this requires the count data to be assembled in a text document (here, “counts.txt”) as well as in an index file (here, “table.txt,” Step D3).
Generate a “counts.txt”-file that contains the counts for each replicate of a given sample (here, MUT_X) as well as the reference sample (here, WT_X) generated in Step D1 as columns in a tab-delimited txt document (Figure 3). As a quality check, it is recommended to check several lines (i.e., genes) for consistency (i.e., similar read counts among replicates of a certain condition). Importantly, the read counts are not yet normalized to the total number of read counts in each sample, and respective variations are expected.
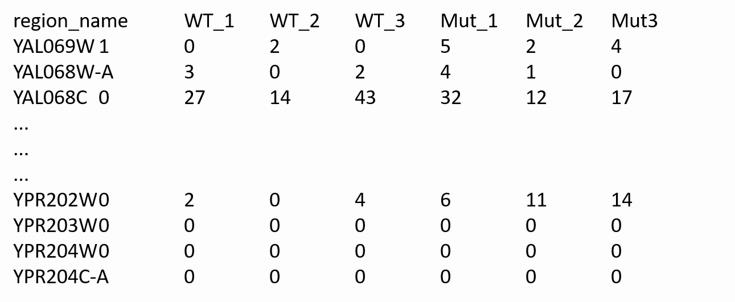
Figure 3. Example of a counts.txt-file in tab-delimited format. The first column designates the names of open reading frames (ORFs), and the first row indicates the names of the wild-type and mutant replicates. The numerical matrix contains the number of reads mapped in each replicate to the respective ORF.Generate a “table.txt”-file for each sample, indexing each column of data (Figure 4) in tab-delimited format.
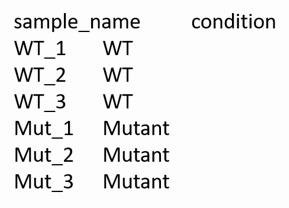
Figure 4. Example of a table.txt-file in tab-delimited format. Replicate names, as designated in counts.txt from Step D3, are indexed by their common condition (e.g., wild type or mutant).In R, load the Dseq2 library, the combined counts-file from Step D3, and the table-file from Step D4.
library(DESeq2)
count_table <- read.delim(‘counts.txt’,sep=’\t’,header=TRUE,row.names=’region_name’)
sample_table <- read.delim(‘table.txt’,sep=’\t’,header=TRUE,row.names=’sample_name’)
Write the RNA-seq expression and p-values to file using DESeq2. The generated .txt file (wt_mutant_p-values.txt) contains the log2-fold expression and p-values for the respective mutant in tab-delimited format and can now be used for further analysis or visualization. For data inspection, an MA-plot is generated (Figure 5). In MA-plots generated by DESeq2, significant hits are colored in red; hence, the first quality check is how many data points are colored in black (i.e., since most genes are not differentially expressed, most data points should be colored in black).
dds <- DESeqDataSetFromMatrix(countData = count_table,colData = sample_table,design = ~ condition)
dds <- DESeq(dds)
res <- results(dds)
resOrdered <- res[order(res$padj),]
plot <- plotMA(res, main = ‘mutant‘, ylim = c(-2,2), xlab = ‘mean count‘)
write.table(as.data.frame(resOrdered),sep=‘\t‘,quote=FALSE,file=‘wt_mutant_p-values.txt‘)
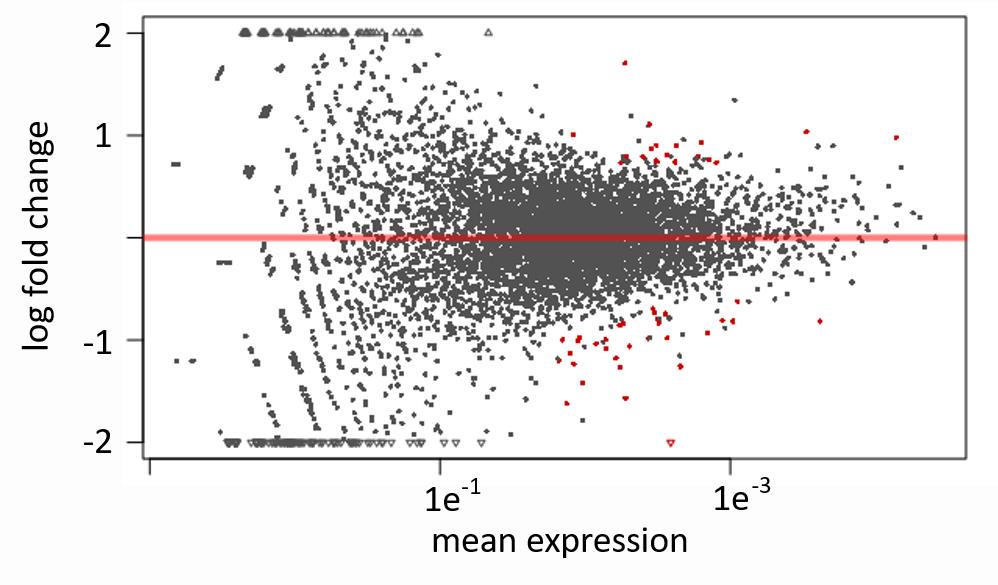
Figure 5. Example of MA-plot analysis as generated by DESeq2. Genes that are statistically significantly up- or downregulated are marked in red above and below the x-axis, respectively.
Recipes
Yeast Extract Peptone Dextrose (YEPD) media
For each liter of YEPD, autoclave a mixture of 20 g Bacto Peptone, 10 g yeast extract, and 950 ml H2O. Add 50 ml 40% (w/v) glucose, mix and cool before use.
For YEPD plates, add 24 g agar to the solution before autoclaving. Place the autoclaved solution on a magnetic stir plate, add a stir bar and 50 ml 40% (w/v) glucose, and cool the solution while stirring. Pour warm media into Petri dishes, allow to cool until solid, and store at 4°C until use.
1 M Tris-HCl solution, pH 7.5
Dissolve 121.14 g Tris in 800 ml H2O.
Adjust the pH to 7.5 with HCl.
Bring the final volume to 1 L with deionized H2O.
Autoclave and store at room temperature.
0.5 M EDTA solution, pH 8.0
Add 18.6 g EDTA to 80 ml H2O (use DEPC-treated H2O).
Mix on a magnetic stirrer until dissolved.
Adjust the pH to 8.0 with NaOH (~2 g NaOH pellets).
Dispense into aliquots and sterilize by autoclaving.
20% SDS solution
Dissolve 20 g SDS in 90 ml H2O (use DEPC-treated H2O).
Heat to 68°C and mix with a magnetic stirrer until dissolved.
Tris-EDTA-SDS (TES) solution
10 mM Tris-HCl pH 7.5
10 mM EDTA pH 8.0
0.5% SDS
3 M NaAc solution, pH 5.2
Add 24.6 g sodium acetate to 80 ml H2O.
Mix on a magnetic stirrer until dissolved.
Adjust the pH to 5.2 with glacial acetic acid.
Bring the volume to 100 ml with H2O.
Acknowledgments
I would like to thank Dr. Pavel Sinitcyn, Dr. Assa Yeroslaviz, and Dr. Rin Ho Kim from the Next-Generation Sequencing Core Facility at MPI Biochemistry for critical reading of the manuscript.
This protocol is based on the RNA-seq expression analysis performed in Braberg et al. (2020).
References
- Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data.
- Andrews, S., Ply, P. T. and Huber, W. (2014). HTSeq – A Python framework to work with high-throughput sequencing data. bioRxiv. doi: https://doi.org/10.1101/002824.
- Bainbridge, M. N., Warren, R. L., Hirst, M., Romanuik, T., Zeng, T., Go, A., Delaney, A., Griffith, M., Hickenbotham, M., Magrini, V., Mardis, E. R., Sadar, M. D., Siddiqui, A. S., Marra, M. A. and Jones, S. J. (2006). Analysis of the prostate cancer cell line LNCaP transcriptome using a sequencing-by-synthesis approach. BMC Genomics 7: 246.
- Batut, B. (2021). Quality Control (Galaxy Training Materials).
- Bolger, A. M., Lohse, M. and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15): 2114-2120.
- Braberg, H., Echeverria, I., Bohn, S., Cimermancic, P., Shiver, A., Alexander, R., Xu, J., Shales, M., Dronamraju, R., Jiang, S., Dwivedi, G., Bogdanoff, D., Chaung, K. K., Huttenhain, R., Wang, S., Mavor, D., Pellarin, R., Schneidman, D., Bader, J. S., Fraser, J. S., Morris, J., Haber, J. E., Strahl, B. D., Gross, C. A., Dai, J., Boeke, J. D., Sali, A. and Krogan, N. J. (2020). Genetic interaction mapping informs integrative structure determination of protein complexes. Science 370(6522).
- Cheung, F., Haas, B. J., Goldberg, S. M., May, G. D., Xiao, Y. and Town, C. D. (2006). Sequencing Medicago truncatula expressed sequenced tags using 454 Life Sciences technology. BMC Genomics 7: 272.
- Collart, M. A. and Oliviero, S. (2001). Preparation of yeast RNA. Curr Protoc Mol Biol Chapter 13: Unit13 12.
- Dillies, M. A., Rau, A., Aubert, J., Hennequet-Antier, C., Jeanmougin, M., Servant, N., Keime, C., Marot, G., Castel, D., Estelle, J., Guernec, G., Jagla, B., Jouneau, L., Laloe, D., Le Gall, C., Schaeffer, B., Le Crom, S., Guedj, M., Jaffrezic, F. and French StatOmique, C. (2013). A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform 14(6): 671-683.
- Grüning, B., Dale, R., Sjodin, A., Chapman, B. A., Rowe, J., Tomkins-Tinch, C. H., Valieris, R., Koster, J. and Bioconda, T. (2018). Bioconda: sustainable and comprehensive software distribution for the life sciences.Nat Methods 15(7): 475-476.
- Emrich, S. J., Barbazuk, W. B., Li, L. and Schnable, P. S. (2007). Gene discovery and annotation using LCM-454 transcriptome sequencing.Genome Res 17(1): 69-73.
- Haque, A., Engel, J., Teichmann, S. A. and Lonnberg, T. (2017). A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med 9(1): 75.
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4): 357-359.
- Langmead, B., Trapnell, C., Pop, M. and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome.Genome Biol 10(3): R25.
- Li, H. and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14): 1754-1760.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16): 2078-2079.
- Logsdon, G. A., Vollger, M. R. and Eichler, E. E. (2020). Long-read human genome sequencing and its applications. Nat Rev Genet 21(10): 597-614.
- Love, M. I., Huber, W. and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2.Genome Biol 15(12): 550.
- R Core Team. (2017). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
- Robinson, J. T., Thorvaldsdottir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G. and Mesirov, J. P. (2011). Integrative genomics viewer. Nat Biotechnol 29(1): 24-26.
- Nagalakshmi, U., Wang, Z., Waern, K., Shou, C., Raha, D., Gerstein, M. and Snyder, M. (2008). The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320(5881): 1344-1349.
- Weber, A. P., Weber, K. L., Carr, K., Wilkerson, C. and Ohlrogge, J. B. (2007). Sampling the Arabidopsis transcriptome with massively parallel pyrosequencing. Plant Physiol 144(1): 32-42.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Bohn, S. (2021). Protocol for RNA-seq Expression Analysis in Yeast. Bio-protocol 11(18): e4161. DOI: 10.21769/BioProtoc.4161.
- Braberg, H., Echeverria, I., Bohn, S., Cimermancic, P., Shiver, A., Alexander, R., Xu, J., Shales, M., Dronamraju, R., Jiang, S., Dwivedi, G., Bogdanoff, D., Chaung, K. K., Huttenhain, R., Wang, S., Mavor, D., Pellarin, R., Schneidman, D., Bader, J. S., Fraser, J. S., Morris, J., Haber, J. E., Strahl, B. D., Gross, C. A., Dai, J., Boeke, J. D., Sali, A. and Krogan, N. J. (2020). Genetic interaction mapping informs integrative structure determination of protein complexes. Science 370(6522).
Category
Systems Biology > Genomics > Sequencing
Microbiology > Microbial biochemistry > RNA
Molecular Biology > RNA > RNA detection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.