- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Methods for the Extraction of Heme Prosthetic Groups from Hemoproteins
(*contributed equally to this work) Published: Vol 11, Iss 18, Sep 20, 2021 DOI: 10.21769/BioProtoc.4156 Views: 4951
Reviewed by: Laxmi Narayan MishraAmit KumarAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2020
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Hemoproteins are widely researched because they contain redox-active heme prosthetic groups (iron + protoporphyrin IX) that enable them to perform a range of vital functions, acting as enzymes, participants in electron transfer reactions, or gas sensing, carrying, and storage proteins. While the heme prosthetic group is almost always essential for hemoprotein function, it is frequently desirable to remove it from the protein to enable biochemical or protein engineering studies. Obtaining high yields of the apo form of the hemoprotein can be challenging since high heme-protein binding affinities necessitate the use of harsh conditions to remove heme. In this Bio-Protocol, we present three chemical extraction methods that can be used to efficiently remove heme: methyl ethyl ketone extraction, acid-acetone precipitation, and on-column heme extraction. We also present protocols that can be used to quantitate the amount of residual heme bound to the protein after performing the extraction procedures.
Keywords: HemoproteinBackground
Many proteins in biology contain heme prosthetic groups with redox properties that enable them to participate in a myriad of cellular processes (Huang and Groves, 2018). Hemoproteins have a range of functions, including among others, acting as enzymes that mediate cellular metabolism (e.g., oxidases, catalases, peroxidases), as participants in electron transfer reactions, and as oxygen sensing, carrying, and storage proteins (e.g., hemoglobin, myoglobin) (Poulos, 2014). Moreover, because heme contains the essential micronutrient iron, many microbial pathogens express hemoproteins that capture iron-laden heme from their hosts during infections (Runyen-Janecky, 2013; Choby and Skaar, 2016). There are four major types of heme (A, B, S, and O), which differ in their substituent pattern around the porphyrin ring. The most common type is heme B, which consists of protoporphyrin IX and a central iron atom (Reedy et al., 2008). Heme B, hereafter referred to simply as heme, is lipophilic and tightly bound by hemoproteins that donate axial ligands to coordinate the iron atom (Baker et al., 2003). It is often desirable to remove heme from a hemoprotein, as it enables study of the apo (heme-free)-protein and provides a route toward the introduction of non-native metalloporphyrins that confer novel spectroscopic or enzymatic properties to the resulting artificial metalloprotein (Fruk et al., 2009). However, removing heme effectively is quite challenging, as it can be tightly bound within the heme pocket of the protein, generally precluding its removal through simple dialysis approaches.
Several protocols have been reported to extract heme from a hemoprotein: (1) methyl ethyl ketone (MEK) extraction (Teale, 1959); (2) acid-acetone precipitation (Rossi-Fanelli et al., 1958); and (3) prosthetic group transfer to a high-affinity apo-hemoprotein (Chien et al., 2017). Acid-acetone precipitation was the first of these methods to be implemented and requires the use of acidified acetone to disrupt heme–protein interactions and precipitate the apo form of the protein. The protein is recovered by redissolving the precipitate in buffer or water, but the harsh conditions utilized in the reaction often result in loss of protein due to poor resolubilization of the apo-protein (Ascoli et al., 1981). In the MEK extraction method, the affinity between heme and the hemoprotein is also weakened by sample acidification. In this procedure, the apo-protein and free heme partition into distinct liquid phases, enabling their separation. MEK extraction is the most widely used of the chemical extraction methods (Sjodt et al., 2018; Ellis-Guardiola et al., 2020) and in our experience, results in more thorough heme-stripping and higher apo-protein recovery than acid-acetone precipitation. For some hemoproteins, MEK or acid-acetone chemical extraction of heme leads to low recovery yields of the apo-protein, presumably because the apo-protein aggregates or does not refold properly. To circumvent these problems, apo-myoglobin can be used to remove heme molecules from the hemoprotein (Muller-Eberhard et al., 1969; Chien et al., 2017). This method enables heme extraction without protein denaturation, but it can be cumbersome to use in practice because it often requires large quantities of the apo-myoglobin reagent, and additional procedures are needed to physically separate myoglobin from the apo-protein after heme has been removed. As an alternative to these methods, our lab has implemented an on-column heme removal protocol that allows for concomitant heme removal and protein purification by immobilized metal affinity chromatography (IMAC). Histidine-tagged hemoproteins are denatured while bound to a cobalt- or nickel-nitrilotriacetic acid (NTA) resin, and the heme molecules are stripped using a solution of ethanol and chaotrope. Finally, the apo-protein is refolded and eluted from the column, producing purified apo-protein in a single step.
Herein, we present three protocols that can be used to chemically extract heme from a hemoprotein: MEK extraction, acid-acetone precipitation, and on-column extraction. These methods have proven effective for heme removal from a wide range of hemoproteins in our laboratory. We caution the reader to test apo-proteins obtained through these procedures to ensure that their native activity and fold are restored upon reconstitution. Although these methods have only been tested on heme B-containing hemoproteins (protoporphyrin IX + iron), we expect them to be applicable to removing other types of non-covalently bound heme molecules, as well as metalloporphyrin molecules in general.
Materials and Reagents
Note: Other brands of these materials may be used if necessary.
Methyl ethyl ketone extraction
SnakeSkinTM dialysis tubing, 3.5K MWCO, 35 mm (Thermo Fisher ScientificTM, Thermo ScientificTM, catalog number: 88244)
SpectrumTM dialysis tubing closures: Spectra/PorTM Standard Type (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: 08-750-24C)
Amicon® Ultra-15 centrifugal filter unit (Millipore Sigma, catalog number: UFC900308)
HPLC vials with conical inserts (Thermo Fisher ScientificTM catalog number: 03-375-17CA)
Glass separatory funnel
15 ml FalconTM tubes (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: 05-527-90)
50 ml FalconTM tubes (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: 14-432-22)
9” Glass Pasteur pipets, FisherbrandTM (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: 13-678-20C)
Pipet bulbs, FisherbrandTM (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: 03-448-21)
Eppendorf® UVette® (Millipore Sigma, catalog number: Z618683)
Tris hydrochloride (Millipore Sigma, catalog number: 10812846001)
Methyl ethyl ketone, HPLC Grade. J.T.BakerTM (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: 02-003-445)
12 N HCl (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: A144-500)
Trifluoroacetic acid, OptimaTM LC/MS Grade, Fisher ChemicalTM (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: A116-05AMP)
Acetonitrile OptimaTM, for HPLC and GC, Fisher ChemicalTM (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: A996-4)
MEK extraction buffer (see Recipes)
Recovery buffer (see Recipes)
Acid-acetone precipitation
FisherbrandTM Premium microcentrifuge tubes: 1.5 ml (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: 05-408-129)
Slide-A-LyzerTM MINI dialysis device, 10 K MWCO, 2.0 ml (Thermo Fisher ScientificTM, catalog number: 88404)
Eppendorf® UVette® (Millipore Sigma, catalog number: Z618683)
Amicon® Ultra-15 centrifugal filter unit (Millipore Sigma, catalog number: UFC900308)
Acetone (HPLC), Fisher ChemicalTM (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: A949-4)
12 N HCl (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: A144-500)
Sodium phosphate monobasic dihydrate (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: S381-500)
Sodium chloride, non-iodized, granular, FCC, 99-100.5%, SpectrumTM (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: 18-606-420)
Imidazole, 99%, ACROS OrganicsTM (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: AC122025000)
Alfa AesarTM guanidine hydrochloride, 98% (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: AAA1354301)
Trifluoroacetic acid, OptimaTM LC/MS Grade, Fisher ChemicalTM (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: A116-05AMP)
Acetonitrile OptimaTM, for HPLC and GC, Fisher ChemicalTM (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: A996-4)
Acid acetone (see Recipes)
Denaturing buffer (see Recipes)
On-column heme extraction
Thermo ScientificTM NalgeneTM Rapid-FlowTM sterile single-use vacuum filter unit (Thermo ScientificTM, catalog number: 09-740-28C)
Test tubes
DWK Life Sciences KimbleTM KontesTM FlexColumnTM economy column (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: K420400-2520)
HPLC vials with conical inserts (Thermo Fisher ScientificTM catalog number: 03-375-17CA)
Parafilm® M (Millipore Sigma, catalog number: P7793)
Beakers
Eppendorf® UVette® (Millipore Sigma, catalog number: Z618683)
HisPurTM cobalt resin (Thermo Fisher ScientificTM, Thermo ScientificTM, catalog number: 89966)
HisPurTM nickel Ni-NTA resin (Thermo Fisher ScientificTM, Thermo ScientificTM, catalog number: 88221)
Sodium phosphate monobasic dihydrate (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: S381-500)
Sodium chloride, non-iodized, granular, FCC, 99–100.5%, SpectrumTM (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: 18-606-420)
Imidazole, 99%, ACROS OrganicsTM (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: AC122025000)
Ethyl alcohol, 200 proof, for molecular biology (Millipore Sigma, catalog number: E7023-4L)
Trifluoroacetic acid, OptimaTM LC/MS Grade, Fisher ChemicalTM (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: A116-05AMP)
Acetonitrile OptimaTM, for HPLC and GC, Fisher ChemicalTM (Thermo Fisher ScientificTM, Fisher ScientificTM, catalog number: A996-4)
Equilibration buffer (see Recipes)
Histidine-tagged Ulp1 SUMO protease solution (see Recipes)
Denaturing buffer (see Recipes)
Heme wash buffer (see Recipes)
High imidazole buffer (see Recipes)
Equipment
General equipment
Clay Adams Nutator Mixer 1105 (Cambridge Scientific, catalog number: 17847)
Uvette® adapter; included in the UVette® starter set (Eppendorf, catalog number 952010077)
Spectrophotometer (Shimadzu, model: UV-1800)
HPLC (AgilentTM 1100 Series equipped with a G1315B Diode Array UV/Vis Detector)
C4 reversed-phase HPLC column (WatersTM, Symmetry300TM C4 5 μm 4.6 × 150 mm, catalog number: 186000288)
Methyl ethyl ketone extraction
Centrifuge (Beckman Coulter, model: Allegra X-14R)
SX4750A swinging bucket rotor (Beckman Coulter, model: SX4750A ARIESTM)
pH meter (Thermo Fisher ScientificTM, Thermo ScientificTM Orion Star A111)
Acid-acetone precipitation
Microcentrifuge (EppendorfTM, model: 5424 R)
Microcentrifuge rotor (EppendorfTM, catalog number: FA-45-24-11)
Software
GraphPad Prism 9
Procedure
Heme removal by methyl ethyl ketone (MEK) extraction
This protocol works best for relatively concentrated protein samples (~0.1-1 mM on a heme basis) and when the sample volume is at least 1 ml. The latter requirement enables facile separation of the organic and aqueous solution phases that are generated during the extraction procedure. The flow chart in Figure 1 describes the removal of heme from hemoproteins by methyl ethyl ketone (MEK) extraction. Purified protein is extracted with MEK, leading to phase separation of the organic, heme-containing layer and the aqueous, protein-containing layer. The aqueous layer is collected and the protein is rescued into recovery buffer.
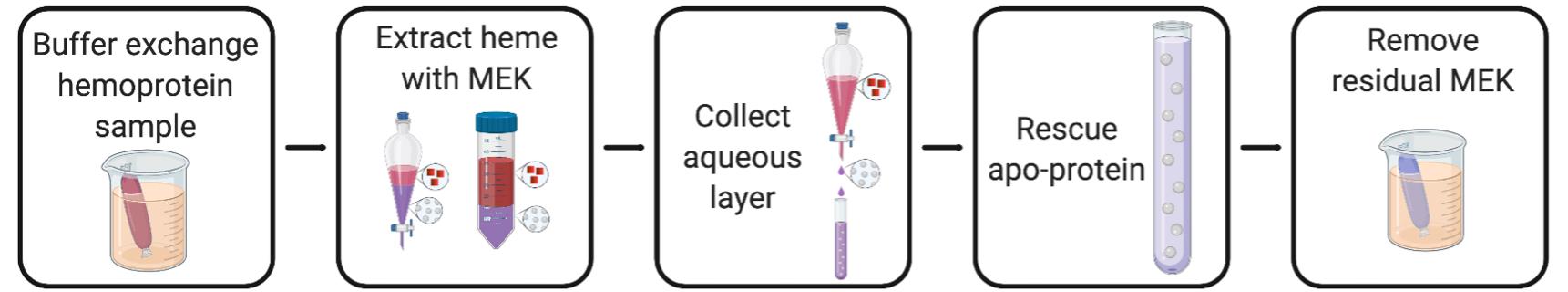
Figure 1. Methyl ethyl ketone extraction. Hemoprotein is buffer-exchanged and transferred to a separatory funnel or 50-ml conical. The sample is extracted with MEK, which forms a heme-containing (red squares) organic layer above the aqueous layer (purple) containing apo-protein (grey spheres) after a short incubation period. The aqueous layer is collected, by opening the separatory funnel or by using a pipet to transfer the lower layer from the 50-mL conical, and rescued into recovery buffer. Residual MEK is removed by buffer-exchanging the sample. The figure was created using BioRender.com.Exchange the hemoprotein solution into MEK extraction buffer and adjust the concentration to 1-10 mg/ml by either diluting the sample in MEK extraction buffer or buffer-exchanging the sample via centrifugal concentration (Amicon® Ultra-15 centrifugal filter unit) or dialysis (SnakeSkinTM dialysis tubing). A practical final volume is 5-10 ml.
Pre-chill 15 ml MEK per 2 L expression culture (or approximately 1 ml MEK per 2 mg protein) and a ~3-fold sample volume of recovery buffer separately on ice.
Dilute 12 N HCl to a final concentration of 2 M HCl in nanopure water.
Adjust the pH of the chilled hemoprotein solution to 2.25-2.45 with 2 M HCl. Monitor the change in pH using a pH meter.
Combine ice-cold MEK and hemoprotein solution at a 1:1 ratio in a glass separatory funnel or 15–50 ml Falcon tube. Mix gently and thoroughly to maximize the heme extraction efficiency.
Allow approximately 15 min for the resulting emulsion to fully undergo phase separation. The upper MEK layer should have a red-pink color due to the presence of solvated heme.
If using a separatory funnel, collect the lower aqueous layer containing the apo-protein by careful elution. If using a Falcon tube, insert a glass Pasteur pipet to the bottom of the tube and carefully pipet the aqueous layer into a fresh tube. Additional extraction and wash steps may be required for certain samples.
Transfer the aqueous protein solution to ice-cold recovery buffer at a 1:1 ratio by volume, and incubate the sample on ice for approximately 10 min to allow the protein of interest to refold.
Buffer-exchange the protein by dialysis (SnakeSkinTM dialysis tubing) against fresh recovery buffer to remove the excess dissolved MEK.
Concentrate and buffer-exchange the protein in its final storage/assay buffer or an appropriate buffer for additional chromatography steps.
Analyze the efficacy of heme extraction as described in the Data Analysis section. Example absorbance spectra and UV/Visible HPLC chromatograms of the Staphylococcus aureus hemoprotein IsdH (iron surface determinant H, residues 326–660) extracted by MEK are shown in Figure 5.
Acid-acetone precipitation
This extraction method gives the best apo-protein recovery when working with a high concentration (~1 mM on a heme basis) of protein and low volumes. This protocol is specifically designed for volumes ≤ 1.5 ml. The flow chart in Figure 2 depicts the workflow of heme extraction by acid-acetone precipitation. The hemoprotein is precipitated in cold acid-acetone and the resulting apo-protein is pelleted, washed with additional acid-acetone, lyophilized to completely remove the solvent, and re-dissolved.

Figure 2. Acid-acetone precipitation. The protein of interest is incubated with cold acid-acetone to precipitate the protein. The resulting apo-protein-containing solid is pelleted, and the heme-containing supernatant is removed. The pellet is lyophilized and re-dissolved in a buffer of choice. The sample is buffer-exchanged to remove any residual acetone. Figure created using BioRender.com.Pre-chill the acid-acetone solution to -20°C and the microcentrifuge rotor to -10°C.
Aliquot 100-µl fractions of purified hemoprotein into 1.5-ml FisherbrandTM Premium microcentrifuge tubes and incubate on ice. Save 15 µl holo-protein for comparison of the heme content before and after acid-acetone precipitation.
Add 1 ml acid-acetone to each 100-µl aliquot of hemoprotein and incubate at -20°C for 20 min. Protein precipitate should be visible after incubation.
Centrifuge the hemoprotein/acid-acetone mixture in a microcentrifuge at 21,000 × g (rcf) for 20 min to pellet the precipitated protein, and then decant or pipet off the supernatant (see Note 8).
Resuspend the pellet in an additional 1 ml acid-acetone and repeat Steps B3 and B4.
Air-dry the protein pellets at room temperature for 30 min.
Re-dissolve the protein in 100 µl buffer of choice. If the protein does not re-dissolve into solution, proceed with Steps B8-B9. If the protein readily dissolves, proceed to Step B10.
Titrate denaturing buffer into the protein pellet suspension until the precipitate is no longer visible.
Dialyze the denatured apo-protein against the buffer of choice at 4°C for a minimum of 4 h using a Slide-A-LyzerTM MINI dialysis device, 3.5 K MWCO, 0.5 ml.
Buffer-exchange the sample into the buffer of choice by centrifugal filtration using an Amicon® Ultra-15 centrifugal filter unit or dialyze against the buffer of choice for a minimum of 4 h to remove excess acetone using a Slide-A-LyzerTM MINI dialysis device, 3.5 K MWCO, 2.0 ml.
Analyze the heme content of the hemoprotein before and after extraction, as described in the Data Analysis section. Example absorbance spectra and HPLC chromatograms of IsdH extracted by acid-acetone precipitation are shown in Figure 5.
On-column heme removal for histidine-tagged hemoproteins
This protocol is highly versatile since it can be performed on purified histidine-tagged hemoproteins or integrated into the IMAC purification of crude, recombinantly expressed histidine-tagged hemoproteins in clarified Esherichia coli BL21 (DE3) lysate. In favorable cases where full protein unfolding is achieved using chaotropic reagents, this method is the easiest to perform, requiring the fewest discrete steps for heme removal with the highest yields of refolded apo-protein (Table 1). Figure 3 depicts a scheme of the steps necessary to remove heme from histidine-tagged hemoproteins concomitant with purification by immobilized metal affinity chromatography (IMAC). The column is equilibrated, and hexahistidine-tagged proteins adhere to the resin, allowing proteins lacking a histidine tag to be separated from the protein of interest. The hemoprotein is denatured while still bound to the column, disrupting the secondary and tertiary structural features that form the heme-protein interface, thus freeing heme molecules to be stripped from the protein of interest by a binary mixture of chaotrope and ethanol. Finally, the apo-protein is re-folded on the column and eluted.
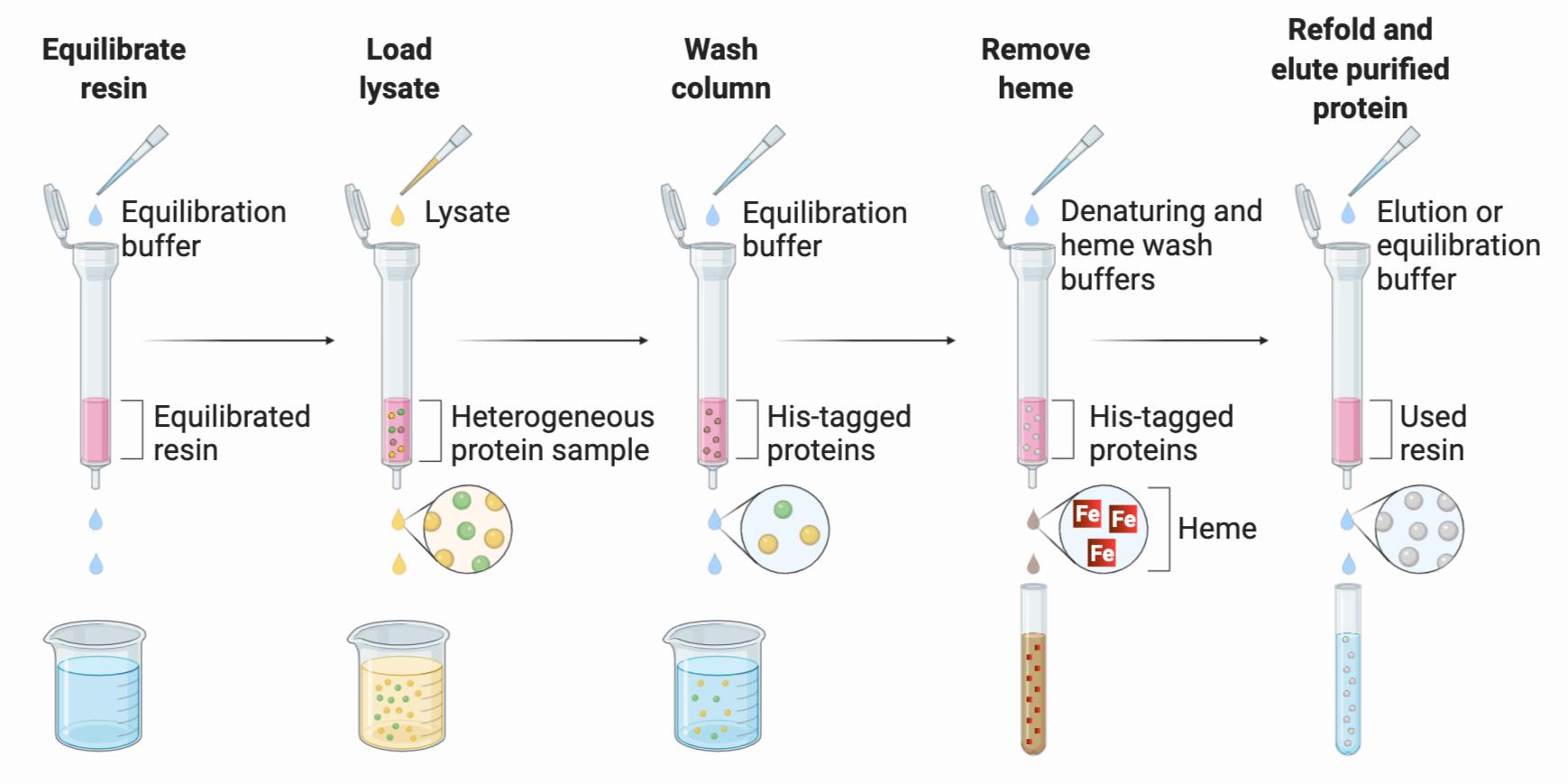
Figure 3. Immobilized metal affinity chromatography and heme removal. Separation of His-tagged hemoproteins (red spheres) from a heterogenous protein sample (brown, yellow, and green spheres) is depicted. Upon heme (red squares) removal, a light brown solution elutes from the column, and the apo-proteins (grey spheres) are re-folded and eluted to obtain a homogenous sample. The figure was created using BioRender.com.Filter all buffers through a Thermo ScientificTM NalgeneTM Rapid-FlowTM sterile single-use vacuum filter unit with a pore size of 0.2 µm; chill to 4°C.
Transfer 2.5 ml HisPurTM cobalt resin or HisPurTM nickel resin per liter cell culture to a DWK Life Sciences KimbleTM KontesTM FlexColumnTM economy column or, if the target protein concentration is known, transfer the minimum amount of resin necessary to fully bind the protein.
Equilibrate the resin with a minimum of 10 column volumes (CV) equilibration buffer.
Transfer the clarified cell lysate containing the protein of interest to the gravity column.
Cap the column and seal it with Parafilm® M to ensure that no leaks occur. Equilibrate the resin and cell lysate at 4°C on a Clay Adams nutator mixer 1105 for a minimum of 15 min to maximize binding of the histidine-tagged hemoprotein to the resin.
Bring the column back to a standing position and collect the lysate that flows through the column in a beaker or test tubes.
Wash the resin with 10 CV equilibration buffer.
To prepare a no-extraction negative control (holo-protein) for determination of the efficacy of heme extraction, divert a portion (1-2 ml) of the resin-bound hemoprotein and skip to Step C12.
Suspend the resin in 10 CV denaturing buffer to denature the protein, and allow a 5-min incubation time to ensure re-binding of any protein dislodged through the introduction of denaturing buffer (see Note 9).
Elute the denaturing buffer. This should be slightly brown in color due to the presence of solvated heme. Record the absorbance of the heme Soret peak using a spectrophotometer. An example absorbance spectrum of the eluate from a 5-ml Ni-NTA column preparation of IsdH is shown (“initial denaturation,” Figure 4).
Wash the resin with 4 × 10 CV heme wash buffer and monitor the eluate of these steps for heme content by recording the absorbance of the heme Soret peak using a spectrophotometer. Repeat the wash steps until the heme content of the eluate is minimized (see Note 10). Example absorbance spectra of the eluted heme washes from a 5-ml preparation of IsdH are shown (“heme wash 1-4”, Figure 4).
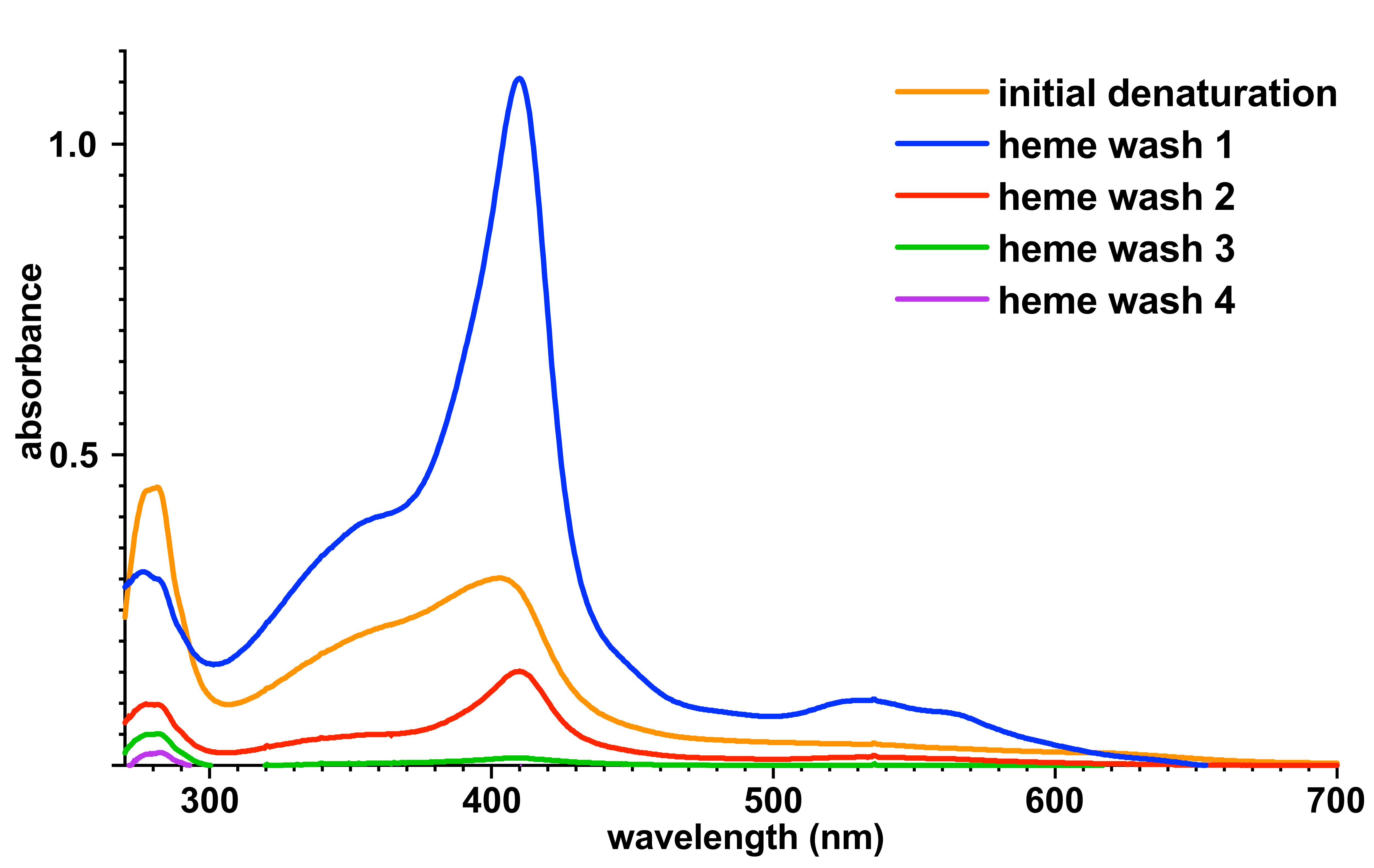
Figure 4. Example UV/Vis spectroscopic analysis of eluted heme fractions. His-tagged IsdH from a 2-L expression culture was bound to 5 ml Ni-NTA resin and on-column heme removal was performed. Initial denaturation was performed with 50 ml denaturing buffer, and heme washes were performed with 4 × 50 ml heme wash buffer. The UV/Vis spectrum for each heme removal elution is shown, with progressively less heme (λSoret = 385-405 nm).Re-fold the bound protein by resuspending the resin in 10 CV equilibration buffer and incubating the slurry for 5 min.
Elute the equilibration buffer and wash the resin with an additional 10 CV equilibration buffer.
Elute any protein still bound to the resin by washing with 2 × 5 CV high imidazole buffer and check the eluate for protein content by recording the A280. Transfer ≥ 50 µl sample into an Eppendorf® UVette® within a UVette® adapter and record the absorbance at 280 nm using a spectrophotometer.
Combine the desired eluted protein fractions and check the heme content of the eluted protein using the methods described in the Data Analysis section. Example absorbance spectra and HPLC chromatograms of apo-IsdH prepared by on-column extraction are shown in Figure 5.
Concentrate and buffer-exchange the protein into its final storage/assay buffer or an appropriate buffer for additional chromatography steps.
Data analysis
Evaluation of the heme removal efficacy by UV/Vis
Transfer ≥ 50 µl apo-protein to a UVette.
Using a UVette adaptor, measure the sample absorbance at A280 and ASoret in a spectrophotometer. Sample absorbance should not exceed 1.5 AU at either wavelength. Dilute the sample with buffer if this threshold is exceeded.
Interpretation of UV/Vis data.
If the molar extinction coefficients for heme absorbance at the specific protein-bound heme Soret (specific to the protein bound to heme, εSoret in M-1cm-1) and the protein backbone (determined experimentally or calculated from the protein primary sequence through ε280 computation) are known:
Quantitate the total heme content by ASoret using Beer’s Law and the known molar extinction coefficient of the protein-bound heme Soret (see Note 11). b corresponds to the pathlength of the cuvette (in cm):
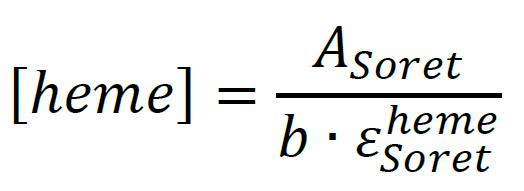
Quantitate the total protein content by A280 using Beer’s Law and the theoretical or experimentally determined molar extinction coefficient of the protein of interest. This equation accounts for the absorbance contribution of heme at 280 nm using the calculated concentration from the step above (see Note 12):
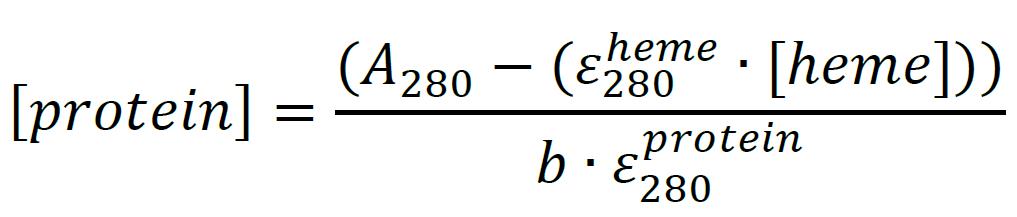
Calculate the absolute heme-loading by comparing the heme content before and after extraction (Figure 5A, Table 1):
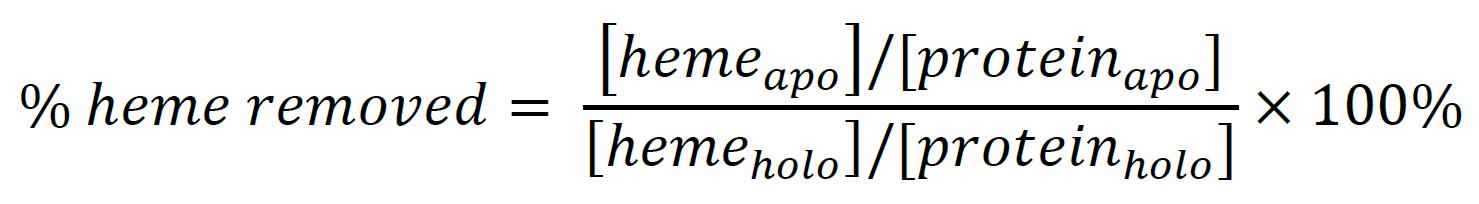
If the extinction coefficients for the protein-bound heme and protein scaffold are unknown, the relative efficiency of heme removal can be approximated by comparison of the apo-protein Soret absorbance with that of the corresponding holo-protein (Figure 5A).
Measure ASoret for both the holo-protein and the apo-protein at the Soret absorbance maximum wavelength λSoret.
Measure A280 for both the holo-protein and the apo-protein to normalize the Soret absorbance:
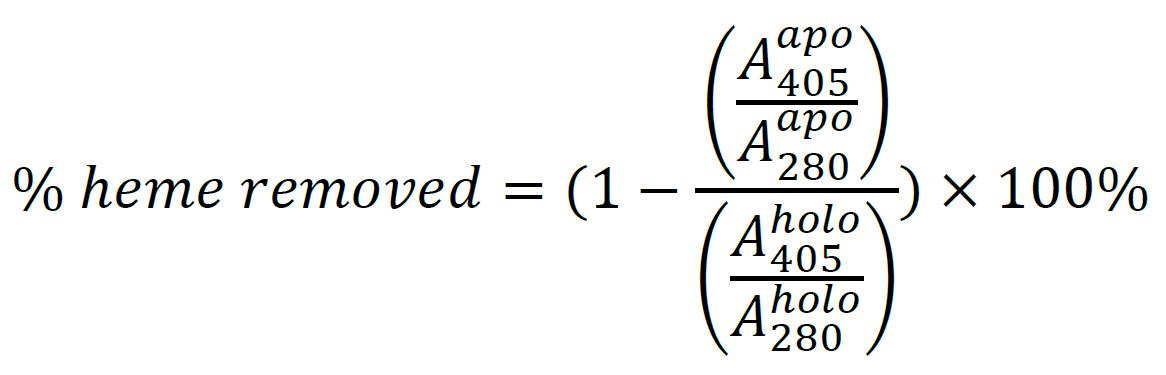
Evaluation of heme removal by HPLC
Quantitate the protein concentration of HPLC samples by A280 using Beer’s Law and the theoretical or experimentally determined millimolar extinction coefficient of the protein of interest.
Dilute the sample to 5-500 µM in the buffer of choice and transfer 15 µl to an HPLC vial.
Set the diode array detector to monitor the absorbance at 280 nm for protein detection and at the heme Soret wavelength (385-405 nm for free heme) for heme detection. Additional wavelengths can be added if desired. See Notes 13 and 14.
Attach a C4 reversed-phase HPLC column to the HPLC and equilibrate by flowing 12 ml 65:35 Buffer A:Buffer B (Buffer A = nanopure H2O with 0.1% trifluoroacetic acid (TFA)); Buffer B = acetonitrile with 0.1% TFA) through the column at 1 ml/min.
Inject 10 µl sample onto the C4 column and analyze using a method optimized for your protein. For IsdH analysis (Figure 5B), samples were analyzed by gradient elution at 1.0 ml/min with a gradient program (0-1 min, 35-35% B; 1-30 min, 35-47.3% B; 30-35 min, 47.3-100% B; 35-36 min, 100–100% B). This method was adapted from Samuel et al. (2017).
Determine the relative extent of heme removal by comparison of the 405 nm chromatograms for the apo- and holo- forms of the protein (Figure 5B, Table 1). Integrate the chromatograms and determine the heme removal efficacy using the following equation (see Note 15):
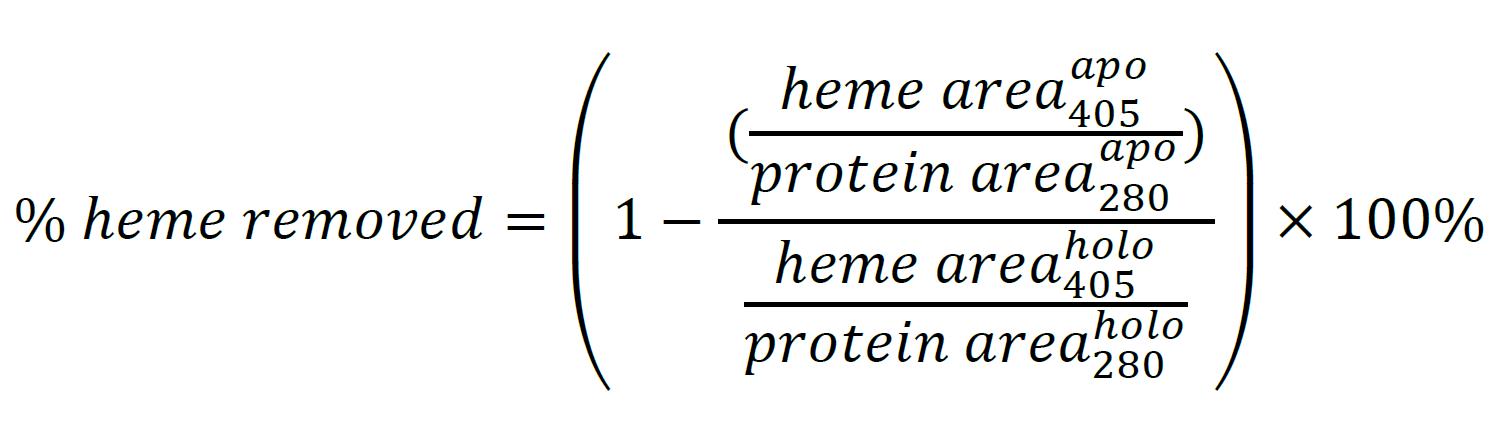
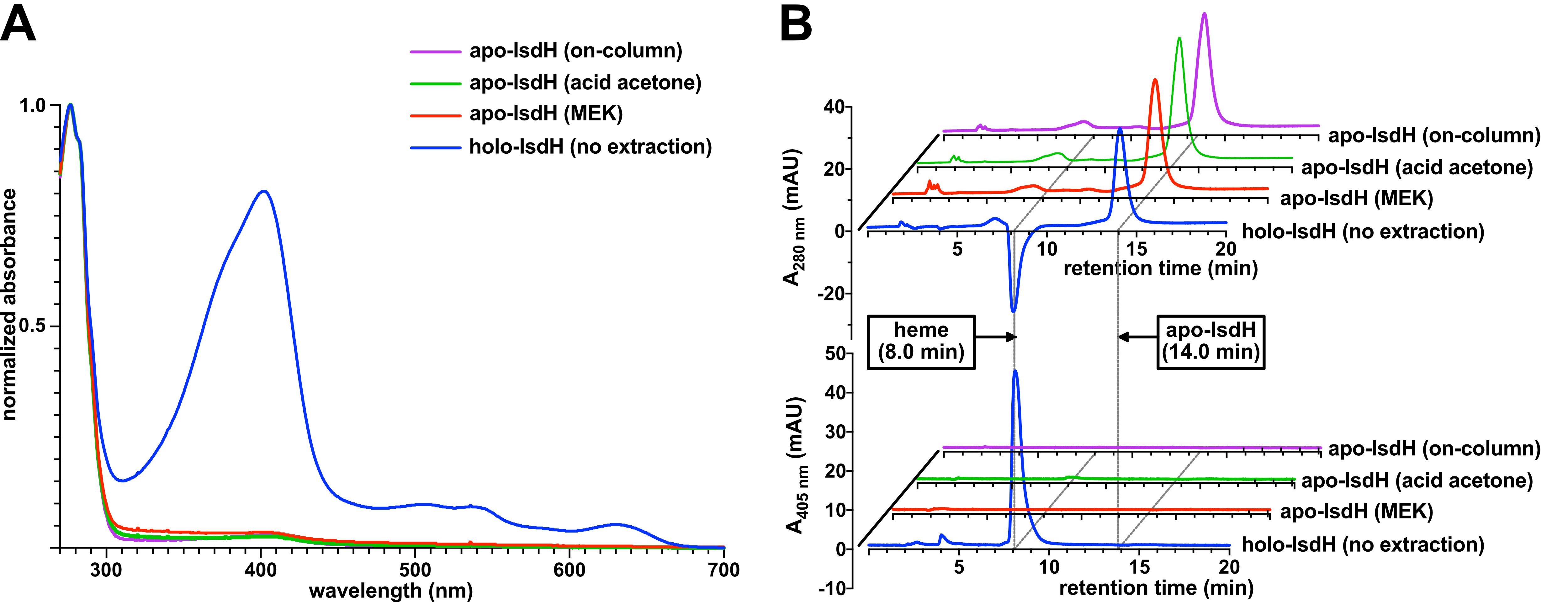
Figure 5. Measurement of the heme content. A. Evaluation of the heme removal efficacy by UV/Vis. Normalized absorbance spectra for holo-IsdH (no extraction) and apo-IsdH (prepared using each protocol) are shown, revealing similar extents of heme removal for each protocol as determined by comparing A405/A280. B. Evaluation of heme removal efficacy using HPLC. HPLC absorbance chromatograms at 280 nm (top) are used to assess the protein content and those at 405 nm (bottom) are used to determine the heme content of samples. We observe a negative A280 peak associated with heme. The retention time of heme is similar to that reported in Samuel et al. (2017).Table 1. Comparison of heme extraction methods to prepare apo-IsdH
1Yields and % heme removal were determined for 2 L-scale E. coli BL21(DE3) culture preparations in Luria-Bertani broth supplemented with 50 µg/ml kanamycin sulfate and 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG).MEK Acid-acetone on-column apo-IsdH yield1 36 mg 38 mg 42 mg % heme removed1 (UV/Vis) 96% 97% 97% % heme removed1
(HPLC)>99% 90% >99%
Validation of the apo-protein integrity through activity measurements
We encourage readers to ensure that the protein activity and fold are retained upon reconstitution or refolding following heme extraction. Given that activity measurements are protein-specific, rather than outlining a specific procedure, we provide example data for the comparison of apo-IsdH activity prepared by MEK extraction (the original published method) and on-column heme extraction. We have previously reported a stopped-flow activity assay for apo-IsdH, which rapidly transfers iron(III) heme from iron(III)-hemoglobin. The reaction is monitored as a decrease in heme absorbance at 405 nm, as heme is transferred from hemoglobin. The heme transfer activity of IsdH F365Y/A369F prepared by MEK extraction or on-column heme extraction was compared in this way (Figure 6). Both heme extraction methods yielded apo-IsdH with virtually identical activities and fitted rate constants (inset, Figure 6), consistent with previously published values (Sjodt et al., 2018). This demonstrates viability of the newly described on-column method for preparing active apo-proteins with a similar high fidelity to those produced by more traditional procedures.
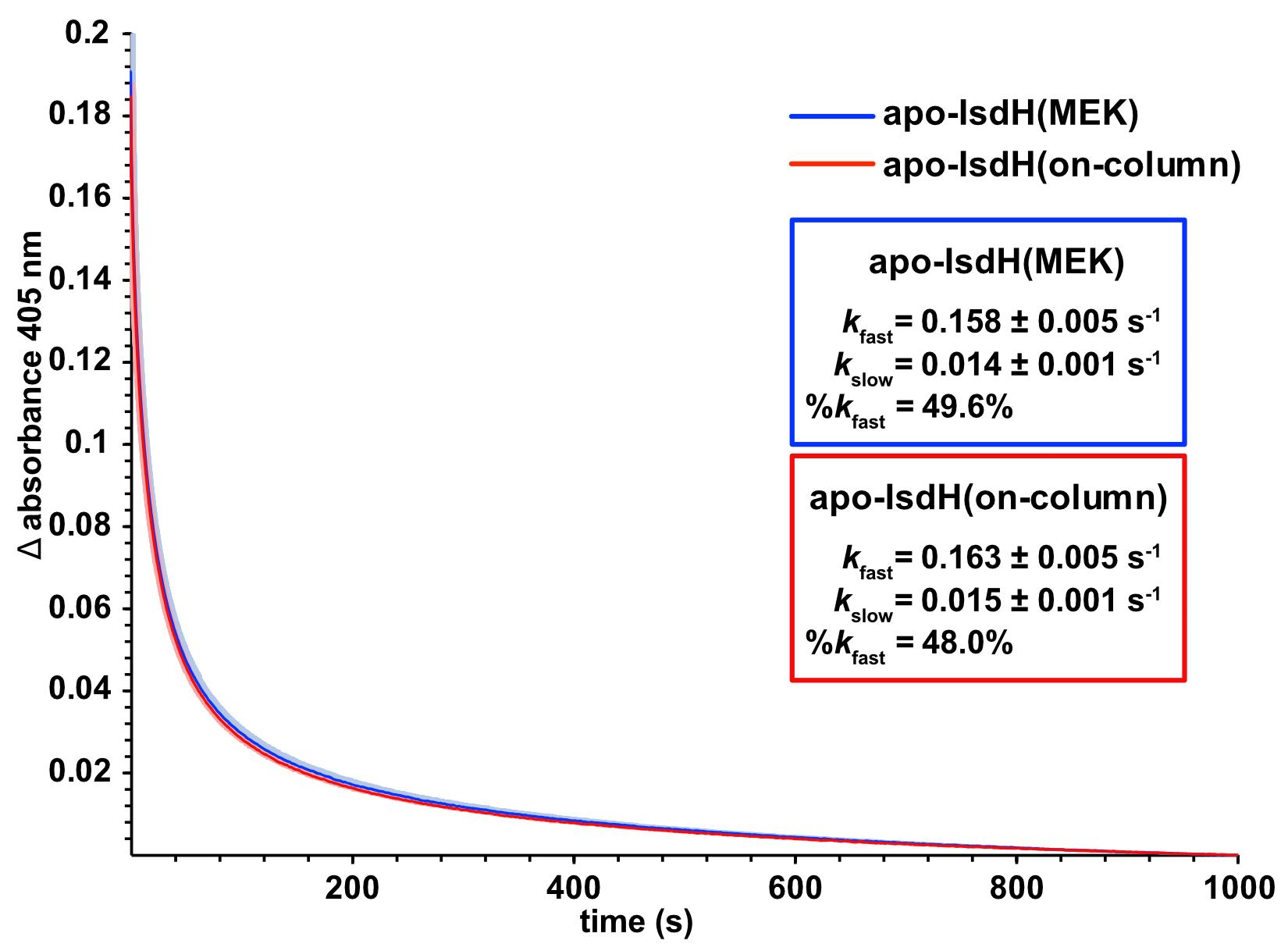
Figure 6. Assay to validate the activity of apo-IsdH prepared by MEK or on-column heme extraction. Stopped-flow UV/Vis spectroscopic measurements monitoring relative change in sample absorbance at 405 nm upon rapidly mixing 5 µM hemoglobin mutant 0.1 with 50 µM apo-IsdH F365Y/A369F in 20 mM NaH2PO4, 150 mM NaCl, 0.45 M sucrose, pH 7.5 at 25°C. The absorbance change was monitored using the short 0.2-cm pathlength setting on the SX20 Stopped-Flow Spectrophotometer (Applied PhotophysicsTM). Curves represent the mean of 4 technical replicates, with the standard error indicated by shading. Rate constants (inset) were calculated from two-exponential fitting of the curves using the equation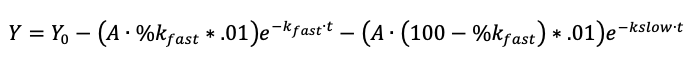 in GraphPad Prism.
in GraphPad Prism.
Notes
Save the lysate and all the washes that flow through the HisPurTM cobalt or nickel resin. If the binding capacity of the resin is exceeded, a portion of the protein of interest will flow through the gravity column without binding. Additionally, proteins with different properties may unexpectedly elute from the column during the wash steps.
The buffers may need to be optimized for the protein of interest. If the protein does not fully bind to the HisPurTM resin, reduction of the imidazole concentration in the equilibration buffer may remedy the issue. Nickel-nitriloacetic acid has a higher binding capacity and exhibits increased tolerance to reductants. Altering the denaturant concentration or changing the denaturant in the denaturing buffer and heme wash buffer may lead to more complete heme removal with certain proteins. Optimization of the alcohol concentration and identity (i.e., isopropanol) in the denaturing heme wash buffer may also lead to more complete heme removal with certain proteins.
The on-column heme removal protocol has been used successfully for the following histidine-tagged proteins: myoglobin, S. aureus IsdH, IsdB, IsdC, and IsdA. Proteins with intrinsic resistance to denaturation by chaotropes may require modifications to this method.
The heme Soret peak wavelength varies according to the iron oxidation state and ligand; thus, care should be taken to ensure that the heme content is monitored at the appropriate wavelength. For a list of the Soret peak wavelengths for hemoglobin and myoglobin, see Antonini and Brunori (1971).
SDS-PAGE analysis of any chromatography eluate determined to contain protein by A280 is recommended before combining the fractions, as it is possible that not all protein-containing fractions will contain the protein of interest.
Additional protein refolding procedures may be required after heme removal if the buffer exchange or dialysis is not sufficient. For an example refolding protocol, see Gennadios et al. (2009).
Running an additional size-exclusion chromatography step after methyl ethyl ketone extraction is recommended, as aggregates can form during the extraction and methyl ethyl ketone exhibits relatively high miscibility with aqueous buffer.
For acid-acetone precipitation, the centrifuge rotor ideally should be chilled to -20°C, but the coldest available rotor setting can be used if a centrifuge cannot cool to -20°C.
Prior preparations showed higher yields when the column was allowed to stand for ~5 min with the guanidine hydrochloride solution rather than allowing it to immediately flow through the column. We surmise that this could be due to the sudden introduction of a high ionic strength solution with chloride counterions that, while having low affinity for Ni2+-NTA and Co2+-NTA, may still be present in high enough concentrations to compete with the hexahistidine tag for binding of Ni2+-NTA or Co2+-NTA. Another reasonable explanation may be that guanidine hydrochloride solution solubilizes protein non-specifically adhered to the IMAC beads.
Performing the washes described in procedure section C, step 4 as four discrete washes yields more efficient heme removal than a single wash of the same volume (i.e., 4 × 50 ml vs. 1 × 200 ml).
This calculation makes the assumption that heme in the sample is bound to protein in its native coordination environment and its original ferric (iron III) oxidation state. After denaturing and renaturing extraction, this may no longer be the case, with non-native coordination modes and alternative oxidation states altering the expected spectral characteristics of the bound heme.
Heme itself has a non-negligible absorbance at 280 nm (ε280 = 18 ± 3 mM-1cm-1 in 20 mM NaH2PO4, 0.15 M NaCl, pH 7.5). Protein concentrations measured by A280 should be corrected for the absorbance contribution of bound heme, especially in cases where >10% of protein is heme-bound.
The HPLC method described here is run at a sufficiently high percentage of acetonitrile to denature IsdH; thus, separate apo-IsdH and heme peaks are observed in the reversed-phase chromatogram (Figure 5B). This may not be the case for all hemoproteins.
Under the analysis conditions given, we are able to reliably detect heme down to ~160 pmol (~100 ng).
Quantitation of the absolute heme and protein content in the sample requires experimental response curves plotting pmol sample injected versus absorbance integral at either 280 nm (for the protein backbone) or λSoret (for heme).
Recipes
Equilibration buffer
50 mM NaH2PO4 (pH 7.5)
300 mM NaCl
10 mM imidazole
Histidine-tagged Ulp1 SUMO protease solution
1 mg/ml histidine-tagged Ulp1 protease
50% v/v glycerol in equilibration buffer
High imidazole buffer
50 mM NaH2PO4 (pH 7.5)
300 mM NaCl
300 mM imidazole
MEK extraction buffer
50 mM Tris HCl (pH 6.0)
Recovery buffer
500 mM Tris HCl (pH 8.0)
Acid acetone
5 mM HCl in acetone
Denaturing buffer
50 mM NaH2PO4 (pH 7.5)
300 mM NaCl
10 mM imidazole
6 M guanidine HCl
Heme wash buffer
80% denaturing buffer
20% ethanol
Acknowledgments
We are thankful for funding support from the National Institutes of Health (AI52217); National Science Foundation (MCB-1716948); and the Department of Energy Office of Science, Office of Biological and Environmental Research program under Award Number DE-FC02-02ER63421. J.S was supported by a National Institutes of Health NIGMS-funded predoctoral fellowship (T32 GM136614).
Competing interests
The authors declare no competing interests.
References
- Antonini, E. and Brunori, M. (1971). Hemoglobin and myoglobin in their reactions with ligands. North-Holland Pub. Co. 21: pp436.
- Ascoli, F., Fanelli, M. R. and Antonini, E. (1981). Preparation and properties of apohemoglobin and reconstituted hemoglobins. Methods Enzymol 76: 72-87.
- Baker, H. M., Anderson, B. F. and Baker, E. N. (2003). Dealing with iron: Common structural principles in proteins that transport iron and heme.Proc Natl Acad Sci U S America 100(7): 3579-3583.
- Chien, S. C., Shoji, O., Morimoto, Y. and Watanabe, Y. (2017). Use of apomyoglobin to gently remove heme from a H2O2-dependent cytochrome P450 and allow its reconstitution.New J Chem 41(1): 302-307.
- Choby, J. E. and Skaar, E. P. (2016). Heme Synthesis and Acquisition in Bacterial Pathogens. J Mol Biol 428(17): 3408-3428.
- Ellis-Guardiola, K., Clayton, J., Pham, C., Mahoney, B. J., Wereszczynski, J. and Clubb, R. T. (2020). The Staphylococcus aureus IsdH Receptor Forms a Dynamic Complex with Human Hemoglobin that Triggers Heme Release via Two Distinct Hot Spots.J Mol Biol 432(4): 1064-1082.
- Fruk, L., Kuo, C. H., Torres, E. and Niemeyer, C. M. (2009). Apoenzyme Reconstitution as a Chemical Tool for Structural Enzymology and Biotechnology.Angewandte Chemie-International Edition 48(9): 1550-1574.
- Gennadios, H. A., Gonzalez, V., Di Costanzo, L., Li, A., Yu, F., Miller, D. J., Allemann, R. K. and Christianson, D. W. (2009). Crystal structure of (+)-delta-cadinene synthase from Gossypium arboreum and evolutionary divergence of metal binding motifs for catalysis. Biochemistry 48(26): 6175-6183.
- Huang, X. and Groves, J. T. (2018). Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins.Chem Rev 118(5): 2491-2553.
- Muller-Eberhard, U., Liem, H. H., Yu, C. A. and Gunsalus, I. C. (1969). Removal of heme from cytochrome P-450CAM by hemopexin and apomyoglobin associated with loss of P-450 hydroxylase activity. Biochem Biophys Res Commun 35(2): 229-235.
- Poulos, T. L. (2014). Heme enzyme structure and function. Chem Rev 114(7): 3919-3962.
- Reedy, C. J., Elvekrog, M. M. and Gibney, B. R. (2008). Development of a heme protein structure-electrochemical function database.Nucleic Acids Res 36(Database issue): D307-313.
- Rossi-Fanelli, A., Antonini, E. and Caputo, A. (1958). Pure Native Globin from Human Hemoglobin - Preparation and Some Physico-Chemical Properties.Biochimica Et Biophysica Acta 28(1): 221-221.
- Runyen-Janecky, L. J. (2013). Role and regulation of heme iron acquisition in gram-negative pathogens.Front Cell Infect Microbiol 3: 55.
- Samuel, P. P., Ou, W. C., Phillips, G. N., Jr. and Olson, J. S. (2017). Mechanism of Human Apohemoglobin Unfolding. Biochemistry 56(10): 1444-1459.
- Sjodt, M., Macdonald, R., Marshall, J. D., Clayton, J., Olson, J. S., Phillips, M., Gell, D. A., Wereszczynski, J. and Clubb, R. T. (2018). Energetics underlying hemin extraction from human hemoglobin by Staphylococcus aureus.J Biol Chem 293(18): 6942-6957.
- Teale, F. W. (1959). Cleavage of the haem-protein link by acid methylethylketone. Biochim Biophys Acta 35: 543.
Article Information
Copyright
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Ellis-Guardiola, K., Soule, J. and Clubb, R. T. (2021). Methods for the Extraction of Heme Prosthetic Groups from Hemoproteins. Bio-protocol 11(18): e4156. DOI: 10.21769/BioProtoc.4156.
Category
Biochemistry > Protein > Activity
Biochemistry > Protein > Isolation and purification
Biophysics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.